Please note that the date you requested in the address for this web page is not an actual date upon which a change occurred to this item of legislation. You are being shown the legislation from , which is the first date before then upon which a change was made.
ANNEXU.K.
IDENTIFICATION AND DETERMINATION OF GLYCEROL l-(4-AMINOBENZOATE) U.K.
A.IDENTIFICATIONU.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method will detect alpha-monoglyceryl 4-aminobenzoate (glycerol 1-(4-aminobenzoate). It will also detect ethyl 4-aminobenzoate (benzocaine INN) which may be present as an impurity.
2.PRINCIPLEU.K.
This identification is done by thin layer chromatography on silica gel with a fluorescent indicator and detection of the free primary amine group by formation of a diazo dye on the plate.
3.REAGENTSU.K.
All reagents should be of analytical purity.
3.1.Solvent mixture: cyclohexane/propan-2-ol/stabilized dichloromethane 48/64/9 (v/v/v).U.K.
3.2.Development solvent: petroleum ether (40-60)/benzene/acetone/ammonium hydroxide solution (minimum 25 % NH3): 35/35/35/1 (v/v/v/v).U.K.
3.3.
:
sodium nitrite: 1 g in 100 ml of 1 M hydrochloric acid (prepared immediately before use);
2-naphthol: 0,2 g in 100 ml of 1 M potassium hydroxide.
3.4.Standard solutions:U.K.
alpha-monoglyceryl 4-aminobenzoate: 0,05 g in 100 ml of mixed solvent 3.1;
ethyl 4-aminobenzoate: 0,05 g in 100 ml of mixed solvent 3.1.
3.5.Silica gel 60 F254 plates, 0,25 mm thick, 200 mm × 200 mm.U.K.
4.APPARATUSU.K.
4.1.Normal apparatus for thin layer chromatography.U.K.
4.2.Ultrasonic vibrator.U.K.
4.3.Millipore filter FH 0,5 μm or equivalent.U.K.
5.PROCEDUREU.K.
5.1. Sample preparation U.K.
Weigh 1,5 g of the product to be analyzed in a 10 ml stoppered graduated flask. Make up to the mark with the solvent 3.1. Stopper and leave for one hour at room temperature in an ultrasonic vibrator (4.2). Filter through a Millipore filter (4.3) and use the filtrate for chromatography.
5.2. Thin layer chromatography U.K.
Deposit 10 μl of sample solution (5.1) and of each standard solution (3.4) on the plate (3.5).
Develop the chromatogram to a height of 150 mm in a tank previously saturated with solvent 3.2. Allow the plate to dry at ambient temperature.
5.3. Development U.K.
5.3.1.Observe the plate under 254 nm UV light.U.K.
5.3.2.Spray the completely dried plate with the solution 3.3 (a).U.K.
Allow to dry at room temperature for 1 minute and immediately spray with the solution 3.3 (b).
Dry the plate in an oven at 60 oC. The spots appear an orange colour. Alpha-monoglyceryl 4-aminobenzoate: RF 0,07; ethyl 4-aminobenzoate: RF 0,55.
B.DETERMINATIONU.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method determines alpha monoglyceryl 4-aminobenzoate. It will also determine ethyl 4-aminobenzoate. It cannot determine more than 5 % (m/m) of alpha monoglyceryl 4-aminobenzoate and 1 % (m/m) of ethyl 4-aminobenzoate.
2.DEFINITIONU.K.
The alpha monoglyceryl 4-aminobenzoate and ethyl 4-aminobenzoate contents measured by this method are expressed as percentage by mass (% m/m) of the product.
3.PRINCIPLEU.K.
The product to be analyzed is suspended in methanol and after appropriate treatment of the sample it is determined by high-performance liquid chromatography (HPLC).
4.REAGENTSU.K.
All reagents should be of analytical purity and should be suitable for HPLC where appropriate.
4.1.Methanol.U.K.
4.2.Potassium dihydrogenorthophosphate (KH2PO4).U.K.
4.3.Zinc di(acetate) (Zn(CH3COO)2 · 2H2O).U.K.
4.4.Acetic acid 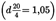 .U.K.
.U.K.
4.5.Tetrapotassium hexacyanoferrate, (K4(Fe(CN)6) · 3H2O).U.K.
4.6.Ethyl 4-hydroxybenzoate.U.K.
4.7.Alpha monoglyceryl 4-aminobenzoate.U.K.
4.8.Ethyl 4-aminobenzoate.U.K.
4.9.Phosphate buffer solution (0,02 M): dissolve 2,72 g of potassium dihydrogenorthophosphate (4.2) in one litre of water.U.K.
4.10.Eluant: phosphate buffer solution (4.9)/methanol (4.1) 61/39(v/v)U.K.
The composition of the mobile phase may be changed in order to achieve a resolution factor R ≥ 1,5.

where
=
retention times, in minutes, of the peaks,
=
peak widths at half height, in milimetres,
=
the chart speed, in millimetres per minute.
4.11.Stock solution of alpha-monoglyceryl 4-aminobenzoate: weigh accurately about 40 mg of alpha-monoglyceryl 4-aminobenzoate and introduce it into a 100 ml graduated flask. Dissolve in 40 ml of methanol (4.1). Make up to the mark with buffer solution (4.9) and mix.U.K.
4.12.Stock solution of ethyl 4-aminobenzoate: weigh accurately about 40 mg of ethyl 4-aminobenzoate and introduce it into a 100 ml graduated flask. Dissolve in 40 ml of methanol (4.1). Make up to the mark with the buffer solution (4.9) and mix.U.K.
4.13.Internal standard solution: weigh accurately about 50 mg of ethyl 4-hydroxybenzoate (4.6), transfer to a 100 ml standard flask, dissolve in 40 ml of methanol (4.1), make up to the mark with the buffer solution (4.9) and mix.U.K.
4.14.Standard solutions: prepare four standard solutions by dissolving in 100 ml eluant (4.10) according to the following table:U.K.
| a These values are given as an indication and correspond to the exact masses of 4.11, 4.12 and 4.13. | ||||||
| NB: These solutions may be prepared in a different way. | ||||||
| Standard Solution | Alpha-monoglyceryl 4-aminobenzoate | Ethyl 4-aminobenzoate | Ethyl 4-hydroxybenzoate | |||
|---|---|---|---|---|---|---|
| (μg/ml)a | ml (4.11)a | (μg/ml)a | ml (4.12) | (μg/ml)a | ml (4.13) | |
| I | 8 | 2 | 8 | 2 | 50 | 10 |
| II | 16 | 4 | 12 | 3 | 50 | 10 |
| III | 24 | 6 | 16 | 4 | 50 | 10 |
| IV | 40 | 10 | 20 | 5 | 50 | 10 |
4.15Carrez I solution: dissolve 26,5 g of tetrapotassium hexacyanoferrate (4.5) in water and make up to 250 ml.U.K.
4.16.Carrez II solution: dissolve 54,9 g of zinc di(acetate) (4.3) and 7,5 ml of acetic acid (4.4) in water and make up to 250 ml.U.K.
4.17.Merck Lichrosorb RP-18, or equivalent, with an average particle size of 5 μm.U.K.
5.APPARATUSU.K.
5.1.The usual laboratory equipment.U.K.
5.2.High-performance chromatography equipment with a variable wavelength UV detector and thermostatted chamber set at 45 oC.U.K.
5.3.Stainless-steel column: length: 250 mm; internal diameter: 4,6 mm; packing: Lichrosorb RP — 18 (4.17).U.K.
5.4.Ultrasonic bath.U.K.
6.PROCEDUREU.K.
6.1. Sample preparation U.K.
6.1.1.Weigh accurately about 1 g of sample into a 100 ml beaker and add 10 ml of methanol (4.1).U.K.
6.1.2.Place the beaker in the ultrasonic bath (5.4) for 20 minutes to produce a suspension. Transfer the suspension thus obtained quantitatively into a 100 ml standard flask with not more than 75 ml of eluant (4.10).U.K.
Add in succession 1 ml of Carrez I solution (4.15) and 1 ml of Carrez II solution (4.16) and mix after each addition. Make up to the mark with eluant (4.10), re-mix and filter through a pleated filter paper.
6.1.3.With a pipette, transfer 3,0 ml of the filtrate obtained in 6.1.2 and 5,0 ml of the internal standard solution (4.13) into a 50 ml standard flask. Make up to the mark with eluant (4.10) and mix. Use the solution thus obtained for carrying out the chromatography analysis described in 6.2.U.K.
6.2. Chromatography U.K.
6.2.1.Adjust the flow rate of the mobile phase (4.10) to 1,2 ml/min and set the column temperature to 45 oC.U.K.
6.2.2.Set the detector (5.2) to 274 nm.U.K.
6.2.3.With a microsyringe, inject at least two times 20 μl of solution (6.1.3) into the chromatograph and measure the areas of the peaks.U.K.
6.3. Calibration curve U.K.
6.3.1.Inject 20 μl of each of the standard solutions (4.14) and measure the peak area.U.K.
6.3.2.For each concentration calculate the ratio between the peak areas of alpha-monoglyceryl 4-aminobenzoate and the peak areas of the internal standard. Plot this ratio on the abscissa and on the ordinate the ratio of the corresponding masses.U.K.
6.3.3.Proceed in the same manner for ethyl 4-hydroxybenzoate.U.K.
7.CALCULATIONU.K.
7.1.From the calibration curve obtained in 6.3 read off the mass ratios (RP1, RP2) corresponding to the ratios between the areas of the peaks calculated in 6.2.3 whereU.K.
=
mass of alpha-monoglyceryl 4-aminobenzoate/mass of ethyl 4-hydroxybenzoate,
=
mass of ethyl 4-aminobenzoate/mass of ethyl 4-hydroxybenzoate.
7.2.From the mass ratios obtained in this way calculate the alpha-monoglyceryl 4-aminobenzoate and ethyl 4-aminobenzoate contents, as mass percentages (% m/m) with the formulae:U.K.


=
quantity of ethyl 4-hydroxybenzoate (internal standard) weighed, in milligrams, in 4.12,
=
quantity of sample, in grams, weighed in 6.1.1.
8.REPEATABILITY(1) U.K.
8.1.For a 5 % (m/m) content of alpha-monoglyceryl 4-aminobenzoate, the difference between the results of two parallel determinations carried out on the same sample must not exceed 0,25 %.U.K.
8.2.For a 1 % (m/m) content of ethyl 4-aminobenzoate the difference between the results of two parallel determinations carried out on the same sample must not exceed 0,10 %.U.K.
9.NOTESU.K.
9.1.Before carrying out an analysis, check whether the sample contains substances likely to overlap with the peak of the internal standard (ethyl 4-aminobenzoate) on the chromatogram.U.K.
9.2.In order to check the absence of any interference, repeat the determination by changing the proportion of methanol in the mobile phase by 10 % relative.U.K.
DETERMINATION OF CHLOROBUTANOL U.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method is suitable for the determination of chlorobutanol (INN) up to a maximum concentration of 0,5 % (m/m) in any cosmetic product, except aerosols.
2.DEFINITIONU.K.
The content of chlorobutanol measured by this method is expressed as percentage by mass (% m/m) of product.
3.PRINCIPLEU.K.
After appropriate treatment of the product to be analyzed the determination is done by gas chromatography using 2,2,2-trichloroethanol as the internal standard.
4.REAGENTSU.K.
All the reagents should be of analytical purity.
4.1.Chlorobutanol (1,1,1-trichloro-2-methylpropan-2-ol).U.K.
4.2.2,2,2-Trichloroethanol.U.K.
4.3.Absolute ethanol.U.K.
4.4.Standard solution of chlorobutanol: 0,025 g in 100 ml ethanol (4.3) (m/v).U.K.
4.5.Standard solution of 2,2,2-trichloroethanol: 4 mg in 100 ml ethanol (4.3) (m/v).U.K.
5.APPARATUSU.K.
5.1.Normal laboratory equipment.U.K.
5.2.Gas chromatograph with electron detector, Ni 63.U.K.
6.PROCEDUREU.K.
6.1. Preparation of sample U.K.
Weigh accurately between 0,1 and 0,3 g (p g) of the sample. Place in 100 ml volumetric flask. Dissolve it in ethanol (4.3), add 1 ml of the internal standard solution (4.5) and make up to the mark with ethanol (4.3).
6.2.Gas chromatography conditionsU.K.
6.2.1.The operating conditions must yield a resolution factor R ≥ 1,5.U.K.
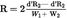
Where
=
retention times, in minutes, of the peaks,
=
peak widths at half height, in millimetres,
=
the chart speed, in millimetres per minute.
6.2.2.As examples, the following operating conditions provide the required resolution:U.K.
| Column | I | II |
|---|---|---|
| Material | Glass | Stainless steel |
| Length | 1,80 m | 3 m |
| Diameter | 3 mm | 3 mm |
| Stationary phase | 10 % Carbowax 20 M TPA on Gaschrom Q 80-100 mesh | 5 % OV 17 on Chromosorb WAW DMCS 80-100 mesh |
| Conditioning | 2 to 3 days at 190 oC | |
| Temperature: | ||
| — injector | 200 oC | 150 oC |
| — column | 150 oC | 100 oC |
| — detector | 200 oC | 150 oC |
| Carrier gas | Nitrogen | Argon/methane (95/5 v/v) |
| Flowrate | 35 ml/min | 35 ml/min |
6.3. Standard curve U.K.
Using five 100 ml volumetric flasks, add 1 ml of the standard solution (4.5) and 0,2, 0,3, 0,4, 0,5, and 0,6 ml of solution 4.4 respectively, and make up to the mark with ethanol (4.3) and mix. Inject 1 μl of each of these solutions into the chromatograph in accordance with the operating conditions described in 6.2.2 and construct a calibration curve by plotting as the abscissa the ratio of the mass of chlorobutanol to that of 2,2,2-trichloroethanol and as the ordinate the ratio of the corresponding peak areas.
6.4.Inject 1 μl of solution obtained in 6.1 and proceed according to the conditions described in 6.2.2U.K.
7.CALCULATIONU.K.
7.1.Calculate from the standard curve (6.3) the quantity ‘a’ expressed as μg of chlorobutanol, in the solution 6.1.U.K.
7.2.The content of chlorobutanol in the sample is calculated according to the formula:U.K.

8.REPEATABILITY(2) U.K.
For a chlorobutanol content of 0,5 % (m/m) the difference between the results of two determinations in parallel carried out on the same sample should not exceed 0,01 %.
Note U.K.
If the result is equal to or exceeds the maximum permitted concentration it is necessary to check the absence of interferences.
IDENTIFICATION AND DETERMINATION OF QUININE U.K.
A.IDENTIFICATIONU.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method is intended to detect the presence of quinine in shampoo and hair lotions.
2.PRINCIPLEU.K.
Identification is done by thin layer chromatography on silica gel. Detection of quinine is by the blue fluorescence of quinine in acidic conditions at 360 nm.
For further confirmation, the fluorescence can be eliminated by bromine vapours, and ammonia vapours will cause a yellowish fluorescence to appear.
3.REAGENTSU.K.
All reagents should be of analytical purity.
3.1.Silica gel plates, without fluorescence indicators, 0,25 mm thick, 200 mm × 200 mmU.K.
3.2.Developing solvent: toluene /diethyl ether /dichloromethane /diethylamine /20/20/20/8 (v/v/v/v).U.K.
3.3.Methanol.U.K.
3.4.Sulphuric acid (96 %;  ).U.K.
).U.K.
3.5.Diethyl ether.U.K.
3.6.Developing agent: carefully add 5 ml of sulphuric acid (3.4) to 95 ml of diethyl ether (3.5) in a cooled container.U.K.
3.7.Bromine.U.K.
3.8.Ammonium hydroxide solution (28 %;  ).U.K.
).U.K.
3.9.Quinine, anhydrous.U.K.
3.10.Standard solution: weigh accurately about 100,0 mg of anhydrous quinine (3.9) into a standard flask and dissolve in 100 ml of methanol (3.3).U.K.
4.APPARATUSU.K.
4.1.Normal equipment for thin layer chromatography.U.K.
4.2.Ultrasonic bath.U.K.
4.3.Millipore filter, FH 0,5 ƒm or equivalent with suitable filtration equipment.U.K.
5.PROCEDUREU.K.
5.1. Preparation of the sample U.K.
Weigh accurately a quantity of the sample which may contain approximately 100 mg of quinine into a 100 ml standard flask, dissolve and make up to the mark with methanol (3.3).
Stopper the flask and leave for one hour at room temperature in an ultrasonic vibrator (4.2). Filter (4.3) and use the filtrate for the chromatography.
5.2. Thin layer chromatography U.K.
Deposit 1,0 μl of standard solution (3.10) and 1,0 μl of sample solution (5.1) on the silica gel plate (3.1). Develop the chromatogram over a distance of 150 mm using solvent 3.2. in a tank previously saturated with solvent (3.2).
5.3. Development U.K.
5.3.1.Dry the plate at room temperature.U.K.
5.3.21.Spray with reagent 3.6.U.K.
5.3.3.Leave the plate to dry for one hour at room temperature.U.K.
5.3.4.Observe under the light from a UV lamp adjusted to a wavelength of 360 nm. Quinine appears as a fluorescent intense blue spot.U.K.
By way of example the table below gives the values of the RF of the main alkaloids related to quinine when developed with solvent 3.2.
| Alkaloid | RF |
|---|---|
| Quinine | 0,20 |
| Quinidine | 0,29 |
| Cinchonine | 0,33 |
| Cinchonidine | 0,27 |
| Hydroquinidine | 0,17 |
5.3.5.For further confirmation that quinine is present, the plate is exposed for approximately one hour to bromine vapour (3.7). The fluorescence disappears. When the same plate is exposed to ammonia vapour (3.8), the spots reappear with a brown colour, and when the plate is again examined under UV light at 360 nm a yellowish fluorescence can be observed.U.K.
Detection limit: 0,1 μg of quinine.
B.DETERMINATIONU.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method describes the determination of quinine. It may be used to determine the maximum permitted concentration of 0,5 % (m/m) in shampoos and 0,2 % in hair lotions.
2.DEFINITIONU.K.
The quinine content determined by this method is expressed as a percentage by mass (% m/m) of the product.
3.PRINCIPLEU.K.
After appropriate treatment of the product to be analyzed the determination is done by high-performance liquid chromatography (HPLC).
4.REAGENTSU.K.
All reagents should be of analytical purity and suitable for HPLC.
4.1.Acetonitrile.U.K.
4.2.Potassium dihydrogenorthophosphate (KH2PO4).U.K.
4.3.Orthophosphoric acid (85 %;  ).U.K.
).U.K.
4.4.Tetramethylaminium bromide.U.K.
4.5.Quinine, anhydrous.U.K.
4.6.Methanol.U.K.
4.7.Orthophosphoric acid solution (0,1 M): weigh 11,53 g of orthophosphoric acid (4.3) and dissolve in 1 000 nl of water in a graduated flask.U.K.
4.8.Potassium dihyrogenorthophosphate solution (0,1 M): weigh 13,6 g of potassium dihydrogenorthophosphate (4.2) and dissolve in 1 000 ml of water in a graduated flask.U.K.
4.9.Tetramethylammonium bromide solution: dissolve 15,40 g of tetramethylammonium bromide (4.4) in 1 000 ml of water in a graduated flask.U.K.
4.10Eluant: orthophosphoric acid (4.7) /potassium dihydrogenorthophosphate (4.8) /tetramethylammonium bromide (4.9)/water/acetonitrile (4.1) 10/50/100/340/90 (v/v/v/v/v).U.K.
The composition of this mobile phase may be changed in order to achieve a resolution factor R ≥ 1,5.

where
=
retention times, in minutes, of the peaks,
=
peak widths at half height, in millimetres,
=
the chart speed, in millimetres per minute.
4.11.Silica treated with octadecylsilane, 10 μm.U.K.
4.12.Standard solutions: weigh accurately approximately 5,0, 10,0, 15,0 and 20,0 mg respectively of quinine anhydrous (4.5) into a set of 100 ml standard flasks. Make up to the mark with methanol (4.6) and shake the contents of the flasks until the quinine dissolves. Filter each sample through a 0,5 μm filter.U.K.
5.APPARATUSU.K.
5.1.Usual laboratory equipment.U.K.
5.2.Ultrasonic bath.U.K.
5.3.High-performance liquid chromatography equipment with a variable wavelength detector.U.K.
5.4.Column: length: 250 mm; internal diameter: 4,6 mm; filling: silica (4.11).U.K.
5.5.Millipore filter FH 0,5 μm, or equivalent, with suitable filtration apparatus.U.K.
6.PROCEDUREU.K.
6.1. Sample preparation U.K.
Weigh accurately into a 100 ml standard flask a quantity of the product sufficient to contain 10,0 mg of anhydrous quinine, add 20 ml of methanol (4.6) and place the flask in an ultrasonic bath (5.2) for 20 minutes. Make up to the mark with methanol (4.6). Mix the solution and then filter an aliquot (5.5).
6.2. Chromatography U.K.
Flowrate: 1,0 ml/min.
Detector wavelength (5.3): 332 nm.
Injection volume: 10 μl of filtered solution (6.1).
Measurement: peak area.
6.3. Calibration curve U.K.
Inject at least three times 10,0 μl of each reference solution (4.12), measure the area of the peaks, and calculate the average area at each concentration.
Produce the calibration curve and verify that it is rectilinear.
7.CALCULATIONU.K.
7.1.From the calibration curve (6.3) determine the quantity in μg of anhydrous quinine present in the volume injected (6.2).U.K.
7.2.The concentration of anhydrous quinine in the sample, as a percentage by mass (% m/m), is obtained by the following formula:U.K.

where
is the quantity, in micrograms, of anhydrous quinine determined in the 10 microlitres of the filtered solution (6.1).
is the mass of the sample in grams (6.1).
8.REPEATABILITY(3) U.K.
For an anhydrous quinine content of 0,5 % (m/m), the difference between the results of two determinations performed in parallel on the same sample must not exceed 0,02 %.
For an anhydrous quinine content of 0,2 % (m/m), the difference between the results of two determinations performed in parallel on the same sample must not exceed 0,01 %.
IDENTIFICATION AND DETERMINATION OF INORGANIC SULPHITES AND HYDROGEN SULPHITES SCOPE AND FIELD OF APPLICATIONU.K.
The method describes the identification and determination of inorganic sulphites and hydrogen sulphites in cosmetic products. It is only applicable to products that have an aqueous or alcoholic phase and for concentrations up to 0,2 % sulphur dioxide.
A.IDENTIFICATIONU.K.
1.PRINCIPLEU.K.
The sample is heated in hydrochloric acid, and sulphur dioxide liberated is identified either by its odour or its effect on an indicator paper.
2.REAGENTSU.K.
All reagents should be of analytical purity.
2.1.Hydrochloric acid (4 M).U.K.
2.2.Potassium iodate starch paper or other suitable alternative.U.K.
3.APPARATUSU.K.
3.1.Normal laboratory equipment.U.K.
3.2.Flask (25 ml) fitted with a short reflux condenser.U.K.
4.PROCEDUREU.K.
4.1.Place about 2,5 g of sample in the flask (3.2) with 10 ml of hydrochloric acid (2.1).U.K.
4.2.Mix and heat to boiling.U.K.
4.3.Test for the emission of sulphur dioxide either by smell or indicator paper (2.2).U.K.
B.DETERMINATIONU.K.
1.DEFINITIONU.K.
The sulphite or hydrogen sulphite content of the sample as determined by the method is expressed as percentage by mass of sulphur dioxide.
2.PRINCIPLEU.K.
After acidification of the sample, sulphur dioxide liberated is distilled into a solution of hydrogen peroxide. Sulphuric acid formed is titrated against a standardized sodium hydroxide solution.
3.REAGENTSU.K.
All reagants should be of analytical purity.
3.1.Hydrogen peroxide 0,2 % (m/v). Prepare daily.U.K.
3.2.Orthophosphoric acid (  ).U.K.
).U.K.
3.3.Methanol.U.K.
3.4.Sodium hydroxide (0,01 M) standardized solution.U.K.
3.5.Nitrogen.U.K.
3.6.Indicator: mixture 1: 1 (v/v) of methyl red (0,03 % m/v in ethanol) and methylene blue (0,05 % m/v in ethanol). Filter the solution.U.K.
4.APPARATUSU.K.
4.1.Normal laboratory equipment.U.K.
4.2.Distillation apparatus (see figure).U.K.
5.PROCEDUREU.K.
5.1.Weigh accurately about 2,5 g of sample into the distillation flask A (see figure).U.K.
5.2.Add 60 ml of water and 50 ml of methanol (3.3) and mix.U.K.
5.3.Place 10 ml of hydrogen peroxide (3.1), 60 ml of water and a few drops of indicator (3.6) in the distillation receiver D (see figure). Add a few drops of sodium hydroxide (3.4) until the indicator turns green.U.K.
5.4.Repeat 5.3 for the wash bottle E (see figure).U.K.
5.5.Assemble the apparatus and adjust the nitrogen (3.5) flow to about 60 bubbles per minute.U.K.
5.6.Run 15 ml of orthophosphoric acid (3.2) from the funnel into the distillation flask A.U.K.
5.7.Heat rapidly to boiling and then simmer gently for a total time of 30 minutes.U.K.
5.8.Detach the distillation receiver D. Rinse the tube and then titrate with sodium hydroxide solution (3.4) until the indicator turns green (3.6).U.K.
6.CALCULATIONU.K.
Calculate the content of sulphite or hydrogen sulphite by mass in the sample by the expresssion:

where
=
molar concentration of sodium hydroxide solution (3.4),
=
volume of sodium hydroxide (3.4) required for titration (5.8), in millilitres,
=
mass of sample (5.1) in grams.
7.REPEATABILITY(4) U.K.
For a content of 0,2 % m/m of sulphur dioxide the difference between two parallel determinations done on the same sample should not be greater than 0,006 %.
Sulphurdioxide distillation apparatus according to Tanner All dimensions in mmU.K.

IDENTIFICATION AND DETERMINATION OF CHLORATES OF THE ALKALI METALS SCOPE AND FIELD OF APPLICATIONU.K.
The method describes the identification and determination of chlorates in toothpastes and other cosmetic products.
A.IDENTIFICATIONU.K.
1.PRINCIPLEU.K.
Chlorates are separated from other halates by thin layer chromatography and identified by the oxidation of iodide to form iodine.
2.REAGENTSU.K.
All reagents should be of analytical purity.
2.1.Reference solutions: aqueous solutions of potassium chlorate, bromate and iodate (0,2 % m/v) prepared freshly.U.K.
2.2.Development solvent: ammonia solution (28% m/v) acetone/butanol (60/130/30 v/v/v).U.K.
2.3.Potassium iodide, aqueous solution (5 % m/v).U.K.
2.4.Starch solution (1 to 5 % m/v).U.K.
2.5.Hydrochloric acid (1 M).U.K.
2.6.Ready-for-use cellulose thin-layer plates (0,25 mm).U.K.
3.APPARATUSU.K.
Normal equipment for thin layer chromatography.
4.PROCEDUREU.K.
4.1.Extract about 1 g of the sample with water, filter, and dilute to about 25 ml.U.K.
4.2.Deposit 2 (μl on the plate (2.6) of the solution (4.1) together with 2 μl aliquots of each of the three reference solutions (2.1).U.K.
4.3.Place the plate in a tank and develop by ascending chromatography about three-quarters of the length of the plate (2.6) with solvent 2.2.U.K.
4.4.Remove from the tank and allow the solvent to evaporate. (NB: This may take up to two hours.)U.K.
4.5.Spray the plate with potassium iodide (2.3) and allow to dry for about five minutes.U.K.
4.6.Spray the plate with starch solution (2.4) and allow to dry for about five minutes.U.K.
4.7.Spray the plate with hydrochloric acid (2.5).U.K.
5.EVALUATIONU.K.
If the chlorate is present a blue spot (possibly a brown spot) will appear after half an hour with RF value approximately 0,7 to 0,8.
| Halates | RF |
|---|---|
| Iodate | 0 to 0,2 |
| Bromate | 0,5 to 0,6 |
| Chlorate | 0,7 to 0,8 |
It should be noted that bromates and iodates give immediate reaction. Care should be taken not to confuse spots from bromates and chlorates.
B.DETERMINATIONU.K.
1.DEFINITIONU.K.
The chlorate content of the sample determined by this method is expressed as percentage by mass of chlorate.
2.PRINCIPLEU.K.
Chlorate is reduced by zinc powder under acid conditions. The formed chloride is measured by potentiometric titration using a silver nitrate solution. A similar determination before reduction permits the possible presence of halides.
3.REAGENTSU.K.
All reagents should be of analytical purity.
3.1.Acetic acid, 80 % (m/m).U.K.
3.2.Zinc powder.U.K.
3.3.Silver nitrate standard solution (0,1 M).U.K.
4.APPARATUSU.K.
4.1.Normal laboratory equipment.U.K.
4.2.Potentiometer equipped with a silver indicator electrode.U.K.
5.PROCEDUREU.K.
5.1. Sample preparation U.K.
Weigh accurately a quantity ‘m’ of approximately 2 g in centrifuge tube. Add about 15 ml acetic acid (3.1) and mix carefully. Wait 30 minutes and centrifuge for 15 minutes at 2 000 rev/min. Transfer the supernatant solution to a 50 ml volumetric flask. Repeat centrifuging twice by adding 15 ml acetic acid (3.1) to the residue. Collect the solution containing chlorate in the same volumetric flask. Fill to the mark with acetic acid (3.1).
5.2. Reduction of chlorate U.K.
Take 20 ml of solution 5.1 and add 0,6 g of zinc powder (3.2). Bring to the boil in a flask fitted with a condenser tube. After 30 minutes boiling, cool and filter. Rinse the flask with water. Filter and combine the filtrate with the rise's.
5.3. Determination of chloride U.K.
Titrate 20 ml solution 5.2 with silver nitrate (3.3) by using the potentiometer (4.2). Titrate in the same way 20 ml of solution 5.1 with silver nitrate (3.3).
NB: If the product contains bromine or iodine derivatives which can release bromides or iodides after reduction, the titration curve will have several inflexion points. In this case the volume of the titrated solution (3.3) corresponding to chloride is the difference between the last and the penultimate inflexion points.
6.CALCULATIONU.K.
The content of chlorate of the sample (% m/m) is calculated by the formula:

where
=
volume in millilitres, of silver nitrate solution (3.3) used to titrate solution 5.2,
=
volume, in millilitres, of silver nitrate solution (3.3) used to titrate 20 millilitres of solution 5.1,
=
molality of silver nitrate standard solution (3.3),
=
mass of sample, in grams.
7.REPEATABILITY(5) U.K.
For a chlorate content of 3 to 5 % m/m the difference between the results of two determinations carried out in parallel on the same sample should not exceed 0,07 % m/m.
IDENTIFICATION AND DETERMINATION OF SODIUM IODATE SCOPE AND FIELD OF APPLICATIONU.K.
The method describes the procedure for identifying and determining rinse of cosmetic products containing sodium iodate.
A.IDENTIFICATIONU.K.
1.PRINCIPLEU.K.
Sodium iodate is separated from other halates by thin layer chromatography and identified by the oxidation of iodide to form iodine.
2.REAGENTSU.K.
All reagents should be of analytical purity.
2.1.Reference solutions. Aqueous solutions of potassium chlorate, bromate and iodate (0,01 % m/v) prepared freshly.U.K.
2.2.Development solvent.U.K.
Ammonia solution (28 % m/v) /acetone /butanol (60/130/30 v/v/v).
2.3.Potassium iodide aqueous solution (5 % m/v).U.K.
2.4.Starch solution (1 to 5 % m/v).U.K.
2.5.Hydrochloric acid (1 M).U.K.
3.APPARATUSU.K.
3.1.Ready-for-use cellulose thin-layer chromatography (0,25 mm) plates.U.K.
3.2.Normal equipment for thin layer chromatography.U.K.
4.PROCEDUREU.K.
4.1.Extract about 1 g of the sample with water, filter, and dilute to about 10 ml.U.K.
4.2.Deposit 2 µl of this solution onto the base line of the plate (3.1) together with 2 µl aliquots of each of the three reference solutions (2.1).U.K.
4.3.Place the plate in a tank and develop by ascending chromatography about three-quarters of the length of the plate with solvent (2.2).U.K.
4.4.Remove the plate from the tank and allow the solvent to evaporate at ambient temparature (NB: this may take up to two hours).U.K.
4.5.Spray the plate with potassium iodide (2.3) and allow to dry for about five minutes.U.K.
4.6.Spray with starch (2.4) and allow to dry for about five minutes.U.K.
4.7.Finally spray with hydrochloric (2.5).U.K.
5.EVALUATIONU.K.
If iodate is present a blue spot (the colour may be brown or become brown on standing) will appear immediately with an RF value approximately 0 to 0,2.
It should be noted that bromates give immediate reactions at RF values approximately at 0,5 to 0,6 and chlorates, after about 30 minutes, at RF values of 0,7 to 0,8 respectively.
B.DETERMINATIONU.K.
1.DEFINITIONU.K.
The sodium iodate content as determined by this method is expressed as a percentage by mass of sodium iodate.
2.PRINCIPLEU.K.
Sodium iodate is dissolved in water and determined by means of high-performance liquid chromatography, using in series, a reverse-phase C18 column and an anion-exchange column.
3.REAGENTSU.K.
All reagents should be of analytical purity and especially suitable for high-performance liquid chromatography (HPLC).
3.1.Hydrochloric acid (4 M).U.K.
3.2.Sodium sulphite aq, 5 % m/v.U.K.
3.3.Sodium iodate stock solution.U.K.
Prepare a stock solution containing 50 mg sodium iodate per 100 ml water.
3.4.Potassium dihydrogenorthophosphate.U.K.
3.5.Disodium hydrogenorthophosphate · 2H2O.U.K.
3.6.HPLC mobile phase: dissolve 3,88 g potassium dihydrogenorthophosphate (3.4) and 1,19 g disodium hydrogenorthophosphate · 2H2O (3.5) in 1 litre water.U.K.
The pH of the resulting solution is 6.2.
3.7.Universal indicator paper, pH 1-11.U.K.
4.APPARATUSU.K.
4.1.Ordinary laboratory apparatus.U.K.
4.2.Circular filter paper, diameter 110 mm, Schleicher and Schüll No 575, or equivalent.U.K.
4.3.High-performance liquid chromatograph with a variable wavelength detector.U.K.
4.4.Columns: length: 120 mm; internal diameter: 4,6 mm; number: two connected in series; first column — Necleosil R 5 C18 or equivalent; second column — Vydac (tm)-301-SB or equivalent.U.K.
5.PROCEDUREU.K.
5.1. Sample preparation U.K.
5.1.1. Fluid samples (shampoos) U.K.
Weigh accurately a test portion of approximately 1,0 g sample in a 10 ml glass stoppered calibrated tube or measuring flask.
Fill up to the mark with water and mix.
If necessary, filter the solution.
Determine the iodate in the solution by means of HPLC as described in section 5.2.
5.1.2. Solid samples (soap) U.K.
Finely divide part of the sample and weigh accurately a test portion of approximately 1,0 g into a 100 ml glass stoppered measuring cylinder. Fill up to 50 ml with water and shake vigorously for one minute. Centrifuge and filter through a filter paper (4.1) or allow the mixture to stand for at least one night.
Shake the jellylike solution vigorously and filter it through a filter paper (4.1).
Determine the iodate in the filtrate by means of HPLC as described in section 5.2.
5.2. Chromatography U.K.
Flowrate: 1 ml/min.
Detector wavelength (4.2): 210 nm.
Injection volume: 10 µl.
Measurement: peak area.
5.3. Calibration U.K.
Pipette respectively 1,0, 2,0, 5,0, 10,0 and 20,0 ml of the sodium iodate stock solution (3.3) into 50 ml volumetric flasks. Fill to the mark and mix.
The solutions thus obtained, contain 0,01, 0,02, 0,05, 0,10 and 0,20 mg sodium iodate per ml respectively.
Inject a 10 µl portion of each standard iodate solution into the liquid chromatograph (4.2) and obtain a chromatogram. Determine the peak area for iodate and plot a curve relating the peak area to the sodium iodate concentration.
6.CALCULATIONU.K.
Calculate the sodium iodate content, in percentage by mass (% m/m), using the formula:

where
is the mass, in grams, of the test portion (5.1),
is the total volume of the sample solution, in millilitres, obtained as described in 5.1,
is the concentration, in milligrams per millilitre of sodium iodate, obtained from the calibration curve (5.3).
7.REPEATABILITY(6) U.K.
For a sodium iodate content of 0,1 % (m/m) the difference between the results of two parallel determinations carried out on the same sample must not exceed 0,002 %.
8.CONFIRMATIONU.K.
8.1. Principle U.K.
In an acidified solution of a cosmetic product, iodate (IO3-) is reduced to iodide (I—) by sulphite and the resulting solution is investigated by means of HPLC. If a peak having a retention time corresponding to the retention time of iodate disappears after treatment with sulphite, the original peak can most probably be attributed to iodate.
8.2. Procedure U.K.
Pipette a 5 ml portion of the sample solution obtained as described in section 5.1 into a conical flask.
Adjust the pH of the solution to a value of 3 or lower with hydrochloric acid (3.1); universal indicator paper (3.7).
Add three drops of sodium sulphite solution (3.2) and mix.
Inject a 10 µl portion of the solution into the liquid chromatograph (4.2).
Compare this chromatogram with the chromatogram obtained as described in paragraph 5 for the same sample.
ISO 5725.
ISO 5725.
ISO 5725.
ISO 5725.
ISO 5725.
ISO 5725.