- Y Diweddaraf sydd Ar Gael (Diwygiedig)
- Pwynt Penodol mewn Amser (08/11/2018)
- Gwreiddiol (Fel y’i mabwysiadwyd gan yr UE)
Commission Implementing Decision (EU) 2019/417Dangos y teitl llawn
Commission Implementing Decision (EU) 2019/417 of 8 November 2018 laying down guidelines for the management of the European Union Rapid Information System ‘RAPEX’ established under Article 12 of Directive 2001/95/EC on general product safety and its notification system (notified under document C(2018) 7334)
You are here:
- Penderfyniadau yn deillio o’r UE
- 2019 No. 417
- Whole Decision
- Blaenorol
- Nesaf

Pa Fersiwn
Nodweddion Uwch
- Dangos Graddfa Ddaearyddol(e.e. Lloegr, Cymru, Yr Alban aca Gogledd Iwerddon)
- Dangos Llinell Amser Newidiadau
Rhagor o Adnoddau
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


Mae hon yn eitem o ddeddfwriaeth sy’n deillio o’r UE
Mae unrhyw newidiadau sydd wedi cael eu gwneud yn barod gan y tîm yn ymddangos yn y cynnwys a chyfeirir atynt gydag anodiadau.Ar ôl y diwrnod ymadael bydd tair fersiwn o’r ddeddfwriaeth yma i’w gwirio at ddibenion gwahanol. Y fersiwn legislation.gov.uk yw’r fersiwn sy’n weithredol yn y Deyrnas Unedig. Y Fersiwn UE sydd ar EUR-lex ar hyn o bryd yw’r fersiwn sy’n weithredol yn yr UE h.y. efallai y bydd arnoch angen y fersiwn hon os byddwch yn gweithredu busnes yn yr UE. EUR-Lex Y fersiwn yn yr archif ar y we yw’r fersiwn swyddogol o’r ddeddfwriaeth fel yr oedd ar y diwrnod ymadael cyn cael ei chyhoeddi ar legislation.gov.uk ac unrhyw newidiadau ac effeithiau a weithredwyd yn y Deyrnas Unedig wedyn. Mae’r archif ar y we hefyd yn cynnwys cyfraith achos a ffurfiau mewn ieithoedd eraill o EUR-Lex. The EU Exit Web Archive legislation_originated_from_EU_p3
Status:
Point in time view as at 08/11/2018.
Changes to legislation:
There are currently no known outstanding effects for the Commission Implementing Decision (EU) 2019/417.
Changes to Legislation
Revised legislation carried on this site may not be fully up to date. At the current time any known changes or effects made by subsequent legislation have been applied to the text of the legislation you are viewing by the editorial team. Please see ‘Frequently Asked Questions’ for details regarding the timescales for which new effects are identified and recorded on this site.
Commission Implementing Decision (EU) 2019/417
of 8 November 2018
laying down guidelines for the management of the European Union Rapid Information System ‘RAPEX’ established under Article 12 of Directive 2001/95/EC on general product safety and its notification system
(notified under document C(2018) 7334)
THE EUROPEAN COMMISSION,
Having regard to the Treaty on the Functioning of the European Union,
Having regard to Directive 2001/95/EC of the European Parliament and of the Council of 3 December 2001 on general product safety(1), and in particular the third subparagraph of Article 11(1) and point 8 of Annex II thereof,
Having regard to Regulation (EC) No 765/2008 of the European Parliament and of the Council of 9 July 2008, setting out the requirements for accreditation and market surveillance on the marketing of products and repealing Regulation (EEC) No 339/93(2),
After consulting the Advisory Committee, set up by Article 15 of Directive 2001/95/EC,
Whereas:
(1) Article 12 of Directive 2001/95/EC establishes a European Union Rapid Information System (‘RAPEX’) for the rapid exchange of information between the Member States and the Commission on measures and action taken on products posing a serious risk to the health and safety of consumers.
(2) Point 8 of Annex II to Directive 2001/95/EC requires the guidelines to be regularly updated in the light of new developments and experience. Commission Decision 2010/15/EU(3) was the first and only update of the guidelines.
(3) In view of new developments and in order to ensure more efficient and effective notification procedures in line with best practice, a further update of the guidelines is required.
(4) Terminology and references have become obsolete as has the means of communication between the Commission and the Member States authorities and between the authorities themselves.
(5) New tools that have been developed over the last years for the proper functioning of RAPEX (wiki's, interface between RAPEX and other market surveillance systems) have to be taken into consideration in the guidelines.
(6) Criteria for the RAPEX notification, following the new developments, have become unclear and need to be clarified.
(7) Cross-border online sales of goods have increased. This development needs to be reflected in the notification techniques as well as in the follow-up instruments to be used.
(8) Regulation (EC) No 765/2008 extends the application of RAPEX provided for in Article 12 of Directive 2001/95/EC also to products covered by that legislation. Extending the application of RAPEX raises some issues that need to be clarified in the guidelines.
(9) Regulation (EC) No 765/2008 applies to consumer products and to professional products such as some medical devices. That Regulation covers a broader scope of risks, other than those related to the health and safety of consumers, such as security and environmental risks. Therefore, a risk can concern not only consumers but also an indeterminate group of people referred to as ‘end-users’.
(10) Article 22 of Regulation (EC) No 765/2008 provides therefore that measures taken against products presenting a serious risk to health and safety or other relevant public interests should be notified via RAPEX.
(11) Directive 2001/95/EC and Regulation (EC) No 765/2008 are complementary and provide a system to improve the safety of non-food products.
(12) RAPEX helps to prevent and restrict the supply of products posing a serious risk to health and safety or, in the case of products covered by Regulation (EC) No 765/2008, also to other relevant public interests. It enables the Commission to monitor the effectiveness and consistency of market surveillance and enforcement activities in the Member States.
(13) RAPEX provides a basis for identifying the need for action at EU level and makes for consistent enforcement of EU product safety requirements and therefore contributes to the smooth functioning of the single market.
(14) The notification procedure established under Article 11 of Directive 2001/95/EC provides for an exchange of information between the Member States and the Commission on measures adopted on products posing a less than serious risk to the health and safety of consumers. It helps to ensure a consistent, high level of consumer health and to preserve the single market.
(15) Article 23 of Regulation (EC) No 765/2008 provides for an information support system where Member States make available to the Commission the information required by the same Article on products presenting a less than serious risk.
(16) According to the applicable legislation, Member States are not obliged to provide such information in the RAPEX system.
(17) Article 16 of the GPSD provides an obligation for Member States and the Commission to make available to the public information relating to risks to consumer health and safety posed by products.
(18) To ensure a coherent system of information for products posing a risk to the health and safety of consumers or, in case of products covered by Regulation (EC) No 765/2008, also to other relevant public interests, it would be desirable that available information concerning dangerous products covered by Article 23 of Regulation (EC) No 765/2008 could also be made available in the RAPEX system.
(19) In order to enable the functioning of the RAPEX system, guidelines should be drawn up on the various aspects of these notification procedures and, in particular, to establish the content of notifications. These should include the information to be contained in the notification, criteria for notifications involving risks that do not or cannot go beyond the territory of the Member State and criteria for the classification of notifications according to the degree of urgency. The guidelines should also lay down operating arrangements, including deadlines for the various steps of the notification and follow-up notification procedures as well as confidentiality rules.
(20) To ensure that notification procedures are properly applied, the guidelines should also set out risk assessment methods with criteria for identifying risks taking into consideration also the management of risks.
(21) In the light of point 2 in Annex II to Directive 2001/95/EC, the new guidelines include a set of risk assessment guidelines for consumer products and refer also to professional products, which specify the criteria for identifying serious risks.
(22) The guidelines should be addressed to all Member States authorities participating in the RAPEX network pursuant to Directive 2001/95/EC and Regulation EC No 765/2008, including market surveillance authorities responsible for monitoring the compliance of products with safety requirements and authorities in charge of external border controls,
HAS ADOPTED THIS DECISION:
Article 1U.K.
The guidelines for the management of the European Union Rapid Information System ‘RAPEX’ established under Article 12 of Directive 2001/95/EC and its notification system are set out in the Annex to this Decision.
Article 2U.K.
Decision 2010/15/EU is repealed.
Article 3U.K.
This Decision is addressed to the Member States.
Done at Brussels, 8 November 2018.
For the Commission
Věra Jourová
Member of the Commission
ANNEXU.K.
GUIDELINES FOR THE MANAGEMENT OF THE EUROPEAN UNION RAPID INFORMATION SYSTEM ‘RAPEX’ ESTABLISHED UNDER ARTICLE 12 OF DIRECTIVE 2001/95/EC (THE GENERAL PRODUCT SAFETY DIRECTIVE) AND ITS NOTIFICATION SYSTEM U.K.
PART I U.K.SCOPE AND ADDRESSEES OF THE GUIDELINES
1. Scope, objectives and update U.K.
1.1. Scope U.K.
The ‘Guidelines for the management of the European Union Rapid Information System ‘RAPEX’ established under Article 12 of Directive 2001/95/EC on general product safety’ (the ‘Guidelines’) are adopted by the Commission(4) under Article 11(1) and Annex II, point 8, of Directive 2001/95/EC (the ‘GPSD’). The Commission is assisted by an advisory committee composed of the representatives from EU Member States and established under Article 15(3) of the GPSD.
Point 8 of Annex II to the GPSD states that: ‘The Commission shall prepare and regularly update, in accordance with the procedure laid down in Article 15(3), guidelines concerning the management of RAPEX by the Commission and Member States.’.
Article 11 of the GPSD prescribes that Member States should inform the Commission of measures taken which restrict the placing on the market of products — or require their withdrawal or recall — to the extent that such information is not eligible for the type of notification Article 12 of the GPSD provides for, nor does it qualify for any other notification under any specific Community legislation.
Article 22 of Regulation (EC) No 765/2008, provides that, where a Member State takes or intends to take a measure that prevents, restricts or imposes specific conditions on the marketing and use of products posing a serious risk to the health, safety and other relevant public interests of the end-users, it must immediately notify such a measure to the Commission using RAPEX.
Article 23 of Regulation (EC) No 765/2008 provides that Member States must make available to the Commission the information at their disposal, and not already provided under Article 22, on products presenting a (less than serious) risk.
Article 16 of the GPSD provides an obligation for Member States and the Commission to make available to the public information relating to risks to consumer health and safety posed by products. It would therefore be opportune that all information on measures adopted against products posing a risk, insofar as product safety is at stake, are contained in the system intended for this purpose. Member States are therefore encouraged to provide RAPEX with the measures adopted against products posing a risk and entering into the scope of application of the GPSD or Regulation (EC) No 765/2008. The information can be provided directly in RAPEX. In case the information has to be notified in another information system according to Regulation (EC) No 765/2008(5), the Member State can generate a RAPEX notification from within the information system (see Part II, Chapters 1.2(h) and 2.2 of these Guidelines).
Whereas the GPSD applies only to consumer products posing a risk to the health and safety of consumers, Regulation (EC) No 765/2008 applies to consumer products but also professional products covered by EU harmonisation legislation (such as certain medical devices and marine equipment). It also covers a broader scope of risk, in addition to those related to the health and safety of consumers, such as security and environmental risks. Therefore, a risk can concern not only consumers but also, where Regulation (EC) No 765/2008 applies, other ‘end-users’.
Risk Assessment Guidelines of Appendix 6 on Part III are an integral part of the RAPEX Guidelines. They are the instruments that enable determining the level of risk of a product and therefore help to identify the measures to be adopted.
The Risk Assessment Guidelines refer to the level of risk as well as to the possible injuries caused by a single product. The risk assessment for a single product must be accompanied by sound risk management. For example, the risk level for a defective household electrical appliance posing a risk of fire may be only ‘low’, meaning that the probability of a single appliance causing a fatal fire during the lifetime of the appliance is less than one in a million. Nevertheless, if millions of the defective appliances have been placed on the market, it is almost inevitable that fatal fires will occur if appropriate measures are not taken.
Member States(6), applicant countries, countries which are parties to the European Economic Area (EEA) Agreement as well as other non-EU countries and international organisations that are granted access to RAPEX (on the conditions defined in Article 12(4) of the GPSD), participate in the system according to the rules provided for in the GPSD and these Guidelines(7).
1.2. Objectives U.K.
The objectives of these Guidelines are to:
(a)
streamline the processes for the notification mechanisms;
(b)
set out the notification criteria for the notification mechanisms;
(c)
define the content of notifications and follow-up notifications sent under the notification mechanism, in particular what data are required and which forms are to be used;
(d)
establish follow-up activities to be taken by Member States upon receipt of a notification and the type of information to be provided;
(e)
describe the handling of notifications and follow-up notifications by the Commission;
(f)
set deadlines for the various types of action taken under the notification mechanisms;
(g)
set out the practical and technical arrangements needed at Commission and Member State level for the notification mechanisms to be employed effectively and efficiently; and
(h)
establish risk assessment methods and, in particular, criteria for identifying serious risks.
1.3. Update U.K.
The Guidelines will be regularly updated by the Commission in accordance with the advisory procedure on the basis of experience and new developments in the product safety area.
2. Addressees of the Guidelines U.K.
The Guidelines are addressed to all Member States authorities acting on product safety and participating in the RAPEX network, including market surveillance authorities responsible for monitoring the compliance of products with safety requirements and authorities in charge of external border controls.
3. Products U.K.
3.1. Products covered by these Guidelines U.K.
These Guidelines cover two sets of products: the products covered by the GPSD and the products covered by Regulation (EC) No 765/2008.
3.1.1. Products covered by the GPSD U.K.
Under Article 2(a) of the GPSD, consumer products for the purpose of these Guidelines are:
(a) ‘products intended for consumers’
—
products that are designed and manufactured for and made available to consumers;
(b) ‘migrating products’(8)
—
products that are designed and manufactured for professionals, which are likely, however, under reasonably foreseeable conditions, to be used by consumers. These are products manufactured for professionals that are made available to consumers, who can purchase and operate them without any special knowledge or training, e.g. a power drill, an angle grinder and a table saw designed and manufactured for professionals, but also supplied on the consumer market (i.e. consumers can readily purchase them in shops and operate them on their own without any special training).
Both products intended for consumers and migrating products can be given to consumers free of charge, can be purchased by consumers and can be provided to consumers in the context of a service. All three situations are covered by RAPEX.
According to Article 2 (a) of the GPSD, products provided to consumers in the context of a service are to be considered as including:
(a)
products supplied to consumers that are taken away and used outside the premises of a service provider, such as cars and lawn-mowing machines rented or leased in rental shops, and tattoo inks and implants (that are not classified as medical devices) implanted beneath the skin of a consumer by a service provider;
(b)
products used on the premises of a service provider, provided that consumers themselves actively operate a product (e.g. start the machine, have the option of stopping it, and affect its operation by changing its position or intensity during use). Sun-beds used in tanning salons and fitness centres are examples of such products. Use of the products by consumers must be active, and involve a significant degree of control. Merely passive use, such as the use of a shampoo by a person whose hair is washed by a hairdresser, or the use of a bus by its passengers, does not qualify as use by consumers.
3.1.2. Products covered by Regulation (EC) No 765/2008 U.K.
Under Regulation (EC) No 765/2008, products for the purpose of RAPEX are to be considered the products according to the scope and definitions contained in Article 15 of the same Regulation whether intended for consumers or for professional users.
3.2. Products not covered by these Guidelines U.K.
These Guidelines do not cover:
(a)
Products that are covered by specific and equivalent notification mechanisms established by other EU legislation, notably:
(i)
food and feed and other products covered by Regulation (EC) No 178/2002 of the European Parliament and of the Council(9);
(ii)
medicinal products covered by Directive 2001/83/EC of the European Parliament and of the Council(10), and Directive 2001/82/EC of the European Parliament and of the Council(11);
(iii)
medical devices covered by Regulation (EU) 2017/745 of the European Parliament and of the Council(12);
(iv)
active implantable medical devices covered by Council Directive 90/385/EEC(13).
(b)
Products that are not covered by the definition of a ‘product’ as laid down in Article 2(a) of the GPSD, notably:
(i)
second-hand products or products supplied as antiques or as products to be repaired or reconditioned prior to being used, provided that the supplier clearly informs the person to whom he supplies the product to that effect (Article 2(a) of the GPSD);
(ii)
equipment used or operated by a professional service provider to supply a service, e.g. equipment on which consumers ride or travel and equipment which is operated by a service provider and not by the consumer (recital 9 of the GPSD);
(c)
Products which do not enter into the definition of product contained in Article 15(4) of Regulation (EC) No 765/2008.
4. Measures U.K.
4.1. Types of measures U.K.
Preventive and restrictive measures can be taken in relation to products posing a risk either on the initiative of the economic operator who placed and/or distributed it on the market (‘voluntary measures’), or as ordered by an authority of a Member State competent to monitor the compliance of products with the safety requirements (‘compulsory measures’).
For the purpose of these Guidelines, the compulsory measures and voluntary measures are defined as follows:
(a) Compulsory measures
:
measures adopted or decided to be adopted by Member State authorities, often in the form of an administrative decision, which oblige an economic operator to take preventive, corrective or restrictive action in relation to a specific product that they made available on the market.
(b) Voluntary measures
:
(i)
preventive and restrictive measures adopted on a voluntary basis by an economic operator, i.e. without any intervention of an authority of a Member State;
(ii)
recommendations and agreements with economic operators in their respective activities concluded by Member State authorities; this includes agreements which are not in written form and result in preventive or restrictive action taken by economic operators in their respective activities in relation to products posing a serious risk that they made available on the market.
4.2. Categories of measures U.K.
Article 8(1)(b) to (f) of the GPSD provides a list of the different categories of measures that are notifiable under RAPEX when the conditions for notification are fulfilled, including the following measures:
(a)
marking a product with appropriate warnings on the risk(s) it may present;
(b)
making the marketing of a product subject to prior conditions;
(c)
warning consumers and end-users of the risks that could be posed by a product;
(d)
temporary ban on the supply, offer to supply and display of a product;
(e)
ban on the marketing of a product and any accompanying measures, i.e. measures required to ensure compliance with the ban;
(f)
withdrawal of a product from the market;
(g)
recall of a product from consumers;
(h)
destruction of a withdrawn or recalled product.
For the purpose of RAPEX, the term ‘withdrawal’ is used exclusively for measures aimed at preventing the distribution, display and offer of a product posing a risk to consumers or other end-users, while the term ‘recall’ is used only for measures aimed at achieving the return of such a product that has already been made available to consumers or other end-users by a producer or distributor.
4.3. Requirements of the measures U.K.
Under Article 12(1) of the GPSD and Article 22 of Regulation (EC) No 765/2008 concerning serious risks, both compulsory and voluntary measures are to be notified in RAPEX.
Preventive and restrictive measures adopted on a voluntary basis by an economic operator, i.e. without any intervention of an authority of a Member State concerning a product posing a serious risk and the related preventive or restrictive measures initiated by an economic operator should be immediately notified to the competent authorities of Member States as indicated in Article 5(3) of the GPSD and in Article 22(2) and (3) of Regulation (EC) No 765/2008.
All categories of preventive and restrictive measures taken in relation to the marketing and use of consumer products posing a serious risk to the health and safety of consumers or, in the case of products covered by Regulation (EC) No 765/2008, posing a serious risk to the health, safety or other relevant public interests of the end-users are subject to the notification obligation under RAPEX.
4.4. Exclusion of generally applicable compulsory measures U.K.
Generally applicable acts adopted at national level and aimed at preventing or restricting the marketing and use of (a) generally described category(ies) of consumer products due to the serious risk they pose to the health and safety of consumers should not be notified to the Commission through the RAPEX application. All such national measures that apply to only generally defined categories of products, such as all products in general or all products serving the same purpose — and not to (categories of) products specifically identified by their brand, specific look, producer, trader, model name or number, etc. — are notified to the Commission under Directive (EU) 2015/1535 of the European Parliament and of the Council(14).
5. Risk Levels U.K.
5.1. Serious risk U.K.
Before an authority of a Member State decides to submit a RAPEX notification, it always performs an appropriate risk assessment (see Part III, Appendix 6 of these Guidelines or the complementary EU general risk assessment methodology for products covered by Regulation (EC) No 765/2008(15)) in order to assess whether the product to be notified poses a serious risk to the health and safety of consumers or, in the case of products covered by Regulation (EC) No 765/2008, a serious risk to the health, safety or to other relevant public interests (for example, security or the environment) of the end-users, and thus whether one of the RAPEX notification criteria is met.
5.2. Less than serious risk U.K.
Notifications sent in accordance with Article 11 of the GPSD or Article 23 of Regulation (EC) No 765/2008 are generally considered as notifications for products posing a less than serious risk. Notifications of such products, contrary to notifications for products presenting a serious risk, do not necessarily involve an obligation for follow-up activities by other Member States unless the nature of the product or of the risk so requires (see Part II Chapter 3.4.6.1).
5.3. Risk assessment method U.K.
Part III, Appendix 6 to these Guidelines sets out a risk assessment method that can be used by Member State authorities to assess the level of risks posed by consumer products to the health and safety of consumers and to decide whether a RAPEX notification is necessary. Equally, you may need to consult the complementary EU general risk assessment methodology as referred to in Chapter 5.1 in case the product concerned is covered by Regulation (EC) No 765/2008.
A specific tool (‘RAG’ or Risk Assessment Guidelines(16)) is available on the RAPEX website and in the RAPEX application to perform risk assessments, which takes accounts of the principles provided for in Appendix 6.
5.4. Assessing authority U.K.
The risk assessment is always performed or checked by the authority of a Member State that either carried out the investigation and took appropriate measures, or which monitored the voluntary action taken with regard to a product posing a risk by an economic operator.
Any unclear issues are resolved by the RAPEX Contact Point (see Part II, Chapter 5.1) with the authority responsible before a notification is transmitted through the RAPEX application.
6. Cross-border effects U.K.
6.1. International event U.K.
Under Article 12 of the GPSD and Article 22 of Regulation (EC) No 765/2008, a Member State submits a RAPEX notification only if it considers that the effects of the risk(s) posed by a product go or can go beyond its territory (‘cross-border effects’ or ‘international event’).
In the light of the free movement of products in the internal market, and the fact that products are imported into the EU through different distribution channels and that consumers buy products during stays abroad and via the internet, national authorities are encouraged to interpret the cross-border effects criterion in a fairly broad sense. An Article 12 of the GPSD or Article 22 notification of Regulation (EC) No 765/2008, therefore, is submitted where:
(a)
it cannot be excluded that a product posing a risk has been sold in more than one EU Member State; or
(b)
it cannot be excluded that a product posing a risk has been sold via the internet; or
(c)
the product originates from a third country and is likely to have been imported into the EU through multiple distribution channels.
6.2. Local event U.K.
Measures adopted in relation to a product posing a serious risk that can only have a local effect (‘Local event’) are not notified under Article 12 of the GPSD. This applies in situations where an authority of a Member State has concrete and strong reasons to exclude the possibility that a product has been and or will be made available (by any means) in other Member States, e.g. measures taken with regard to a local product manufactured and distributed only in one Member State. In its evaluation, the authorities of the Member State have to take carefully into consideration the possibility that a product could be sold online or through new emerging distribution channels.
A notification in relation to a product posing a serious risk involving a local event only requires to be submitted to the Commission insofar as it involves information likely to be of interest to Member States from the product safety standpoint, and in particular if they are in response to a new type of risk which has not yet been notified, a new type of risk arising from a combination of products or a new type or category of products.
Such notification is to be submitted under Article 11 with reference to the second subparagraph of Article 11(1), of the GPSD.
PART II U.K.EU RAPID INFORMATION SYSTEM ‘RAPEX’ ESTABLISHED UNDER ARTICLE 12 OF THE GENERAL PRODUCT SAFETY DIRECTIVE
1. Introduction U.K.
1.1. Objectives of RAPEX U.K.
Article 12 of the GPSD establishes an EU Rapid Information System (‘RAPEX’).
RAPEX plays an important role in the area of product safety. It complements other actions taken both at national and at EU level to ensure a high level of product safety in the EU.
RAPEX data helps to:
(a)
prevent and restrict the supply of dangerous products;
(b)
monitor the effectiveness and consistency of market surveillance and enforcement activities carried out by Member State authorities;
(c)
identify needs and provide a basis for action at EU level; and
(d)
make for consistent enforcement of the EU product safety requirements and therefore contribute to the smooth functioning of the single market.
1.2. Components of RAPEX U.K.
RAPEX consists of several complementary components, which are crucial for its effective and efficient operation. The most important are:
(a)
the legal framework that regulates how the system operates (i.e. the GPSD and the Guidelines);
(b)
the online application (‘the RAPEX application’), which allows Member States and the Commission to exchange information rapidly via a web-based platform;
(c)
the RAPEX Contact Points network, which consists of the single RAPEX Contact Points responsible for operating RAPEX in all Member States (see Part II, Chapter 5.1);
(d)
the national RAPEX networks established in all Member States, which include the RAPEX Contact Point (see Part II, Chapter 5.1) and all the authorities involved in ensuring product safety;
(e)
the Commission RAPEX team in the department responsible for the GPSD, which examines and validates documents submitted through the RAPEX application, and maintains and ensures correct operation of RAPEX;
(f)
the RAPEX website(17), which provides summaries of RAPEX notifications as well as weekly updates;
(g)
RAPEX publications, such as RAPEX statistics, RAPEX annual reports and other promotional materials; and
(h)
the interface between RAPEX and ICSMS, which consists on a link between both systems that facilitates the encoding of RAPEX notifications based on investigation data already available in ICSMS. By filling in the appropriate fields in ICSMS, a RAPEX notification can be automatically submitted.
2. Notification criteria U.K.
RAPEX applies to measures which prevent, restrict or impose specific conditions on the marketing and use of products posing a serious risk to the health and safety of consumers or, in the case of products covered by Regulation (EC) No 765/2008, to measures which prevent, restrict or impose specific conditions on the marketing and use of products posing a serious risk to the health, safety or other relevant public interests (for example, security or the environment) of the end-users.
2.1. Mandatory participation in RAPEX: Article 12 of the GPSD and Article 22 of Regulation (EC) No 765/2008 U.K.
Under the GPSD and Regulation (EC) No 765/2008, the participation of Member States in RAPEX is mandatory. According to Article 12 of the GPSD and Article 22 of Regulation (EC) No 765/2008 Member States have a legal obligation to notify the Commission both compulsory and voluntary measures when the following four notification criteria are met:
(a)
the product falls under the scope of application of the GPSD or under the scope of application of Regulation (EC) No 765/2008;
(b)
the product is subject to measures that prevent, restrict or impose specific conditions on its possible marketing or use (‘preventive and restrictive measures’);
(c)
the product poses a serious risk to the health and safety of consumers or, in case of products covered by Regulation (EC) No 765/2008, also to other relevant public interests of the end-users;
(d)
it cannot be ruled out that the effect of the serious risk to the health and safety of consumers or, in case of products covered by Regulation (EC) No 765/2008, also to other relevant public interests of the end-users, goes beyond the territory of the notifying Member State.
2.2. Non-mandatory participation in RAPEX: Article 11 of the GPSD and Article 23 of Regulation (EC) No 765/2008 U.K.
According to Article 11 of the GPSD, Member States should inform the Commission of measures taken which restrict the placing on the market of products — or require their withdrawal or recall — insofar such information does not qualify for an Article 12 nor any other notification set out in any specific Community legislation.
For the sake of simplification and efficiency gains, Member States may also make use of the RAPEX application to notify measures taken against products which would not qualify for submitting an Article 12 notification in the terms outlined herein.
Where the following four notification criteria are met, Member States have a legal obligation to notify the Commission under Article 11 of the GPSD:
(a)
the product concerned is a consumer product;
(b)
it is subject to restrictive measures adopted by national authorities (compulsory measures);
(c)
it poses a less than serious risk to the health and safety of consumers and the effects of which can or do go beyond the territory of one Member State or, it poses a serious risk to the health and safety of consumers and the effect of which do not or cannot go beyond its territory yet the measures adopted involve information likely to be of interest to other Member States from a product safety standpoint(18);
(d)
The measures adopted do not have to be notified under any other notification procedure established by EU law.
Notwithstanding the fact that Article 11 of the GPSD does not contain an explicit obligation to notify voluntary measures adopted against products posing a less than serious risk, Article 16 of the GPSD requires Member States and the Commission to make information relating to risks to consumer health and safety available to the public. Therefore, for the sake of coherence in the notification system and to effectively implement the obligations both Member States and the Commission have according to Article 16 of the GPSD, Member States are recommended to notify in RAPEX also voluntary measures adopted by the producers and distributors against products posing a less than serious risk.
According to Article 23 of Regulation (EC) No 765/2008 Member States provide the Commission with information at their disposal, and not already provided under Article 22, on products presenting a (less than serious) risk. Contrary to Article 22 of this Regulation, Article 23 does not oblige Member States to submit a notification to RAPEX with this information. Article 16 of the GPSD obliges, though, the Commission and the Member States to make public the information they may have relating to risks to consumer health and safety. For the sake of coherence and to effectively implement the obligations contained in Article 16 of the GPSD, the most pragmatic solution could be for RAPEX to contain all measures adopted against products presenting serious and less than serious risks to consumer health and safety both for GPSD products and products covered by Regulation (EC) No 765/2008, and in the latter case, also to other relevant public interests of the end-users. Therefore, when measures are adopted and provided through ICSMS according to Article 23 of Regulation (EC) No 765/2008, Member States are encouraged to notify such information in RAPEX. This can be done either by submitting a separate notification in RAPEX or through ICSMS.
A link between both systems facilitates the encoding of notifications based on investigation data already available in ICSMS. (See Part II, Chapter 1.2(h)).

A notification scheme is included in Part III, Appendix 3 of these Guidelines providing further clarification on the notification criteria referred to in Part II Chapter 2 of these Guidelines.
3. Notifications U.K.
3.1. Types of notification U.K.
3.1.1. Notifications U.K.
The Authorities of the Member States are required to submit a notification to the RAPEX system in the following cases:
(a)
where all the RAPEX notification criteria laid down in Article 12 of the GPSD(19) are met, a Member State prepares and submits to the Commission a RAPEX notification classified in the RAPEX application as an ‘Article 12 notification’.
(b)
where all the RAPEX notification criteria are met and, in addition, a product poses a life-threatening risk and/or there have been fatal accidents, and in other cases where a RAPEX notification requires emergency action by all Member States, the notifying Member State prepares and submits to the Commission a RAPEX notification classified in the RAPEX application as a ‘Notification requiring emergency action’.
(c)
where all RAPEX notification criteria laid down in Article 22 of Regulation (EC) No 765/2008(20) are met, a Member State prepares and submits to the Commission a RAPEX notification classified in the RAPEX application as an ‘Article 22 notification’.
Where all notification criteria laid own in Article 11 of the GPSD(21) are met, a Member State prepares and submits to the Commission a notification, which, when notified in RAPEX is classified as an ‘Article 11 notification’.
Moreover, Member States are encouraged to submit a notification where the criteria laid down in Article 23 of Regulation (EC) No 765/2008 are met(22).
Following the abovementioned reasoning in Part II Chapter 2, Member States are encouraged to prepare and submit, either directly or indirectly, to the Commission a notification classified in RAPEX as an ‘Article 23 notification’ when the criteria laid down in the same article are met.
Before sending a notification to the Commission, the RAPEX Contact Point (see Part II, Chapter 5.1) of the notifying Member State checks that all notification criteria are met.
3.1.2. Notifications for information U.K.
If the criteria laid down in these Guidelines for the notifications listed in Part II Chapters 2.1 and 2.2 of these Guidelines are not met, the RAPEX Contact Point (see Part II, Chapter 5.1) may choose to use the RAPEX application to send the information concerned for information purposes. Such notifications are classified in RAPEX as ‘Notifications for information’ and they may be sent in the following situations:
(a)
Where all the RAPEX notification criteria laid down in Article 12 of the GPSD or in Article 22 of Regulation (EC) No 765/2008 are met but a notification does not contain all the information (mainly on product identification and distribution channels) necessary for other Member States to ensure follow-up(23) to such a notification. A notification where the product name, brand and picture are missing and thus the notified product cannot be correctly identified and it cannot be distinguished from other products of the same category or type that are available on the market, is an example of a notification that can be distributed through the RAPEX application as ‘Notification for information’. Assessment as to whether a notification contains sufficient information for other Member States to ensure follow-up activities is always on a case-by-case basis.
(b)
Where a Member State is aware of the fact that a consumer product that is available on the EU market poses a serious risk to the health and safety of consumers or, in the case of products covered by Regulation (EC) 765/2008, is aware of the fact that a consumer or a professional product poses a serious risk to the health and safety or other relevant public interests of the end-users, but preventive and restrictive measures have not yet been taken by the producer or distributor or adopted or decided to be adopted by an authority of a Member State. If information on such a product is distributed through the RAPEX application before measures are taken, the notifying Member State subsequently informs the Commission (as soon as possible and not later than the deadlines specified in Appendix 4 to these Guidelines) of the final decision taken with regard to the notified product (mainly, what type of preventive or restrictive measures were taken or why such measures were not taken). Where the notifying Member State takes measures at a later stage, it informs the Commission, who will update the notification in application of Article 12 of the GPSD or Article 22 of Regulation (EC) No 765/2008.
(c)
Where a Member State decides to notify preventive and restrictive measures taken in relation to a consumer product posing a serious risk to the health and safety of consumers which has only local effects (‘local event’). If, however, as explained in Part I, Chapter 6.2, a notification by ‘local event’ involves information on product safety likely to be of interest for other Member States, it should be sent as if it were a notification under Article 11 of the GPSD.
(d)
Where a notification concerns a product whose safety aspects (especially the level of risk posed to the health and safety of consumers) are subject to discussion at EU level to ensure a common approach between Member States to risk assessment and/or enforcement action(24).
(e)
Where a decision cannot be taken with certainty that one or more of the notification criteria are met, but a notification involves information on product safety likely to be of interest for other Member States.
When sending a ‘Notification for information’, the RAPEX Contact Point (see Part II, Chapter 5.1) clearly states the reasons for so doing.
3.2. Content of notifications U.K.
3.2.1. Scope of data U.K.
Notifications sent to the Commission through the RAPEX application include the following types of data:
(a)
Information enabling the notified product to be identified, i.e. product category, product name, brand, model and/or type number, barcode, batch or serial number, customs code, description of the product and its packaging accompanied by pictures showing the product, its packaging and labels. Detailed and accurate product identification is a key element for market surveillance and enforcement, as it allows national authorities to identify the notified product, to distinguish it from other products of the same or similar type or category that are available on the market and to find it on the market and take or agree on appropriate measures.
(b)
Information establishing the product's origin, i.e. country of origin, name, address and contact details, such as telephone number and e-mail address, of a manufacturer and exporters. In particular, Member States provide all available information on manufacturers and exporters located in third countries that cooperate closely with the EU on product safety. The following documents are also to be attached to the form where available: copies of orders, sales contracts, invoices, shipping documents, customs declarations, etc. These documents should be transmitted in pdf format or any other format accepted by the application. Detailed information on third country producers allows the Commission to promote more effective enforcement in those countries and helps to reduce the number of products posing a risk to consumers exported into the EU.
(c)
Wherever possible, information about where exactly the product has been made available (a major store, local shop or market, online, etc.).
(d)
Information on the safety requirements applicable to the notified product, including the reference number and name of the applicable legislation and standards.
(e)
A risk description of the notified product, including a description of the results of laboratory or visual tests, test reports and certificates proving non-compliance of the notified product with the safety requirements, a complete risk assessment with conclusions and information on known accidents or incidents (see Part I Chapter 3.3.1 of these Guidelines).
(f)
Information on the supply chains of the notified product in the Member States and, in particular, information on the countries of destination, plus information on importers and also, if available, on distributors of the notified product in Europe.
(g)
Information on measures taken, in particular, the type (compulsory or voluntary), category (e.g. withdrawal from the market, recall from consumers), scope (e.g. national, local), and date of entry into force and duration of the measure (e.g. permanent, temporary).
(h)
Indication of whether a notification, part of it and/or attachment(s) are covered by confidentiality. Requests for confidentiality are always accompanied by a justification clearly stating the reasons for such a request.
(i)
Information on whether the product is counterfeit, when available. For this purpose, the Commission will provide Member States with any specific tools available at European level to facilitate the identification of counterfeit products.
(j)
Information on reported accidents related to the product, indicating when possible the reasons for the accident (risk related to the use made by the user or inherent to the product).
(k)
Additional information on whether the notification has been submitted in the context of a coordinated enforcement activity at European level.
(l)
Information on whether the authorities of a Member State envisage sending other notifications related to the same product or similar products. This should be indicated in the original notification.
Member States are encouraged to look for and provide information on the supply chains of the notified product in non-EU countries that cooperate closely with the EU on product safety.
3.2.2. Completeness of data U.K.
Notifications should be as complete as possible. The elements to be contained in the notification are listed in Appendix 1 to these Guidelines and are included in the RAPEX application. All fields of the notification template should be completed with the required data. Where the required information is not available at the time a notification is submitted, this is clearly indicated and explained on the form by the notifying Member State. Once the missing information becomes available, the notifying Member State updates its notification. The updated notification is examined by the Commission before being validated and distributed through the system.
RAPEX Contact Points provide all national authorities that participate in the RAPEX network with instructions on the scope of data required to complete the notification. This helps to ensure that the information provided by these authorities to the RAPEX Contact Point is correct and complete (see Part II, Chapter 5.1).
Where part of the information required by these Guidelines is not yet available, Member States should nonetheless comply with the established deadlines and not delay sending a RAPEX notification on a product that poses a life-threatening risk to the health and safety of consumers or other end-users and/or where a RAPEX notification requires emergency action by Member States.
Before submitting a notification, the RAPEX Contact Point checks (to avoid any unnecessary duplication) that the product concerned has not already been notified through the RAPEX application by another Member State. If the product has already been notified, rather than creating a new notification, the RAPEX Contact Point submits a follow-up notification to the existing notification and provides any additional information that may be relevant for authorities in other Member States, such as additional vehicle identification numbers, a detailed list of importers and distributors, additional test reports, etc. (See also Part II, Chapter 5.1).
3.2.3. Updating of data U.K.
The notifying Member State informs the Commission (as soon as possible and not later than by the deadlines specified in Appendix 4 to these Guidelines) of any developments that require changes to a notification transmitted through the RAPEX application. In particular, Member States inform the Commission of any changes (e.g. following a ruling by a court during an appeal procedure) to the status of the notified measures, to the risk assessment and to new decisions regarding confidentiality.
The Commission examines the information provided by the notifying Member State and updates the information concerned in the RAPEX application and on the RAPEX website, where necessary.
3.2.4. Responsibility for the information transmitted U.K.
Responsibility for the information provided lies with the notifying Member State(25).
The notifying Member State and the national authority responsible ensure that all data provided through the RAPEX application are accurate so as to avoid any confusion with similar products of the same category or type that are available on the EU market.
The authority(ies) involved in the notification procedure (e.g. by performing the risk assessment of the notified product or by providing information on distribution channels) take responsibility for the information provided through the RAPEX application. The RAPEX Contact Point checks and validates all notifications received from the authorities responsible before transmitting them to the Commission (See also Part II, Chapter 5.1).
Any action taken by the Commission, such as examining notifications, validating and distributing them through the RAPEX application and publishing them on the RAPEX website, does not imply any assumption of responsibility for the information transmitted, which remains with the notifying Member State.
3.3. Actors and roles involved in the notification process U.K.
The parties involved in the notification process and their responsibilities therein are the following:
3.3.1. Economic operators U.K.
Economic operators are not directly involved in the submission of notifications in the RAPEX application.
However, in case of a product posing a risk, economic operators shall immediately inform the competent authorities in all Member States where the product was made available. The conditions and details for providing such information are laid down in Annex I to the GPSD.
Such information will be dealt with by the Member State where the notifying producer/distributor is established (‘Main Member State’).
The transmission of information on products posing a risk can be submitted by economic operators through the ‘Product Safety Business Alert Gateway’, a tool available on the RAPEX website (see Part II Chapter 5.3.2). Economic operators should include a detailed description of the risk of the product and can make use of the ‘RAG tool’ available for this purpose (see Part I Chapter 5.3).
Risk assessments carried out by economic operators are not binding on Member State authorities who are responsible for carrying out their own risk assessment. It is therefore possible for an authority of a Member State to come to a different conclusion regarding the risk assessment provided in an alert submitted via the ‘Business Gateway’.
3.3.2. Member States authorities U.K.
Member States authorities notify the Commission through the RAPEX application about both compulsory and voluntary measures taken on their own territory against products posing a risk.
Member States establish the roles for the creation, submission and follow-up of notifications in RAPEX.
3.3.3. Authorities in charge of external border controls U.K.
Measures adopted by the authorities in charge of external border controls that prevent the marketing in the EU of a consumer product posing a serious risk to the health and safety of consumers (e.g. decisions to stop the import at the EU border) should be notified to the Commission through the RAPEX application in the same manner as measures adopted by market surveillance authorities that restrict the marketing or use of a product.
3.3.4. European Commission U.K.
The Commission may inform the RAPEX Contact Points (see Part II, Chapter 5.1) regarding products posing serious risks, imported into or exported from the Community and the European Economic Area(26).
The Commission may transmit information to the Member States about products of EU and non-EU origin posing a risk that, according to the information available, are likely to be on the EU market. This mainly concerns information that the Commission receives from third countries, international organisations, businesses or other rapid alert systems.
This information might be circulated amongst Member States by means other than the RAPEX application.
3.4. Workflow U.K.
3.4.1. Creation of a notification U.K.
3.4.1.1. By a national authority U.K.
According to the national arrangements, different national authorities involved in the RAPEX process (local/regional market surveillance authorities, external border control authorities, etc.) may be allowed to create a notification.
3.4.1.2. By the Commission U.K.
In certain cases, the Commission may create a notification as explained in point 3.3.4.
3.4.2. Submission of notifications to the Commission U.K.
The RAPEX Contact Point is responsible for the submission of all notifications for validation by the Commission. (See Part II, Chapter 5.1).
3.4.3. Examination of notifications by the Commission U.K.
The Commission checks all notifications received through the RAPEX application before transmitting them to Member States to ensure that they are correct and complete.
3.4.3.1. Correctness U.K.
When assessing the correctness of a notification, the Commission checks in particular that:
(a)
The notification meets all the relevant requirements set out in the GPSD or in Article 22 of Regulation (EC) No 765/2008 and in these Guidelines;
(b)
the notified product has not already been notified (to avoid any unnecessary duplication, including between ICSMS and RAPEX);
(c)
the notification submitted for validation by the notifying Member State is classified in accordance with the criteria set out in Part II Chapter 2 of these Guidelines;
(d)
the information provided including the risk assessment takes due account of the applicable legislation and the relevant standards;
(e)
the correct notification procedure has been used.
3.4.3.2. Completeness U.K.
Once a notification is confirmed as correct, the Commission checks that it is complete. Part II, Chapters 3.2.1 and 3.2.2 of these Guidelines act as a point of reference. Special attention is given to the parts of a notification concerning product identification, risk description, measures, traceability and distribution channels.
The Commission is not responsible for performing a risk assessment of the product, but only for checking that the notification includes an appropriate risk assessment containing all the elements listed in Part II Chapter 3.2.1 of these Guidelines (with the exceptions referred to in point 3.4.3.3). See also Part I Chapter 5.1 of these Guidelines.
3.4.3.3. Validation of notifications without a detailed risk assessment U.K.
Member States should submit a risk assessment for every notification but in certain cases, the Commission may validate notifications that are submitted without a detailed and individual risk assessment:
(a)Notifications of products posing chemical risksU.K.
The risk level of a product may be considered to be serious if it contains a chemical substance either banned or in a concentration above the limit established by European legislation. Therefore, in cases where measures are taken against products containing a chemical substance subject to a restriction contained in EU Legislation, a notification may be submitted without a detailed risk assessment.
(b)Notifications of cosmetic productsU.K.
Validation of notifications that do not include a detailed risk assessment may equally be possible for cosmetic products containing banned or restricted substances, which are backed up by an EU scientific committee opinion supporting that such presence of substances above the established limits poses a risk to the health and safety of consumers. For this specific product sector, other factors (e.g. concentration or time of exposure) may need to be taken into consideration.
Nevertheless, if measures have been taken against a product containing not authorised chemical substances for which no scientific opinion has been issued confirming that the product poses a risk, a proper risk assessment may be required depending on a case-by-case analysis to prove that the product poses a serious or less than serious risk. In cases where the risk assessment is needed, if such risk assessment is not provided, these cases shall only be validated ‘for information’ in RAPEX.
As regards products that are subject to restrictive measures by market surveillance authorities based on the presence of a chemical substance mentioned in the list of ingredients which is subject to restrictions contained in EU Legislation and where there is no scientific data assessing the risk, notifications need to be assessed on a case-by-case basis. In case where the risk assessment is needed, if such risk assessment is not provided, these cases shall only be validated ‘for information’ in RAPEX.
(c)Notification of other productsU.K.
Where there is well-documented evidence that certain features of certain products consistently lead to a specific risk and risk level (e.g., the presence of any drawstrings or functional cords in the head, neck or upper chest on garments intended for young children always implies a serious risk), no further risk assessment is required for that given product.
3.4.3.4. Requests for additional information U.K.
Should, during examination, the Commission have questions regarding a notification, it may suspend validation of the notification and ask the notifying Member State for additional information or clarification. This additional information is provided by the notifying Member State by the deadline specified in the Commission's request for information.
3.4.3.5. Investigation U.K.
Where necessary, the Commission may carry out an investigation to assess the safety of a product. This investigation may be conducted in particular where there are serious doubts as to the risks posed by the product notified via the RAPEX application. These doubts can either arise during the examination of a notification by the Commission, or be brought to the attention of the Commission by a Member State (e.g. through a follow-up notification) or by a third party (e.g. a producer).
As part of such investigations, the Commission may, in particular:
(a)
ask any Member State to provide information or clarification;
(b)
ask for an independent risk assessment and independent testing (laboratory or visual) of the product under investigation;
(c)
consult the Scientific Committees, the Joint Research Centre or any other institution specialising in the safety of consumer products;
(d)
convene the GPSD Committee, Consumer Safety Network and/or RAPEX Contact Points meetings, as well as consult the relevant Working Groups to discuss developments in an investigation.
Where an investigation concerns a product notified through the RAPEX application, the Commission may suspend validation of a notification or, where such a notification has already been validated and distributed through the RAPEX application, temporarily remove the overview published on the RAPEX website. After an investigation, and depending on the outcome, the Commission (after consulting the notifying Member State, where necessary) may in particular validate and distribute through the RAPEX application the previously suspended notification, uphold the validated notification in the RAPEX application (with any changes) or permanently withdraw the notification from RAPEX.
The Commission informs all Member States of the following:
(a)
its decision to launch an investigation, clearly stating the reasons for its decision;
(b)
its decision to close an investigation, presenting its conclusions and changes to the investigated notification(s) (if any);
(c)
all the relevant developments during an investigation.
3.4.4. Validation and distribution of notifications U.K.
The Commission validates and distributes through the RAPEX application, by the deadlines specified in Appendix 5 to these Guidelines, all notifications assessed as correct and complete during the examination.
Where, during an examination, a request for additional information or clarification was sent to the notifying Member State (followed by a reminder, if necessary), the Commission may take the following decisions:
(a)
where the additional information or clarification requested has been provided, the Commission re-examines the notification and may validate it with the changed classification where necessary (e.g. from a ‘Notification for information’ to an ‘Article 12 notification’) or keep it on hold until further clarification;
(b)
where the additional information or clarification requested has not been provided within a specified deadline or it is insufficient, the Commission takes a decision on the basis of the information provided and, depending on the circumstances, may either validate it after changing the classification (e.g. from an ‘Article 12 notification’ to ‘Notification for information’) or decide not to validate it.
Once a common approach to risk assessment and/or enforcement has been agreed between Member States, depending on the circumstances and the views of the Member States, the Commission may take one of the following actions:
(a)
keep the notifications concerned in the RAPEX application;
(b)
change the classification of the notifications stored in the RAPEX application;
(c)
withdraw notifications from RAPEX(27).
3.4.5. Publication of notifications U.K.
3.4.5.1. Disclosure of information as a general rule U.K.
The public has the right to be informed about products posing a risk. To meet this obligation, the Commission publishes overviews of new notifications on the RAPEX website(28).
For external communication reasons, the RAPEX website will in future be called ‘Safety Gate’.
Member States equally provide the public with information in the national languages on products posing a serious risk to consumers and on measures taken to address this risk. Such information may be distributed via the internet, on paper, by electronic media, etc.
The information made available to the public is a summary of a notification and includes in particular the elements which allow the identification of the product, as well as the information about the risks and measures taken to prevent or restrict those risks. The Commission and the Member States may decide to disclose other elements of the notifications to the public, only when this information, due to its nature, is not confidential (professional secrets) and does not need to be protected.
The following notifications are made available on the RAPEX website, in line with the requirements laid down in Article 16 of the GPSD:
(a)
notifications submitted falling under the scope of Article 12 of the GPSD;
(b)
notifications submitted falling under the scope of Article 22 of Regulation (EC) No 765/2008;
(c)
notifications submitted falling under the scope of Article 11 of the GPSD for products posing less than serious risk, the cross-border effect of which has also been recognised. As Chapter 3.4 provides for, the cross-border effect ascertains whether such a scenario is to be notified under Article 11;
(d)
notifications submitted falling under the scope of Article 23 of Regulation (EC) No 765/2008 concerning products presenting risks that are less than serious and regardless of whether the measures taken were compulsory or voluntary(29);
(e)
notifications submitted for information only if the notifying Member State so requests by ticking the ad hoc box in RAPEX, especially when voluntary measures are adopted and the products concerned are sufficiently identified. The publication of these notifications might need to be considered from the standpoint of securing an appropriate risk management.
3.4.5.2. Exceptions to the general rule U.K.
Member States and the Commission should not disclose to the public any information about a product notified through the RAPEX application if such disclosure undermines the protection of court proceedings, monitoring and investigation activities or professional secrecy, except for information relating to the safety properties of products which must be made public if circumstances so require to protect the health and safety of consumers, or, in case of products covered by Regulation (EC) No 765/2008, also to protect other relevant public interests of the end-users(30).
3.4.5.3. Requests for confidentiality U.K.
A notifying Member State may request confidentiality of a notification. Such a request clearly indicates the part(s) of the notification that should be kept confidential.
Furthermore, each request for confidentiality is accompanied by a justification clearly stating the reasons(31).
Requests for confidentiality are subject to examination by the Commission. The Commission checks that the request is complete (i.e. that it states which parts of the notification are covered by confidentiality and that it contains a justification) and justified (i.e. that it is in line with the provisions of the GPSD and these Guidelines). A decision as to the validity of the request is taken by the Commission after consulting the respective RAPEX Contact Point. (See Part II, Chapter 5.1).
3.4.5.4. Handling of notifications covered by confidentiality U.K.
Article 16(2) of the GPSD states that the protection of professional secrecy or confidentiality shall not prevent the dissemination to the competent authorities of information relevant for ensuring the effectiveness of market monitoring and surveillance activities. Notifications covered partially or fully by confidentiality are examined by the Commission and, after being validated and distributed through the RAPEX application, they are subject to the usual follow-up activities by the Member States. The confidentiality of a notification or parts of it does not prevent it from being handled and distributed through the RAPEX application to the competent national authorities.
The only significant difference in the handling and follow-up procedures is that the Commission and Member States should not disclose any parts of a notification that are confidential to the public. These parts have to remain confidential and thus they should not be published in any form. Member State authorities that receive confidential information through the RAPEX application ensure that it is protected when performing their activities.
3.4.5.5. Withdrawal of request for confidentiality U.K.
The notifying Member State withdraws its request for confidentiality immediately after the authority in that Member State becomes aware that the justification for such a request is no longer valid, and informs the Commission accordingly. The Commission informs all Member States of the withdrawal of confidentiality on receipt of such a request by the notifying Member State.
A notification that is no longer covered by full or partial confidentiality is made available to the public in line with the ‘general rules’ applying to publication of notifications set out in these Guidelines.
3.4.6. Follow-up to notifications U.K.
3.4.6.1. Follow-up to the different types of notification U.K.
Member States ensure appropriate follow-up to ‘Article 12 notifications’, ‘Article 12 notifications requiring emergency action’, notifications under Article 22 of Regulation (EC) No 765/2008 and to information on products posing a risk sent by the Commission (Chapter 3.3.4) as soon as possible and by the deadlines specified in Appendix 4 to these Guidelines at the latest.
Notifications for information as well as notifications under Article 11 of the GPSD and notifications under Article 23 of Regulation (EC) No 765/2008 (notification for less than serious risks) do not require any specific follow-up activities. These notifications often do not contain the data needed for effective and efficient enforcement regarding the notified product (e.g. the notified product and/or measures are not sufficiently identified) or the level of the risk is not considered to be serious.
Although there is no specific need for a follow-up in the referred cases, it is still important that Member States verify whether they disagree with the consideration of the risk as less than serious so they may eventually make a follow-up upon the information of a different risk assessment. Member States are therefore encouraged to ensure follow-up to such notifications where the notified product is likely to have been made available to consumers on their market and product identification allows measures to be taken.
3.4.6.2. Objectives of the follow-up activities U.K.
On receipt of a notification, a Member State examines the information provided in the notification and takes appropriate action in order to:
(a)
establish whether the product was marketed on its territory;
(b)
assess what preventive or restrictive measures should be taken with regard to the notified product found on its market, taking into account the measures taken by the notifying Member State and any special circumstances that could justify different types of measures or no action being taken;
(c)
perform additional risk assessment and testing of the notified product, if necessary;
(d)
collect any additional information that may be relevant for other Member States (e.g. information on distribution channels of the notified product in other Member States).
3.4.6.3. Follow-up techniques U.K.
To ensure efficient and effective follow-up, best practice follow-up techniques should be employed by national authorities, including:
(a)Checks on the marketU.K.
National authorities organise regular (planned and random) checks on the market in order to establish whether consumer products notified through the RAPEX application are made available to consumers. When the Member State is mentioned as a country of destination, reinforced checks on the market shall be carried out, notably by contacting the economic operator(s) indicated in the notification.
(b)Cooperation with business associationsU.K.
National authorities provide, when necessary, business associations with overviews of the most recent notifications and enquire whether any of the notified products were produced or distributed by their members. National authorities provide businesses only with summaries of notifications, such as the weekly overviews published on the RAPEX website. Whole notifications should not be transmitted to third parties, as certain information (e.g. details of the risk description or information on distribution channels) is often confidential and should be protected.
(c)Publication of RAPEX data via the internet or other electronic and paper mediaU.K.
National authorities regularly alert consumers and businesses about consumer products notified through the RAPEX application via their websites and/or other media, e.g. referring consumers and business to the RAPEX website. Information published in this way allows consumers to check whether they have and use products posing a risk and often provides the authority with useful feedback.
(d)Online checksU.K.
National authorities regularly perform online checks to try to identify whether products notified via RAPEX are available on online markets. Online check techniques may include web-crawling, data mining, data scraping, etc.
National authorities apply various follow-up techniques in parallel and ideally do not limit their activities to only one of them.
The Member State in which a manufacturer, a representative or an importer of the notified product is established (‘Main Member State’) ensures appropriate follow-up to notifications distributed through the RAPEX application. The ‘Main Member State’ often has better legal and technical means of obtaining information on the notified case, which will help other Member States to undertake effective follow-up activities.
3.4.7. Withdrawal/removal of notifications U.K.
3.4.7.1. Permanent withdrawal of a notification from RAPEX U.K.
Notifications distributed through the RAPEX application are kept in the system for an unlimited period of time. The Commission may, however, in the situations presented in this Chapter, permanently withdraw a notification from RAPEX.
3.4.7.1.1.Situations where withdrawal of a submitted or validated notification is possibleU.K.
(a)
There is proof that one or more of the notification criteria(32) are not met and thus a notification is not justified. This concerns cases in particular where it is established that the original risk assessment was performed incorrectly and that the notified product does not pose a risk. It also covers situations where the notified measures were successfully challenged in court or in other proceedings and they are no longer valid.
(b)
No measures have been taken with regard to a product notified through the RAPEX application (for information) before it was decided to adopt measures or take action(33).
(c)
After a discussion held at EU level, Member States agree that it is not useful to exchange information on certain safety aspects that have been notified through the RAPEX application(34).
(d)
There is proof that products covered by a notification are no longer marketed and there is proof that all items that had been made available have already been withdrawn from the market and retrieved in all Member States.
Withdrawal of a notification that has been submitted or validated cannot be requested on the basis of the fact that the notified product has been subject to changes needed for it to comply with all the applicable safety requirements, unless proof is provided that all the products (items) concerned that had been made available have been withdrawn and retrieved in all Member States and that they are no longer marketed.
3.4.7.1.2.Request for permanent or temporary withdrawal by Member StatesU.K.
The Commission may withdraw notifications from RAPEX only at the request of the notifying Member State, as the latter takes full responsibility for the information transmitted through the system. Other Member States, however, are encouraged to inform the Commission of any facts that may justify withdrawal.
3.4.7.1.3.Content of the request for permanent or temporary withdrawalU.K.
Every request for withdrawal is accompanied by a justification stating the reasons and by all available documents supporting those reasons. The Commission examines each request and checks the justification and the supporting documents in particular. The Commission may request additional information, clarification or the opinion of the notifying Member State and/or other Member States before taking any decision.
3.4.7.1.4.Decision to withdrawU.K.
Should, on the basis of the justification provided, the Commission decide to withdraw a notification from RAPEX, it removes it from:
(a)
the RAPEX application (or makes it otherwise invisible to all users of the system);
(b)
the RAPEX website (if necessary).
The Commission informs all Member States of the withdrawal of a notification by mail or through other equally effective means and, if necessary, also the public by publishing a corrigendum on the RAPEX website.
3.4.7.2. Temporary removal of a notification from the RAPEX website U.K.
3.4.7.2.1.Situations where temporary removal is possibleU.K.
Where justified, the Commission may temporarily remove a notification from the RAPEX website, especially where the notifying Member State suspects that a risk assessment submitted in a notification has been performed incorrectly and thus the notified product may not pose a risk. A notification can be temporarily removed from the RAPEX website until the risk assessment of the notified product has been clarified.
3.4.7.2.2.Request for temporary removal by Member StatesU.K.
The Commission may temporary remove notifications from the RAPEX application only at the request of the notifying Member State, as the latter takes full responsibility for the information transmitted through the application. Other Member States, however, are encouraged to inform the Commission of any facts that may justify such removal.
3.4.7.2.3.Content of the request for temporary removalU.K.
Every request for temporary removal is accompanied by a justification stating the reasons and by all available documents supporting those reasons. The Commission examines each request and checks the justification and the supporting documents in particular. The Commission may request additional information, clarification or the opinion of the notifying Member State and/or other Member States before taking any decision.
3.4.7.2.4.Decision to removeU.K.
Should, on the basis of the justification provided, the Commission decide to remove a notification from the RAPEX website, it informs all Member States by e-mail or by other equally effective means and, if necessary, also the public by publishing a corrigendum on the RAPEX website.
3.4.7.2.5.Re-publishing of a notification temporarily removedU.K.
The notifying Member State immediately informs the Commission when the reasons for the removal of a notification from the RAPEX website are no longer valid. In particular, it informs the Commission of the results of any new risk assessment to enable the Commission to determine whether to maintain a notification in the RAPEX application and to re-publish it on the RAPEX website or to withdraw it permanently from RAPEX (following a request from the notifying Member State).
The Commission may re-publish a notification on the RAPEX website following a justified request from the notifying Member State after the risk assessment has been clarified.
The Commission informs the other Member States of the re-publishing of a notification on the RAPEX website by e-mail or by other equally effective means and also the public by replacing the corrigendum with a new one on the RAPEX website.
3.4.8. Notifications older than ten years U.K.
The Commission will place all notifications older than ten years in a separate section of the RAPEX website. These notifications will still be available for public consultation.
3.5. Timing and deadlines for notifications U.K.
3.5.1. Timing of the notification U.K.
Article 12(1) of the GPSD and Article 22 of Regulation (EC) No 765/2008 require Member States to immediately notify the Commission through the RAPEX application of preventive and restrictive measures concerning products posing serious risks. This provision applies to both compulsory and voluntary measures, although the timing of the notification is different.
(a)Compulsory measuresU.K.
These measures are notified through the RAPEX application immediately after being adopted or after the decision to adopt them has been taken, even if an appeal against them at national level is likely, if they are already under appeal or they are subject to publication requirements.
This approach is consistent with the objective of RAPEX, i.e. to ensure the rapid exchange of information between Member States and the Commission in order to prevent the supply and use of products that pose a risk.
(b)Voluntary measuresU.K.
Under Article 5(3) of the GPSD and Article 22 of Regulation (EC) No 765/2008, economic operators are obliged to notify the competent Member State authorities of voluntary action and measures taken to prevent risks to consumers posed by products they have made available on the market (ideally by means of a ‘Business Gateway’ notification). The authority of a Member State receiving this kind of notification uses this information as the basis for a notification (if all the notification criteria are met) and sends it immediately after receipt of the ‘Business Gateway’ notification.
Where voluntary measures are adopted in the form of an agreement between an economic operator and an authority of a Member State or on the basis of a recommendation from an authority to a producer or distributor, a notification is submitted immediately after the conclusion of such an agreement or the adoption of such a recommendation.
To ensure common application of the notification obligation, Part III, Appendix 4 to these Guidelines lays down specific deadlines for submitting notifications to the Commission via the RAPEX application(35).
3.5.2. Deadlines (36) U.K.
Member States notify the Commission of preventive and restrictive measures adopted as soon as possible and by the deadlines specified in Part III, Appendix 4 to these Guidelines at the latest. Appropriate arrangements are in place at national level concerning the transmission of information between national authorities in charge of product safety and the RAPEX Contact Point to ensure that the deadlines are met. (See Part II, Chapter 5.1).
The deadlines provided apply irrespective of any appeal procedure or official publication requirement.
3.5.3. Emergency situations U.K.
All notifications concerning products posing a serious risk requiring emergency action are preceded by a telephone call from the RAPEX Contact Point to the Commission RAPEX Team's mobile telephone number to facilitate immediate action and follow-up. This rule applies in particular to notifications transmitted at weekends or during holiday periods. (See also Part II, Chapter 5.1).
4. Follow-up activities U.K.
4.1. Communication of follow-up activities U.K.
Member States notify the Commission of any findings subsequent to their follow-up activities in relation to RAPEX notifications (i.e. ‘Article 12 notifications’ and ‘Notifications requiring emergency action’ as well as notifications under Article 22 of Regulation (EC) No 765/2008) and information on products posing a risk sent by the Commission (Chapter 3.3.4).
In addition, Member States are encouraged to notify the Commission of any follow-up activities regarding notifications for less than serious risks and for information.
4.2. Content of follow-up notifications U.K.
4.2.1. Scope of data U.K.
Findings resulting from follow-up activities are communicated to the Commission in the form of follow-up notifications. To harmonise the type of information and to keep the workload to a minimum, Member States submit follow-up notifications in particular in the following situations:
(a)A notified product has been found on the marketU.K.
A follow-up notification is sent when national authorities find the notified product on the market or at the external border. This follow-up notification contains the full details of the product in question (e.g. name, brand, model number, bar code, batch number) plus information on the total number of items found on the market. Furthermore, the following details of the measures taken are communicated: type (compulsory or voluntary), category (e.g. withdrawal from the market, recall from consumers), scope (e.g. country-wide, local), date of entry into force and duration (e.g. permanent, temporary). If the notified product was found on the market but no measures were adopted, specific reasons justifying no measures being taken should be given in the follow-up notification.
To reduce the burden on the national authorities as regards their follow-up practice, Member States do not need to inform the Commission (unless the Commission asks to be informed) of the conclusions of follow-up activities by means of a follow-up notification when the notified product is not found on the market.
(b)Different risk assessmentU.K.
A follow-up notification is sent when the conclusions of a risk assessment performed by an authority of the reacting Member State differ from the conclusions set out in the original notification. This follow-up notification contains a detailed risk description (including the results of tests, a risk assessment and information on known accidents and incidents), accompanied by supporting documents (test reports, certificates, etc.). Furthermore, the reacting Member State should prove that the risk assessment submitted with its follow-up notification was performed on the same product as the one notified, i.e. the same brand, name, model number, batch number, origin, etc.
(c)Additional informationU.K.
A follow-up notification is sent when national authorities collect additional information (during their follow-up activities) that may be useful for market surveillance and enforcement in other Member States.
Member States are encouraged to collect additional information that may be relevant for authorities both in other Member States and in third countries that cooperate closely with the EU on product safety. Details include product origin (e.g. information on the country of origin, manufacturer and/or exporters) and information on the supply chains (e.g. information on the countries of destination, importers and distributors). The country carrying out the follow-up activities attaches all available supporting documents to the follow-up notification, such as copies of orders, sales contracts, invoices, customs declarations, etc.
Member States may also indicate whether certain follow-up actions have been performed although the product has not been found in their territory.
4.2.2. Completeness of follow-up notifications U.K.
The RAPEX Contact Point of the reacting Member State, together with the responsible authority, ensures that all data provided in their follow-up notification is accurate and complete and that there is no confusion with other similar products that are available on the EU market. (See also Part II, Chapter 5.1).
The standard follow-up notification template is provided in Part III, Appendix 2 to these Guidelines. Should certain relevant information not be available when a follow-up notification is submitted, the reacting Member State indicates this on the follow-up form. Once this information becomes available, the reacting Member State may request that its follow-up notification be updated. The updated follow-up notification is examined by the Commission before it is validated and distributed through the system.
The RAPEX Contact Point provides all authorities in its own Member State that participate in the RAPEX network with instructions on the scope of the data required to complete the follow-up notification template correctly. This helps to ensure that information provided by these authorities to the Contact Point is correct and complete. (See Part II, Chapter 5.1).
4.2.3. Updating of validated follow-up notifications U.K.
The reacting Member State informs the Commission (as soon as possible and by the deadlines specified in Part III, Appendix 4 to these Guidelines at the latest) of any developments that may require changes to a follow-up notification distributed through the RAPEX application. In particular, Member States inform the Commission of changes in the status of the measures taken or in the risk assessment submitted with their follow-up notification.
The Commission examines the information provided by the reacting Member State and if necessary updates the information concerned.
4.2.4. Responsibility for follow-up notifications U.K.
Responsibility for the information provided in follow-up notifications lies with the notifying Member State(37).
The authority(ies) involved in the follow-up activities (e.g. by carrying out the risk assessment or by adopting restrictive measures) take responsibility for the information provided in follow-up notifications. The RAPEX Contact Point checks and validates all follow-up notifications prepared by the respective authorities before transmitting them to the Commission. (See also Part II, Chapter 5.1).
Any action taken by the Commission, such as examining and validating follow-up notifications, does not imply any assumption of responsibility for the information transmitted, which remains with the Member State submitting the follow-up notification.
4.2.5. Response to follow-up notifications U.K.
Member States may respond to any follow-up notifications regarding their own notification(s) by starting a discussion on the online collaborative space put at the disposal of Member States for the exchange of information (see Part II Chapter 5.3.2). This ensures that the response is visible to all members of RAPEX.
4.3. Actors and roles involved in follow-up activities U.K.
The parties involved in the follow-up notification process and their responsibilities therein are the following:
4.3.1. Economic operators (38) U.K.
Economic operators are not directly involved in the submission of follow-up notifications. However, economic operators must cooperate with national authorities and provide them with any information concerning a product which is the subject of an existing notification in order to facilitate the creation and submission of follow-up notifications via the RAPEX application.
4.3.2. Market surveillance authorities U.K.
Market surveillance authorities notify the European Commission through the RAPEX application about any follow-up activities or other information regarding notifications.
4.3.3. European Commission U.K.
The European Commission examines and validates follow-up notifications according to the specifications included in Part II, Chapter 4.2.
4.4. Workflow U.K.
4.4.1. Creation and submission of a follow-up notification by a Member State U.K.
The RAPEX Contact Point is responsible for the submission of follow-up notifications via the RAPEX application. (See Part II, Chapter 5.1).
4.4.2. Examination of follow-up notifications by the Commission U.K.
4.4.2.1. Correctness and completeness U.K.
The Commission checks all follow-up notifications received through the RAPEX application before they are validated and transmitted to the Member States. These checks focus on the correctness and completeness of the information provided.
The Commission checks if a follow-up notification meets all the relevant requirements set out in the GPSD and in these Guidelines and if the correct procedure was applied. Once the correctness of a follow-up notification is confirmed, the Commission checks its completeness. Chapter 4.2.2 of these Guidelines is to be used as a point of reference for this examination.
The Commission pays special attention to follow-up notifications containing risk assessments. It verifies, in particular, that the risk description is complete, clearly presented and well documented, and that the risk assessment clearly relates to the product covered by a notification.
4.4.2.2. Requests for additional information U.K.
Before validating a follow-up notification, the Commission may request the reacting Member State to provide additional information or clarification within a given deadline. Validation of a follow-up notification may be conditional upon receipt of the data requested.
The Commission may request the opinion of any Member State and, in particular, the notifying Member State on a validated follow-up notification. The Member State submits its opinion to the Commission within a deadline specified by the latter. Furthermore, the notifying Member State informs the Commission whether any changes to the notification (e.g. to the risk assessment) or to its status (e.g. permanent withdrawal from the system) are necessary.
4.4.3. Validation and distribution of follow-up notifications U.K.
All follow-up notifications assessed as correct and complete are validated and distributed by the Commission according to the deadlines specified in Appendix 5 to these Guidelines.
The Commission does not validate follow-up notifications with a risk assessment different from that of the notification they refer to, if the risk assessment is not complete, clearly presented and well documented, or if it is not shown that the risk assessment was performed in relation to the product covered by the notification.
4.4.4. Permanent withdrawal of a follow-up notification from RAPEX U.K.
Follow-up notifications distributed through the RAPEX application are kept in the system as long as the notification to which they are attached. The Commission may permanently withdraw a validated follow-up notification from the RAPEX application if a notification to which this follow-up notification is attached has been withdrawn from RAPEX (in accordance with Part II, Chapter 3.4.7.1.1 of these Guidelines). Furthermore, the Commission may withdraw a validated follow-up notification where it clearly provides incorrect information, and in particular where:
(a)
the product found on the market by the reacting Member State is different from the product covered by the notification;
(b)
the measures adopted by the reacting Member State are successfully challenged in court or in other proceedings and subsequently withdrawn;
(c)
the risk assessment performed by the reacting Member State is proven to be incorrect or relates to a different product from the one covered by the notification.
The provisions of Chapters 3.4.7.1.2 and 3.4.7.1.3 apply.
Once the Commission decides to withdraw a follow-up notification it is removed from RAPEX (or otherwise made invisible to users of the system).
The Commission informs all Member States of the withdrawal of a follow-up notification via the online collaborative space referred to in Part II, Chapter 5.3.2 or through other equally effective means.
4.5. Deadlines for submitting follow-up notifications U.K.
Member States submit follow-up notifications to the Commission as soon as possible and by the deadlines specified in Appendix 4 to these Guidelines at the latest.
Appropriate arrangements are established at national level concerning the transmission of information between all competent authorities and the RAPEX Contact Point to ensure that the deadlines are met. (See Part II, Chapter 5.1).
The deadlines apply irrespective of any appeal procedure or official publication requirement.
4.6. Requests for confidentiality U.K.
A reacting Member State may request confidentiality in its follow-up notification. Such requests clearly state which part(s) of the follow-up notification should be kept confidential. Furthermore, all requests for confidentiality are accompanied by justification clearly stating the reasons.
Requests for confidentiality are examined by the Commission to determine that they are justified (i.e. in line with the provisions of the GPSD and these Guidelines) and complete (i.e. it states which parts of the form that it covers and if it contains a justification). The final decision on confidentiality is taken by the Commission after consultation of the responsible RAPEX Contact Point. (See Part II, Chapter 5.1).
The Commission and the Member States treat follow-up notifications with requests for confidentiality in the same way as the other follow-up notifications. The confidentiality of a follow-up notification or parts of it does not prevent it from being distributed through the RAPEX application to the competent national authorities. However, neither the Commission nor the Member States should disclose any parts of a follow-up notification that are confidential to the public. This information is confidential and therefore cannot be published in any form.
The Member State submitting the follow-up notification withdraws its request for confidentiality immediately after it becomes aware that the reasons for such a request are no longer valid. The Commission informs all Member States of the withdrawal of the confidentiality after the receipt of such a request from the reacting Member State.
5. RAPEX networks U.K.
5.1. RAPEX National Contact Points U.K.
Each Member State establishes a single RAPEX Contact Point to operate RAPEX at national level. The Member States decide within which national authority to set up the RAPEX contact point. Each Member State also organises its national RAPEX network to ensure the efficient flow of information between the national contact point and the various authorities participating in RAPEX. (See Part I Chapter 5.4 and Part II, Chapter 1.2).
5.1.1. Organisation U.K.
Each Member State gives the national Contact Point the resources and information it needs to perform its tasks and, in particular, to operate the system with effective back-up/business continuity.
The RAPEX Contact Point has a separate email account for RAPEX, accessible to all officials in that contact point (e.g. rapex@ …). Professional or private email accounts of officials in charge of the RAPEX Contact Point should not be used as the email account of the RAPEX Contact Point. The RAPEX Contact Point also has a direct phone number through which it can be reached during and outside working hours.
5.1.2. Tasks U.K.
The main tasks of the RAPEX Contact Point are to:
(a)
organise and steer the work of the national RAPEX network, in accordance with the rules set out in these Guidelines;
(b)
train and assist all authorities in the network in the use of RAPEX;
(c)
ensure that all RAPEX tasks stemming from the GPSD and these Guidelines are performed correctly and, in particular, that all required information (i.e. notifications, follow-up notifications, additional information, etc.) is provided to the Commission without delay;
(d)
transmit information between the Commission and the national market surveillance authorities and authorities in charge of external border controls;
(e)
check and validate the completeness of the information received from all authorities before transmission to the Commission through the RAPEX application;
(f)
check before submitting a notification whether a product has already been notified or information on that product has been exchanged through the RAPEX application (to avoid any duplication);
(g)
participate in RAPEX Contact Point Working Group meetings and other events on the operation of RAPEX;
(h)
suggest possible improvements to the operation of the system;
(i)
inform the Commission immediately of any technical problems with the functioning of the RAPEX application;
(j)
coordinate all national activities and initiatives carried out in relation to RAPEX;
(k)
explain to stakeholders how RAPEX operates and clarify their obligations, particularly for the business notification obligation set out in Article 5(3) of the GPSD.
5.2. RAPEX networks established at EU and national levels U.K.
5.2.1. The RAPEX Contact Point Network U.K.
The Commission organises and steers the work of the RAPEX Contact Point Network. This network consists of all RAPEX Contact Points appointed in the Member States and European Economic Area (EEA) countries.
The Commission regularly convenes meetings of the RAPEX Contact Point Network to discuss the operation of the system (e.g. to communicate the latest developments concerning RAPEX, to exchange experience and ‘know-how’), and to improve cooperation between the RAPEX Contact Points.
5.2.2. RAPEX networks established at national level U.K.
The RAPEX Contact Points organise and steer the work of their own ‘RAPEX national network’. The network consists of:
(a)
the RAPEX Contact Point;
(b)
market surveillance authorities responsible for monitoring the safety of products; and
(c)
authorities in charge of external border controls.
RAPEX Contact Points are encouraged to provide for the organisation and operation of the RAPEX national network so as to ensure that all the authorities involved are aware of their roles and responsibilities as regards the operation of RAPEX. This should be consistent with the information contained in these Guidelines.
The RAPEX Contact Points are encouraged to facilitate regular and continuous exchange of information and discussion with their national network in order to discuss with all the authorities involved how RAPEX is organised, how it operates and, if necessary, to give training courses.
5.3. RAPEX internal communication tools, practical and technical arrangements for RAPEX and best practice U.K.
5.3.1. Languages U.K.
The use of languages in notifications and follow-up notifications, as well as communications between the RAPEX Contact Points and the Commission, must take account of the objectives of RAPEX and must ensure a rapid exchange of information between Member States and the Commission on products posing serious risks.
To facilitate the work of the network, Member States authorities are encouraged to use the existing EC eTranslation webpage to ensure all Member States understand what is being communicated through RAPEX.
A link to this translation tool to submit documents or extracts from texts for translation from and into all EU languages(39) is available in the collaborative space. (See Part II, Chapter 5.3.2).
5.3.2. RAPEX online tools U.K.
(a)RAPEX systemU.K.
The Commission has established and maintains a web-based application for use as a communication tool for the purpose of RAPEX. Member States use this system to create and submit notifications and follow-up notifications through the RAPEX application, and the Commission uses it to validate and distribute the documents it receives.
The Commission provides access to the system to all RAPEX Contact Points, competent national authorities and the relevant Commission departments. The Commission lays down the rules for granting access to the system and gives access to as many users as possible, taking into account needs and technical limitations.
Where the RAPEX system is temporarily not operational (for reasons other than regular and planned maintenance work), Member States should only submit notifications of serious risks to the Commission (i.e. ‘Article 12 notifications’, ‘Article 12 notifications requiring emergency action’ or ‘Article 22 of Regulation (EC) No 765/2008) notifications’.
The submission of other notifications and follow-up notifications is suspended until the RAPEX system is re-established. While the system is not operational, RAPEX notifications should be sent to the Commission by email to: just-rapex@ec.europa.eu or to another email address communicated in advance. If email transmission is not possible, RAPEX notifications are sent to the Commission by any other means considered appropriate(40).
(b)‘Product Safety Business Alert Gateway’U.K.
The ‘Product Safety Business Alert Gateway’ (also known as the ‘Business Gateway’) is intended to simplify the practical aspects of the obligation on producers and distributors, or their authorised representative, under Article 5(3) of the GPSD to notify the competent national authorities of the Member States if they know or ought to know, on the basis of the information in their possession and as professionals, that a product they have placed on the market is dangerous.
The ‘Business Gateway’ consists of two elements: (i) the notification template and (ii) the online database. The notification template is reserved for use by producers and distributors to inform the competent national authorities of the Member States that a product they have placed on the market is dangerous, in line with their obligation under Article 5(3) of the GPSD. The online database is intended for use by Member States national authorities responsible for receiving notifications of dangerous consumer products submitted by producers and distributors. The competent national authority may use the information provided to submit a RAPEX notification if all criteria for this are met.
(c)Collaborative spaceU.K.
The Commission also manages a collaborative space to exchange information between the Commission and the Member States competent national authorities. This includes the EU Consumer Product Safety platform, open to the RAPEX Contact Points and their colleagues working on product safety issues in the competent national authorities for all RAPEX-related issues. Requests for access to the space must be made by the RAPEX Contact Points in the relevant Member State and authorised by the Commission.
This space also includes a section, managed by the Commission, containing useful tips and information on the functioning of RAPEX and input from the Member States.
(d)‘RAG tool’(41) U.K.
The Commission has developed this tool available on the RAPEX website to facilitate the risk assessment of products notified through the RAPEX system, in accordance with the principles laid down in Appendix 6.
5.3.3. Contact details U.K.
The Commission provides the RAPEX Contact Points with the contact details of the Commission's RAPEX team, including names, email addresses and telephone numbers.
The RAPEX Contact Points provide the Commission with their contact details, including the names of officials working within the Contact Point, the name and address of the authority where the RAPEX Contact Point is established, the email addresses and phone numbers of officials. Any changes to the contact details are immediately communicated to the Commission by the RAPEX Contact Point. The Commission publishes and updates a list of contact details of the RAPEX Contact Points on the RAPEX website.
Member States process contact details including personal data in application of the EU's data protection legislation. On the information exchange through RAPEX, Member States should process personal data ensuring that it circulates and is distributed only as far as it is strictly necessary.
5.3.4. Operation of RAPEX outside regular working hours U.K.
RAPEX operates non-stop. The Commission and the RAPEX Contact Points ensure that officials responsible for operating RAPEX can always be contacted (by phone, e-mail or other equally effective means) and that they can take whatever action is necessary, including in an emergency and outside regular working hours, such as weekends and holidays.
The Commission provides the RAPEX Contact Points with an emergency telephone number, which should be used to contact the Commission RAPEX team outside of working hours, with priority over any other communication channels.
The RAPEX Contact Points provide the Commission with their contact details, including the phone numbers of officials who can be contacted during and outside working hours. Any changes to the contact details are immediately communicated to the Commission by the RAPEX Contact Points.
PART III U.K.APPENDICES
1. Fields and information included in notifications (42) U.K.
Fields that will be published on the web are shaded.
| * Indicates a mandatory field. | |
| Notification Form | |
|---|---|
| Section 1: General information | |
| Case number | |
| Creation date | |
| Validation/distribution date | |
| Notification type * | |
| Notifying country | |
| Full Contact details of the Notifying Authority * | |
| Section 2: Product | |
| Professional / Consumer Product | |
| Product category * | |
| OECD Portal category (if known) | |
| Product (what the product is) * | |
| Name * | |
| Brand * | |
| Type/number of model: * | |
| Batch number/Bar code * | |
| Customs code * | |
| Product and packaging description * | |
| Total number of items covered by the notification (if known) * | |
| Photos: | |
| Section 3: Regulations and standards applicable | |
| Legal provisions (directive, decision, regulation, etc.) * | |
| Standards * | |
| Proof of conformity * | |
| Is the product counterfeit? * | |
| Certificates | |
| Section 4: Traceability | |
| Country of origin (where the product manufactured) * | |
| Countries of destination * | |
| Full Contact details of the manufacturer or its representative(s) * | |
| Full Contact details of the exporter(s) * | |
| Full Contact details of the importer(s) * | |
| Full Contact details of the distributor(s) * | |
| Full Contact details of the retailer(s) * | |
| Is the product (also) sold online? | |
| Please give details: URL | |
| Section 5: Risk assessment | |
| Risk category * | |
| Risk level | |
| Summary of test results * | |
| Description of the technical issue that leads to the highest risk level | |
| Risk description (how the technical defect leads to the risk) * | |
| EU Legal provisions and /or Standards against which the product was tested and did not comply with * | |
| Information on known incidents and accidents * | |
| Section 6: Measures | |
| Type of measures adopted * | |
| If Voluntary: | Type of economic operator taking notified measure(s) * |
| Name of economic operator taking notified measure(s) * | |
| If Compulsory: | Name of authority ordering the notified measure(s) * |
| Type of economic operator to whom the measure(s) were ordered * | |
| Category of measures * | |
| Date of entry into force * | |
| Duration * | |
| Scope * | |
| Has the notification been sent by a producer or a distributor under Article 5(3) of the GPSD? * | |
| URL link to company recall page (if available) | |
| Section 7: Confidentiality | |
| Is the notification confidential? * | |
| Scope of confidentiality | |
| Justification | |
| Section 8: Other | |
| Additional information | |
| Justification for sending ‘Notification for information’ | |
| Annexes | |
| Photos (products, packaging and label) | |
| Certificates | |
| Test report and risk assessment | |
| Notification sent by an economic operator through ‘Business Gateway’ | |
| Adopted measures | |
2. Fields and information included in follow-up notifications (43) U.K.
Fields that will be published on the web are shaded.
| * Indicates a mandatory field | |
| Section 1: General information | |
|---|---|
| Case number | |
| Validated notification type | |
| Notifying country | |
| Creation date | |
| Validation/distribution date | |
| Submission number | |
| Follow up notification number | |
| Reacting country | |
| Full contact details of the notifying authority | |
| Validated notification product category | |
| Notified product | |
| Notified name | |
| Product (what the product is) | |
| Name (on the product or the packaging) | |
| Brand (on the product or the packaging) | |
| Type/number of model | |
| Batch number/Bar code (or other information to identify which products are affected) | |
| Photos (products, packaging and label) | |
| Section 2: Type of follow-up notification | |
| Product found * | |
| Total number of items found (if known) * | |
| Measures adopted / Measures not adopted | |
| Type of measures adopted * | |
| If Voluntary: | Type of economic operator taking notified measure(s) * |
| Name of economic operator taking notified measure(s) * | |
| If Compulsory: | Name of authority ordering the notified measure(s) * |
| Type of economic operator to whom the measure(s) were ordered * | |
| Category of measures * | |
| Date of entry into force * | |
| Duration * | |
| Scope * | |
| Adopted measures | |
| URL link to company recall page (if available): | |
| Different risk assessment * | |
| Risk category * | |
| Summary of the test results (description of technical defects) * | |
| Indication of legal provisions and standards (with clauses) against which the product was tested * | |
| Different risk assessment * | |
| Information on known incidents and accidents * | |
| Attachments (certificates, test report and risk assessment …) | |
| Additional information * | |
| Complementary information on distribution channels and/or product's origin | |
| Complementary information on the risk assessment | |
| Other complementary information | |
| Section 3: Confidentiality | |
| Is the follow-up confidential? * | |
| Scope of confidentiality | |
| Justification | |
| Annexes | |
| Photos (product, packaging and label) | |
| Test reports and risk assessments | |
| Certificates | |
| Adopted measures | |
3. Notification scheme U.K.
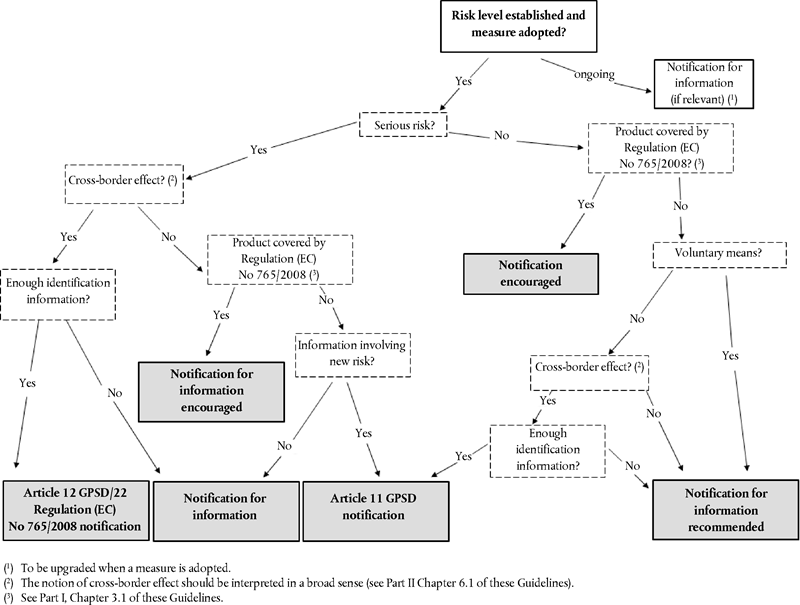
4. Deadlines for member states U.K.
Member States are required to act within the deadlines indicated unless duly justified
| Notification procedure | Action | Deadline | ||
|---|---|---|---|---|
| Notifications | Send ‘Article 12 notification requiring emergency action’ | Within 3 days after:
| ||
| Send ‘Article 12 notification’ or Article 22 Regulation (EC) No 765/2008 notification | Within 10 days after:
| |||
| Confirm measures if the notification was sent before deciding to adopt measures | Within 45 days after submission of the notification | |||
| Update to a notification | Within 5 days after receipt of the information on developments requiring changes to a notification | |||
| Follow-up notifications | Ensure follow-up activities to: | ‘Article 12 notification requiring emergency action’ | Within 20 days after receipt of a notification | |
| ‘Article 12 notification’ and to ‘Notification sent by the European Commission’ as well as Article 22 of Regulation (EC) No 765/2008 notification | Within 45 days after receipt of a notification | |||
| Send follow-up notification to: | ‘Article 12 notification requiring emergency action’ | Within 3 days after:
| ||
| ‘Article 12 notification’ and to ‘Notification sent by the European Commission’ as well as Article 22 of Regulation (EC) No 765/2008 notification | Within 5 days after:
| |||
| Update to a follow-up notification | Within 5 days after receipt of information or developments requiring changes to a follow-up notification | |||
| Notification procedure established under Article 11 of the GPSD | Notifications | Send ‘Article 11 notification’ | Within 10 days after adoption of ‘Compulsory measures’ | |
| Update to the notification | Within 5 days after receipt of information on developments requiring changes to the notification | |||
5. Deadlines for the Commission U.K.
| Notification procedure | Action | Deadline | |
|---|---|---|---|
| EU Rapid Information System ‘RAPEX’ established under Article 12 of the GPSD | Notifications | Validate ‘Article 12 notification requiring emergency action’ | Within 3 days after receipt of a notification |
| Validate ‘Article 12 notification’ as well as Article 22 of Regulation (EC) No 765/2008 notification | Within 5 days after receipt of a notification | ||
| Validate ‘Notification for information’ | Within 10 days after receipt of a notification | ||
| Follow-up notifications | Validate follow-up notification sent to ‘Article 12 notification requiring emergency action’ | Within 3 days after receipt of a follow-up notification | |
| Validate follow-up notification sent to ‘Article 12 notification’ and to ‘Notification sent by the European Commission’ as well as Article 22 of Regulation (EC) No 765/2008 notification | Within 5 days after receipt of a follow-up notification | ||
| Validate follow-up notification sent to ‘Notification for information’ | Within 10 days after receipt of a follow-up notification | ||
| Notification procedure established under Article 11 of the GPSD | Notifications | Validate ‘Article 11 notification’ | Within 10 days after receipt of a notification |
| Follow-up notifications | Validate follow-up notifications sent to ‘Article 11 notification’ | Within 10 days after receipt of a follow-up notification | |
RISK ASSESSMENT GUIDELINES FOR CONSUMER PRODUCTS (44) U.K.
1. Introduction U.K.
Consumer products may cause harm when used, e.g. a hot flat-iron that can cause burns, scissors or knives that can cause cuts, or a household cleaner that can damage the skin. This kind of damage is not a usual occurrence because general knowledge or instructions teach how to use consumer products safely. Nevertheless, the risk of damage remains.
This risk can be assessed in different ways. A range of methods have been used to quantify risk for consumer products, such as a nomograph method(45), a matrix method(46), and the method previously recommended for the EU's RAPEX rapid alert system(47). While the general principles for risk assessment have always been agreed, how to quantify risks has been under permanent development. This has led to diverging results and ensuing discussions, as well as to consideration of what the best possible practice might be.
The purpose of these risk assessment guidelines is therefore to improve the situation and, within the framework of the Directive on General Product Safety(48), to provide a transparent and practicable method for appropriate use by Member States' competent authorities when they assess the risks of non-food consumer products. These guidelines are based on a risk assessment method developed for other purposes, adapted to the specific requirements of non-food consumer products.
A certain amount of training will of course be needed before these guidelines can be put into practice, but expertise in risk assessment will greatly facilitate this task. This will be backed by exchanges of views between risk assessors, since expertise and experience accumulated through the years is invaluable.
In building up a risk assessment method in small, manageable steps, these guidelines help to focus on the relevant issues of a product, its user(s) and its use(s), and to identify possible divergences of views between risk assessors from the onset, thus avoiding time-consuming discussions. They should thus lead to consistent and robust risk assessment results based on evidence and science, and consequently to widely acceptable consensus on the risks that the many non-food consumer products may present.
A quick overview and a flow chart on how to prepare a risk assessment pursuant to these guidelines is provided in section 5 — ‘Consumer products’ mean non-food consumer products throughout these guidelines.
These guidelines do not set out to replace other guidelines that may address very specific products or may be specifically provided for in legislation, such as in the area of chemicals, cosmetics, pharmaceuticals or medical devices. It is highly recommended to use this specific guidance, since it is tailor-made, but it will always be for the risk assessor to decide how best to assess the risks of a product.
Nor are these guidelines to be used by manufacturers ‘just to avoid serious risks’ when designing and manufacturing products. Consumer products have to be safe, and these guidelines aim at helping authorities to identify serious risks when, despite the best efforts of the manufacturer, a product is not safe.
2. Risk assessment — an overview U.K.
2.1. Risk — Combination of hazard and probability U.K.
Risk is generally understood as something that threatens the health or even the lives of people, or that may cause considerable material damage. Nevertheless, people take risks while being aware of the possible damage, because the damage does not always happen. For example:
Climbing a ladder always includes the possibility of falling off and injuring oneself. ‘Falling off’ is therefore ‘built into the ladder’; it is an intrinsic part of using a ladder and cannot be excluded. ‘Falling off’ is thus called the intrinsic hazard of a ladder.
This hazard, however, does not always materialise, since many people climb ladders without falling off and injuring themselves. This suggests that there is a certain likelihood (or probability), but no certainty, of the intrinsic hazard materialising. Whereas the hazard always exists, the probability of it materialising can be minimised, for example by the person climbing the ladder being careful.
Using a household cleaner with sodium hydroxide to free blocked sewage water pipes always entails the possibility of very severe damage to the skin, if the product comes into contact with skin, or even of permanent blindness if drops of the product get into the eye. This is because sodium hydroxide is very corrosive, meaning that the cleaner is intrinsically hazardous.
Nevertheless, when the cleaner is handled properly, the hazard does not materialise. Proper handling may include wearing plastic gloves and protective glasses. Skin and eyes are then protected, and the probability of damage is much reduced.
Risk is thus the combination of the severity of possible damage to the consumer and the probability that this damage should occur.
2.2. A risk assessment in three steps U.K.
It takes three steps to determine the risk:
1.
Anticipate an injury scenario in which the intrinsic product hazard harms the consumer (see table 1). Determine how severe the consumer's injury is.
A yardstick for quantifying the intrinsic product hazard is the extent of the adverse effect that it can cause to the health of a consumer. The risk assessor therefore anticipates an ‘injury scenario’ that describes step by step how the hazard leads to the injury of a consumer (see table 2). In short, the injury scenario describes the accident that the consumer has with the product in question, and the severity of the consumer's injury caused by that accident.
An injury can vary in severity, depending on the hazard of the product, on the way the product is used by the consumer, on the type of consumer who uses the product, and much more (see section 3). The more severe the injury, the more severe the hazard that caused it, and vice versa. The ‘severity of the injury’ is therefore a means of quantifying the hazard. These guidelines propose 4 levels of severity, from injuries that are normally completely reversible to very serious injuries that cause more than approximately 10 % of permanent disability or even death (see table 3).
2.
Determine the probability of the consumer being injured in practice by the intrinsic product hazard.
While the injury scenario describes how the consumer is injured by the hazard, the scenario only happens with a certain probability. The probability can be expressed as a fraction, such as ‘> 50 %’ or ‘> 1/1 000’ (see left-hand side of table 4).
3.
Combine the hazard (in terms of severity of the injury) with the probability (in terms of a fraction) to obtain the risk.
This combination can be made by looking up both values in the appropriate table (see table 4); the table will provide the level of risk in terms of ‘serious’, ‘high’, ‘medium’ and ‘low’ risk.
Where different injury scenarios are foreseeable, the risk for each of those scenarios should be determined the highest risk being labelled as ‘the risk’ of the product. The highest risk is normally crucial because only action on the highest risk can effectively provide a high level of protection.
On the other hand, an identified risk may be lower than the highest risk, but require specific risk reduction action. It is then important also to take measures against that risk so that all risks are effectively reduced.
Once the three steps have been carried out, the risk assessment is basically complete. A flow chart on building a risk assessment is at the end of section 5.
2.3. Some useful tips U.K.
Seek informationU.K.
As can be seen from the examples of Chapter 2.1, each of the three steps of a risk assessment (see point 2.2) requires anticipation of what might happen and how likely it is to happen, since the product under consideration will normally not have caused an accident, and thus the risk will not have materialised (yet). Previous experience with similar products will help in this exercise, as will any other information about the product, such as design, mechanical stability, chemical composition, operation, instructions for use, including possible risk management advice, type of consumers it is intended for (and those for which it is not), test reports, accident statistics, the EU Injury Database (IDB)(49), information about consumer complaints, about the behaviour of different consumers when they are using the product, and about product recalls. Product requirements laid down in legislation, in product standards or in checklists (such as in ISO 14121: Safety of machinery — Risk assessment) can also be useful sources of information.
Nevertheless, the products to be assessed may be quite specific and thus these sources may not contain the information required. The information collected may also be incomplete, inconsistent, or not fully plausible. This may be the case in particular for accident statistics, when only the product category is registered. The absence of an accident history, a small number of accidents or low severity of accidents should not be taken as a presumption of low risk. Product-specific statistics also have to be viewed with great care, since the product may have changed over time, be it in design or composition. The information must always be critically assessed.
Feedback from expert colleagues can be particularly useful, since they can draw from their real-life experience and pro vide suggestions that are not immediately obvious when assessing a product risk. They may also give advice when assessing the risk for different types of consumers, including vulnerable consumers such as children (see table 1), since the latter may handle a product differently. They may also help to assess the risk for different injuries that a product may cause, and the way in which those injuries emerge through the use of the product. They can also judge whether an injury scenario is ‘totally unperceived’, too unlikely, and then guide the risk assessor towards more realistic assumptions.
Thus, feedback from experienced colleagues, although not an obligation, can be helpful in several aspects. A risk assessor from an authority could seek advice from colleagues in that same authority, in other authorities, in industry, in other countries, in scientific groupings, and elsewhere. Conversely, any risk assessor in industry could use his contacts with authorities and others when a new or improved product is to be assessed before it is placed on the market.
New information obtained should of course be used to update any existing risk assessment.
Make a sensitivity analysis of your risk assessmentU.K.
If all information searches and queries to expert colleagues do not provide the required, very specific data, a so-called sensitivity analysis might help. In this analysis a lower and a higher value than previously chosen is assumed for each parameter of the risk assessment, and taken through the entire risk assessment procedure. The resulting risk levels will show how sensitive the risk level reacts to the input of lower and higher values. In this way the range in which the real risk of the product will be can be estimated.
If the most likely value of each parameter can be estimated, then those most likely values should be taken through the procedure, and the resulting risk level will be the most likely risk.
An example of a sensitivity analysis is illustrated in section 6.
Let others check your risk assessmentU.K.
Feedback from colleagues will also help when finalising the risk assessment. They will be able to provide advice on the assumptions and estimations made during the three steps referred to in point 2.2. They will feed in their experience and thus help to generate a more robust, more solid, more transparent and ultimately more acceptable risk assessment. It is therefore recommended that, ideally, advice be sought from expert colleagues, possibly in the form of a group discussion, before concluding a risk assessment. These groups, of perhaps 3 to 5 members, should include a combination of expertise appropriate to the product under assessment: engineers, chemists, (micro-)biologists, statisticians, product safety managers, and others. Group discussion will be particularly useful when a product is new on the market and has never been assessed before.
Risk assessments should be solid and realistic. However, since they require a number of assumptions, different risk assessors may come to different conclusions in view of the data and other evidence they have been able to find or because of their diverging experience. It is thus necessary for risk assessors to talk to one another in order to reach agreement or, at least, consensus. The step-by-step risk assessment described in these guidelines, however, should make such discussions more productive. Each step in a risk assessment must be clearly described in detail. Thus, any point of disagreement can be quickly identified, and consensus can more easily be reached. This will make risk assessments more acceptable.
Document your risk assessmentU.K.
It is important to document your risk assessment, describing the product and all the parameters that you chose while developing it, such as test results, the type(s) of consumers you chose for your injury scenario(s), and the probabilities with the underlying data and assumptions. This will enable you to demonstrate unambiguously how you estimated the level of risk, and it will also help you to update your assessment while keeping track of all changes.
Several hazards, several injuries — but only one riskU.K.
When several hazards, several injury scenarios or differing severities of injuries or probabilities have been identified, each of those should be carried through the entire risk assessment procedure in order to determine the risk for each. As a result, the product may have several risk levels. The overall risk of the product is then the highest risk level identified, because action on the highest risk level is normally the most effective way of risk reduction. Only in special cases may a less-than-highest risk be considered particularly important, since it may require specific risk management measures.
As an example of several risks, a hammer may have a weak head and a weak grip, each of which may break when the hammer is used, and the consumer may be injured. If the relevant scenarios lead to different risk levels, the highest risk should be reported as ‘the risk’ of the hammer.
It could be argued that:
the apparently most significant hazard should be decisive, since it would lead to the most severe injuries. In the example of the hammer in point 2.1, this could be the hammer head breaking, since pieces of the broken head could fly into one's eye, possibly blinding the user. The hammer grip breaking, on the other hand, would never split into small pieces that could do as much damage to the eyes;
However, this would be a hazard assessment, not a risk assessment. A risk assessment also looks at the probability of an injury actually happening. Thus, the ‘most significant hazard’ might cause an injury that is much less likely than a lesser hazard, and therefore present a lower risk. Conversely, a scenario leading to a less severe injury may be much more likely than a scenario resulting in death, and the less severe injury may therefore present a higher risk;
the highest probability for an injury scenario to happen should be the decisive factor for ‘the risk’ of the product. In the example of the hammer in point 2.1, if the hammer grip is very weak, the most likely injury scenario would be from the grip breaking, and that should therefore be decisive.
However, this would not consider the seriousness of eye injuries that the hammer head breaking could cause. Looking at probability alone would not therefore give the whole picture.
In conclusion, risk is a balanced combination of both the hazard and the probability of the injury that the hazard can cause. Risk describes neither the hazard, nor the probability, but both at the same time. Taking the highest risk as ‘the risk’ of the product will ensure the most effective product safety (apart from specific risks requiring specific risk management, as referred to at the beginning of this section.
Can risks cumulate?U.K.
Several injury scenarios leading to several risks can be developed for virtually every product. For example, an angle grinder may present the risk of an electric shock, because electrical wires may be too exposed, and the risk of fire, because the machine may overheat and ignite during normal use. If both risks are considered to be ‘high’, do they add up to the grinder posing an overall ‘serious risk’?
Where several risks are linked to the same product, one of them is obviously more likely to materialise and causes an injury. The overall likelihood of an injury is therefore greater. This does not mean that the overall risk is automatically higher, however:
The overall probability is not calculated by simply adding up probabilities. More complex calculations are necessary, and these always result in a probability that is lower than the sum of all probabilities.
There is difference of a factor of 10 between two succeeding probability levels (table 4). This means that a lot of different scenarios of the same level would be needed to result in higher overall probability (and possibly risk).
Probability values are estimations which may not be totally accurate, as they often err on the ‘safe’ side in order to ensure a high level of protection. It is therefore more useful to look at a more accurate estimation of the probability of a scenario leading to the highest risk than to add up rough estimations of probabilities of all sorts of scenarios.
With a little effort hundreds of injury scenarios could be developed. If risks were simply added together, the overall risk would depend on the number of injury scenarios generated and could increase ‘endlessly’. This does not make sense.
Thus, risks are not simply cumulated. However, if more than one relevant risk exists, action to manage the risks may need to be taken more rapidly or may need to be more pronounced. For example, with two risks, a product may need to be immediately taken off the market and recalled, whereas, with a single risk, halting sales could be sufficient.
Risk management depends on many factors, not only on the number of risks that a product may present at one and the same time. Thus, consideration is given to the link between risk and risk management (section 4).
Compliance with limit values in legislation and standardsU.K.
In market surveillance, consumer products are often tested against limit values or requirements laid down in legislation and in product safety standards. A product that complies with the limit value(s) or requirement(s)(50) is presumed to be safe in terms of the safety characteristics covered by those value(s) or requirement(s). This assumption can be made because the risks of a product from its intended and reasonably foreseeable use are taken into account when establishing the limit value(s) or requirement(s). Manufacturers thus need their products to comply with these values or requirements, because they then only have to look at risks with their products that are not be covered by those limit value(s) or requirement(s).
An example of a limit value in:
legislation is the limit of 5 mg/kg benzene in toys which must not be exceeded, as per point 5 of Annex XVII, to the REACH Regulation(51), as amended by Commission Regulation (EC) No 552/2009(52);
a standard is the small parts cylinder: small parts of a toy for children under 36 months must not fit entirely into the cylinder described in the Toys Standard(53). If they do, they present a risk;
The product is presumed not to be safe where it fails to comply with established limit values. For limit values laid down in:
legislation, such as on cosmetics or restrictions on marketing and use, the product must not be made available on the market;
standards, the manufacturer may nevertheless try to provide evidence that his product is as safe as if it were compliant with the standard's limit value by way of a fully-fledged risk assessment on his product. However, this may require more effort, and may be impossible in cases such as the small parts cylinder referred to in the first bullet of this list, than actually manufacturing the product in compliance with the standard's limit value.
Non-compliance with limit values does not automatically mean that the product presents a ‘serious risk’ (which is the highest risk level covered by these guidelines). Therefore, to ensure appropriate risk reduction measures, a risk assessment will be required for those parts of a product that do not comply with or are not covered by legislation or a standard.
Furthermore, some products, such as cosmetics, require a risk assessment even when they are compliant with the limit values laid down in legislation. This risk assessment should provide evidence of the safety of the whole product(54).
In conclusion, compliance with limit values in legislation or in standards provides presumption of safety, but such compliance may not be sufficient.
Specific risk assessment guidelines in specific casesU.K.
For chemicals there are specific instructions on how to prepare a risk assessment(55), and therefore they are not dealt with in detail in these guidelines. Nevertheless, they follow the same principles as for ‘normal’ consumer products:
hazard identification and assessment — this is the same as determining the severity of the injury, as described in section 2.2;
exposure assessment — in this step, exposure is expressed as the likely dose of the chemical that the consumer may take up via oral, inhalation or dermal routes, separately or jointly, when using the product as anticipated in the injury scenario. This step is the same as determining the probability that the injury will indeed occur;
risk characterisation — this step basically consists of comparing the dose of the chemical that the consumer is likely to take up (= exposure) with the derived no-effect level (DNEL) of that chemical. Should the exposure be sufficiently lower than the DNEL, in other words, should the risk characterisation ratio (RCR) be clearly below 1, risk is considered to be adequately controlled. This is the same as determining the risk level. Risk management measures may not be needed if the level of risk is sufficiently low.
Since a chemical may possess several hazards, risk is normally determined for the ‘leading health effect’, which is the health effect (or ‘endpoint’ such as acute toxicity, irritation, sensitisation, carcinogenicity, mutagenicity, toxicity for reproduction) considered to be the most important.
For cosmetics, there is also specific guidance(56), and there may be specific guidance for other products or purposes.
It is highly recommended to use such specific guidance, since it is tailored to the specific cases in question. Nevertheless, where the data required by the specific guidance do not exist or cannot be estimated the present guidelines may be used for a preliminary risk assessment. This risk assessment will have to be carried out with due care and attention in order to avoid any misinterpretation.
3. Building a risk assessment step by step U.K.
This section describes in detail what points have to be taken into account and what questions have to be asked when preparing a risk assessment.
3.1. The product U.K.
The product should be identified unambiguously. This includes the product name, the brand, the model name, the type number, a possible production lot number, any certificate that may come with the product, a child-resistant fastening if there is one, the identity of the person who placed it on the market, and the country of origin. A picture of the product, the packaging and the marking plate (if appropriate) and a test report(s) identifying the product hazard(s) can also be considered to be part of the product description.
In particular cases, the hazard may be limited to a distinct part of the product, which can be separate from it and also separately available to consumers. In such cases, it is sufficient only to assess the distinct part of the product. Recharge able batteries of notebook computers which may overheat are an example of this.
The description of the product includes any label that may be relevant for risk assessment, in particular warning labels. Instructions for use may also contain relevant information on the risk of the product and how to keep it as low as possible, for example by using personal protective equipment or by excluding children from using the product. An example of this is a chain saw.
Products may also need to be self-assembled by consumers before use, such as self-assembled furniture. Are the assembly instructions clear enough for the ready-to-use product to meet all the relevant safety requirements? Or could consumers make mistakes when putting the product together that could lead to unforeseen risks?
A risk assessment should always consider the entire life time of a product. This is particularly important when a new product has been developed and its risks are assessed. Will age and usage change the type or the extent of the hazard? Will new hazards appear with increasing product age or perhaps through reasonably foreseeable inappropriate use? How long is the ‘time to product failure’? What is the product's lifetime, including shelf life? How long is the product used in practice by the consumer before it becomes waste?
Additional considerations may need to be taken into account when a product becomes unusable after a certain time period, even though it has never been used. Examples are electric blankets or heating pads. The electric cords in the products are usually thin and become fragile after ten years, even if the product has never been used. The heating cords can come into contact with each other, can cause a short-circuit and set the bedclothes on fire.
Finally, the packaging of the product should also be included in any risk assessment.
3.2. The product hazard U.K.
Hazard is the intrinsic property of the product that may cause an injury to the consumer who uses the product. It can appear in different forms:
mechanical hazard, such as sharp edges that can cut fingers, or tight openings in which someone can trap their fingers;
choking hazard, such as from small parts that come loose from a toy, which may be swallowed by a child and make the child choke;
suffocation hazard, such as from the drawstrings of an anorak hood which may lead to strangulation;
electrical hazard, such as from live electrical parts that can cause an electric shock;
heat or fire hazard, such as a heater fan that overheats, catches fire and causes burns;
thermal hazard, such as the hot outer surface of an oven that can cause a burn;
chemical hazard, such as a toxic substance that can poison a consumer immediately upon ingestion, or a carcinogenic substance that can cause cancer in the long term. Some chemicals may damage the consumer only after repeated exposure;
microbiological hazard, such as a bacteriological contamination of cosmetics which may cause a skin infection;
noise hazard, such as ring tones from toy mobile phones that are much too loud and can damage children's hearing capacity;
other hazards, such as explosion, implosion, sonic and ultrasonic pressure, fluid pressure, or radiation from laser sources.
For the purpose of these guidelines, hazards have been grouped, linked to the size, shape and surface of a product, to potential, kinetic or electric energy, to extreme temperatures, and others, as shown in table 2. The table is for guidance only, and any risk assessor should adapt the scenario to the product under consideration. Of course not every type of hazard applies to every product.
Nevertheless, table 2 should help risk assessors to look for and identify all possible hazards in consumer products that are being assessed. Where a product has several hazards, each hazard should be taken separately with its own risk assessment and the highest risk identified as ‘the risk’ of the product. Of course, risks requiring specific risk management measures should also be reported, to ensure that all risks can be reduced.
Note that a single hazard may lead to several injuries in the same scenario. For example, malfunctioning brakes on a motor cycle could cause an accident and result in damage to the driver's head, hands and legs, and could even cause burns if the petrol bursts into flames in the accident. In this case, all injuries would belong to the same injury scenario, and the severity of all injuries together would have to be estimated. Of course, these injuries together are very serious. Several injuries in different scenarios should, however, not be added.
In the daily practice of market surveillance, it may be sufficient to assess the risk from even a single hazard. If the risk from that hazard provides for risk management action, that action can be taken without further ado. Nevertheless, the risk assessor should be sure that the risk identified is (one of) the highest risk(s), to ensure that the risk management action is sufficiently effective. This is always the case when the risk is serious, since this is the highest possible risk level proposed in these guidelines. In cases of less than serious risk, however, further risk assessments might be necessary and possibly specific risk management at a later stage. In conclusion, experience with risk assessment in market surveillance practice will limit the number of required risk assessments to a minimum.
Hazard identification by tests and standardsU.K.
Hazards are often identified and quantified by tests. These tests and how to carry them out may be laid down in European or international product standards. Compliance of a product with a ‘harmonised’ European standard (‘EN …’), of which the references have been published in the Official Journal, provides presumption of safety (albeit only for the safety characteristics covered by the value(s) or standard(s)). It can be presumed in such cases that the product presents only a minimum risk and a high level of protection with regard to the specific hazard tested.
Nevertheless, there may be instances where presumption of safety is not the case, and in such cases a particularly well-documented risk assessment will have to be prepared, including a call for amendment to the harmonised standard.
On the other hand, if a product fails the test, a risk can normally be assumed, unless the manufacturer can provide evidence that the product is safe.
Products may still present a risk even though they do not cause injuriesU.K.
Products may not be hazardous but can nevertheless cause a risk, due to not being fit for their intended use. Examples of this can be observed in the area of personal protective equipment or life-saving equipment, such as reflective jackets that car drivers put on after an accident. These jackets are meant to get the attention of oncoming drivers and traffic participants to warn them of the accident, in particular at night. However, they might not be seen if the reflector stripes are too small or do not reflect sufficiently, and do not therefore protect users as they should. These jackets therefore pose a risk even though they are not hazardous in themselves. Another example is a sunscreen product which displays ‘high protection’ (sun protection factor of 30) on the label but provides only ‘low protection’ (factor of 6). This can lead to severe sunburn.
3.3. The consumer U.K.
The abilities and behaviour of the consumer using the product may greatly influence the level of risk. It is therefore of prime importance to have a clear idea of the type of consumer pictured in the injury scenario.
It may be necessary to generate injury scenarios with different types of consumers in order to identify the highest risk and thus ‘the risk’ of the product. It is not enough, for example, to consider only the most vulnerable consumers, because the probability of their suffering adverse effects in the scenario may be so low that the risk is lower than in an injury scenario with a non-vulnerable consumer.
Consideration should also be given to people who are not actually using the product, but who may be in the vicinity of the user. For example, a chain saw may cause splinters to fly around and hit a bystander in the eye. Thus, although the risk from the chain saw may be effectively managed by the user him- or herself wearing protective equipment and complying with any other risk management measures specified by the manufacturer, bystanders may be under serious threat. Consequently, warnings should be given, for example in the chain saw instructions for use, about the risks to bystanders and how to minimise such risks.
Thus, when developing an injury scenario, the following aspects should be taken into account regarding the type of consumer and how they use the product. This is not a complete list, but it should encourage risk assessors to describe their injury scenarios with the necessary level of detail. It should be noted that ‘consumer’ also means people who are not actually using the product, but who may be affected by virtue of being nearby:
Intended/non-intended user: The intended user of a product may use the product with ease because he goes by the instructions or because he is familiar with this kind of product, including its apparent and non-apparent hazard(s). The hazard of the product may not then materialise, and the product risk could be minor.
The non-intended user may not be familiar with the product and may not recognise the hazard(s). He therefore runs the risk of injury, and the consumer risk is thus higher.
Thus, the risk may be different for an intended and a non-intended user, depending on the product and the way it is used.
Vulnerable consumers: Several categories of vulnerable and very vulnerable consumers can be distinguished: children (0 to 36 months, > 36 months to < 8 years, 8 to 14 years) and others such as the elderly (see table 1). They all have less capacity to recognise a hazard, for example children who, when touching a hot surface, notice the heat only after some 8 seconds (and then are already burnt), whereas adults notice heat immediately.
Vulnerable consumers may also have problems taking account of warning labels, or may have particular problems using a product they have never used before. They may also act in a way that makes them more exposed, for example young children crawling and mouthing. Children may also be attracted to products because of their appeal, which makes them a high risk in the hands of children. On the other hand, supervision by parents or other adults should normally prevent children from running straight into trouble.
Furthermore, consumers who are not usually vulnerable may become vulnerable in specific situations, for example when the instructions or warnings on a product are in a foreign language that the consumer does not understand.
Finally, in the particular case of chemicals, children may be more susceptible to the toxicity of chemicals than the average adult. Therefore, children should not be treated as if they were ‘small adults’.
In conclusion, a product that is normally safe for an average adult may not be safe for vulnerable consumers. This has to be taken into account when determining the severity and probability of an injury (see section 3.5) and thus the risk.
Intended and reasonably foreseeable use: Consumers may use a product for other purposes than the one for which it is intended, although the instructions are clearly understandable, including any warnings. Therefore, as warnings may not be fully effective, other uses than the intended ones also have to be taken into account in a risk assessment. This aspect is particularly important for the manufacturer of a product, since he has to ensure that the product is safe under any reasonably foreseeable conditions of use.
Reasonably foreseeable use may have to be based on experience, because there may be no information available in official accident statistics or other sources of information. It may then be difficult to draw the line between ‘reasonably foreseeable’ and ‘totally unperceived’ scenarios. Nevertheless, even ‘totally unperceived’ scenarios can be considered under these guidelines, even when they lead to very severe injuries, because such scenarios will always have very low probability. This possibly safeguards against such scenarios having too much of an influence in determining the overall risk of the product.
Frequency and duration of use: Different consumers may use a product often or not so often, and for longer or shorter periods of time. This depends on the attractiveness of the product and the ease with which it can be used. Daily or long-term use could make a consumer entirely familiar with a product and its specifics, including its hazards, instructions and warning labels, thus making the risk minor. On the other hand, daily or long-term use may make the consumer too used to the product and lead to user fatigue where he recklessly ignores instructions and warnings, thus increasing the risk.
Finally, daily or long-term use may also accelerate product ageing, and any parts that cannot withstand such frequent use may quickly fail and cause a hazard, and possibly an injury, which also increases the risk.
Hazard recognition and protective behaviour and equipment: Some products are known for their hazards, such as scissors, knives, do-it-yourself drilling machines, chain saws, roller blades, bicycles, motor bikes and cars. In all these cases, the product hazard is clearly known or readily recognisable, or described in the instructions, which will include risk management measures. The consumer can then act carefully or use personal protective equipment such as gloves, helmets or seat-belts, thereby using the product in a way that minimises the risk.
In other cases, the product hazard may not be so readily recognisable, such as a short-circuit within an electric iron, warning labels may be overlooked or misunderstood, and consumers will only rarely be able to take preventive measures.
Consumer behaviour in the event of an incident: Where the hazard impinges on the consumer it may cause injury. It is thus important for a risk assessment to consider how the consumer may react. Will he put the product to one side calmly and take preventive action, such as combating a fire caused by the product, or will he throw it away in a panic? Vulnerable consumers, especially children, may after all not behave the same as other, non-vulnerable consumers.
The consumer's cultural background and the way a product is used in his home country may influence the risk of a product. Manufacturers in particular have to take account of these cultural differences when launching a new product on a market. Manufacturers' experience in this area can thus be a valuable source of information for authorities preparing a risk assessment.
3.4. Injury scenario: Steps leading to injury(ies) U.K.
Most injury scenarios consist of the following three main steps:
1.
the product has a ‘defect’ or can lead to a ‘dangerous situation’ during its foreseeable lifetime;
2.
the ‘defect’ or ‘dangerous situation’ results in an accident;
3.
the accident results in an injury.
These three main steps can be divided into further steps to show how the product hazard can lead to injury and the like. Nevertheless, these ‘steps to injury’ have to be clear and concise, and not exaggerate the detail or the number of steps. With experience, it will be increasingly easier to identify the conditions for the occurrence of any given injury and the ‘shortest path to injury’ (or ‘critical path to injury’).
It is probably easiest to start with a scenario with the consumer for whom the product is intended where the consumer uses the product as per the instructions or, if there are none, according to normal handling and use. If this assessment produces the highest risk level, there is normally no need to carry out further assessments, and appropriate risk reduction measures can be taken. Similarly, where an incident is reported in a specific consumer complaint, a single injury scenario may be sufficient to conclude as to appropriate risk reduction measures.
Otherwise, further scenarios could be developed to include vulnerable consumers, in particular children (see table 1), slight or more pronounced deviations from normal use, use under different climate conditions, such as very cold or very hot, unfavourable conditions of use, such as without proper daylight or illumination, use as suggested when the product was sold (for instance, a lamp sold in a toy shop should also be assessed for its risk when used by a child), use over the entire life-time (including wear and tear), etc. Each scenario should be considered through the entire risk assessment procedure.
Where the product displays several hazards, injury and thus risk scenarios should be developed for each of them. Nevertheless, a plausibility check as to whether an injury scenario might lead to a risk requiring action can limit the number of injury scenarios.
From all the scenarios generated, the scenario providing the highest risk (= ‘the risk’ of the product) will normally be decisive for the risk reduction measures to be taken, because action on the highest risk reduces the risk most effectively. An exception to the rule might be a specific, less-than-highest risk stemming from a different hazard, which could be managed by specific measures and should, of course, also cover the highest risk.
As a rule of thumb, injury scenarios can lead to the highest risk level when:
the injury(ies) considered are in the highest severity levels (levels 4 or 3);
the overall probability of an injury scenario is quite high (at least > 1/100).
Table 4 provides further guidance in this respect. This might help to limit the number of scenarios.
Of course, the number of injury scenarios remains the responsibility of the risk assessor, and it depends on the number of factors that need to be taken into account when determining ‘the risk’ of the product. It is therefore impossible to give a specific number of injury scenarios that may be necessary in a specific case.
To help develop a suitable number of scenarios, these guidelines provide a table with typical injury scenarios (table 2). These should be adapted to the specific product, consumer type and other circumstances.
3.5. Severity of injury U.K.
The injury that a hazard can cause to the consumer can have different degrees of severity. The severity of the injury thus reflects the effect the hazard has on the consumer under the conditions described in the injury scenario.
The severity of the injury can depend on:
the type of hazard (see list of hazards of section 3.2 in table 2). A mechanical hazard, such as sharp edges, can cause cuts to the fingers; these are immediately noticed, and the consumer will take action to heal his injuries. On the other hand, a chemical hazard may cause cancer. This normally passes unnoticed, and the illness may appear only after many years, and is considered to be very severe since cancer is very difficult to cure, if at all;
how powerful the hazard is. For example, a surface heated to 50 °C may cause slight burns, whereas a surface at 180 °C will cause severe burns;
how long the hazard impinges on the consumer. A short contact time with an abrasion hazard may scratch the consumer's skin only superficially, whereas a longer time may take off large parts of the skin;
what body part is injured. For example, penetration by a sharp point into the skin of the arm is painful, but penetration into an eye is a more serious and perhaps a life-affecting injury;
what impact the hazard has on one or several body parts. An electrical hazard may cause an electric shock with unconsciousness and, subsequently, a fire which may damage the lungs when the unconscious person inhales the smoke;
the type and behaviour of the consumer. A product labelled with a warning message can be used, without harm, by an adult consumer, because the consumer adjusts to using the product. On the other hand, a child or other vulnerable consumer (see table 1) who cannot read or understand the warning label may be very seriously injured.
To quantify the severity of injury(ies), table 3 in these guidelines shows how to classify injuries into four categories, depending on the reversibility of an injury, i.e. whether recovery from an injury is possible and to what extent. This categorisation is for guidance only, and a risk assessor should change the category if necessary, and report it in the risk assessment.
Where several injury scenarios are considered in the risk assessment, the severity of each injury should be classified separately, and considered throughout the entire risk assessment process.
An example: A consumer uses a hammer to knock a nail into a wall. The hammer head is too weak (due to incorrect material) and it breaks, one of the pieces flying into the eye of the consumer so hard that it causes blindness. The injury is thus an ‘eye injury, foreign body in eye: permanent loss of sight (one eye)’, which is a level 3 injury in table 3.
3.6. Probability of injury U.K.
The ‘probability of injury’ is the probability that injury scenario may indeed materialise during the expected lifetime of the product.
This probability is not easy to estimate; but when a scenario is described in distinct steps, each step can be given a certain probability, and multiplying these partial probabilities together gives the overall probability of the scenario. This stepwise approach should make it easier to estimate the overall probability. Of course, where several scenarios are developed, each scenario requires its own overall probability.
Where an injury scenario is nevertheless described in a single step, the probability of the scenario can also only be determined in a single overall step. This would only be a ‘guesstimate’, however, which could be severely criticised and thus call the entire risk assessment into question. A more transparent assignment of probabilities to a several-step scenario is therefore preferable, especially as the partial probabilities can be built on undisputable evidence.
These guidelines distinguish between 8 levels of probability to classify overall probability: from < 1/1 000 000 to > 50 % (see left-hand side of table 4). The following example of a hammer head that breaks when the user knocks a nail into a wall should illustrate how to assign a probability to each step, and how to classify overall probability:
| Step 1: | The hammer head breaks when the user tries to knock a nail into a wall because the material of the hammer head is too weak. The weakness was determined in a test, and with the reported weakness the probability of the hammer head breaking during the otherwise expected lifetime of the hammer is put at 1/10. |
| Step 2: | One of the pieces of the hammer hits the user when it breaks. The probability of this happening is put at 1/10, since the area of upper body exposed to the pieces flying off is considered to be 1/10 of the half-sphere in front of the wall. Of course, if the user were standing very close to the wall, his body would take a larger share of the half-sphere, and the probability would be higher. |
| Step 3: | The piece hits the user on the head. The head is estimated to be about 1/3 of the upper body, and the probability is therefore 1/3. |
| Step 4: | The piece hits the user in the eye. The eyes are considered to be about 1/20 of the area of the head, and therefore the probability is 1/20. |
Multiplying the probabilities of these steps together gives an overall probability for the scenario of 1/10 × 1/10 × 1/3 × 1/20 = 1/6 000. This translates into > 1/10 000 (see left-hand side of table 4).
Once the overall probability has been calculated for an injury scenario, it should be checked for plausibility. This requires rather a lot of experience, thus suggesting that the assistance of persons experienced in risk assessment should be sought (see section ‘Let others check your risk assessment’). As experience is gained with these guidelines estimating probability should become easier, and an increasing number of examples will become available to facilitate this task.
Assigning probabilities to different injury scenarios for the same product may lead to the following:
When the product is used by more vulnerable consumers in a scenario, the probability may have to be raised in general because more vulnerable consumers can be injured more easily. This applies in particular to children, since children do not normally have the experience to take preventive action, on the contrary (see also ‘Vulnerable consumers’ in section 3.3).
When the risk is readily recognisable, including through warning labels, the probability may have to be lowered because the user will use the product more carefully in order to avoid injury as far as possible. This may not apply to an injury scenario with a (young) child or other vulnerable user (see table 1) who cannot read.
When accidents have been reported that fit into the injury scenario, the probability for that scenario could increase. In cases where accidents have only rarely been reported, or are not known at all, it may be useful to ask the manufacturer of the product whether he is aware of any accident or adverse effect caused by the product.
When a fairly large number of conditions are needed for the injury to occur, the overall probability of the scenario would normally be lower.
When the conditions needed for the injury to occur are easily met, this may increase the probability.
When the test results of the product fail by a large margin to come within the limit values required (by the relevant standard or legislation), the probability of the injury (scenario) occurring may be higher than if the product performed close to the limit values.
The ‘probability of injury’ in this instance is the probability that the injury scenario may actually happen. Probability does not therefore describe the general exposure of the population to the product, calculated, for example, by considering the millions of product items sold on the market and then considering that a few of them might fail. Considerations of this kind do, however, play a role when determining the appropriate risk reduction measures (see section 4).
Also, accident statistics, even if product-specific, have to be considered with care when used for to estimate probability. The circumstances of the accident may not be reported in sufficient detail, the product may have changed over time, or the manufacturer may be different, and so on. In addition, light accidents may not have been reported to those collecting the data for the statistics. None the less, accident statistics can shed light on injury scenarios and their probability.
3.7. Determination of risk U.K.
Once the severity of the injury and the probability have been determined, if possible for several injury scenarios, the risk level then needs to be looked up in table 4. Table 4 combines both the severity of the injury and the probability, and the highest risk is ‘the risk’ of the product. Risks requiring specific risk management measures should also be reported, to ensure that all risks are reduced to a minimum.
These guidelines distinguish between 4 levels of risk: serious, high, medium and low. The risk level between neighbouring severities of injury or probability normally changes by 1 level. This is consistent with the general experience that risk does not increase incrementally when input factors change gradually. However, where the severity of injury increases from level 1 to level 2 (on the right-hand side of table 4), some risk levels increase by 2 levels, namely from medium to serious and from low to high. This is due to the fact that these guidelines include 4 graduations of severity of injury, whereas the original method (see Introduction) included 5. Nevertheless, 4 graduations are considered normal for consumer products, since they make for a sufficiently robust estimation of severity; 5 levels would be too sophisticated since neither the severity of the injury nor the probability can be determined with very high precision.
At the end of the risk assessment, be it for an individual injury scenario or for the overall risk of the product, the plausibility of the risk level and uncertainties in the estimates should be considered. This may mean verifying that the risk assessor has used the best information available to make his estimations and assumptions. Feedback from colleagues and other experts can also be helpful.
A sensitivity analysis can also be very valuable (see example in section 6.3). How does the risk level change when the severity of injury or probability changes by 1 level up or down? If the risk level does not change at all, it is quite plausible that it has been estimated correctly. If it changes, however, the risk level may be borderline. It is then necessary to reconsider the injury scenarios and the assigned severity of injury(ies) and probability(ies). At the end of the sensitivity analysis the risk assessor should be confident that the risk level is sufficiently plausible and that he can document it and pass the information on.
4. From risk to action: how to manage risk responsibly U.K.
Once the risk assessment is complete it will normally be used to decide whether action needs to be taken to reduce the risk and thus prevent harm to a consumer's health. Although action is separate from risk assessment, some points are raised here to illustrate the possible follow-up of identified risks.
Within market surveillance, action will often be taken in contact between the authority and the manufacturer, importer or distributor. This can help the authority to determine the most effective and efficient way of managing the risk.
With a serious risk in a consumer product, measures to reduce the risk may include withdrawal from the market or recall. Lower levels of risk normally lead to less rigorous measures. It may then be sufficient to add warning labels on the product or to improve the instructions to make the product safe. Thus, whatever the level of risk, the authority should consider whether to take action, and if so, what action.
Nevertheless, there is no automatic link from risk to action. When a product shows several less-than-serious risks, and its overall risk is thus not serious, urgent action may be necessary since any of the risks may materialise quite quickly. The pattern of risks in the product may indicate a lack of quality control in production(57).
It is also important to take account of exposure of the population as a whole. Where there are a large number of products on the market and the product is therefore used by a large number of consumers, even a single less-than-serious risk may require quick action to avoid adverse effects to the health of consumers.
Less-than-serious risks may also require action when the product concerned could cause fatal accidents, even though such accidents may be extremely unlikely. This could be the case with a fastening on a beverage container, which could come loose and be swallowed by a child, causing the child to choke to death. A simple change of design to the lid could eliminate the risk, and no further action might be required. Even a selling-off period may be granted if the risk of a fatal accident were indeed extremely small.
Other risk-related aspects may be the public perception of risk and its likely consequences, cultural and political sensitivities and how it is portrayed in the media. These aspects may be especially relevant when the consumers concerned are vulnerable, in particular children. It will be up to the national market surveillance authority(ies) to determine what measures are required.
Taking action to counteract a risk may also depend on the product itself and the ‘minimum risks compatible with the product's use, considered to be acceptable and consistent with a high level of protection’(58). This minimum risk will probably be much lower for toys, where children are involved, than for a chain-saw, which is known to be so high-risk that solid protective equipment is required to keep the risk at a manageable level.
Finally, even if there is no risk, action may be necessary, for example, when a product is non-compliant with the applicable regulation/legislation (e.g. incomplete markings).
In conclusion, there is no automatic link from risk to action. Surveillance authorities will take account of a range of factors such as those indicated in section 3.3. The principle of proportionality always has to be considered, and action has to be effective.
5. How to prepare a risk assessment — in brief U.K.
1.Describe the product and its hazard.U.K.
Describe the product unambiguously. Does the hazard concern the entire product or only a (separable) part of the product?
Is there only one hazard within the product? Are there several hazards? See table 2 for guidance. Identify the standard(s) or legislation applicable to the product.
Identify the standard(s) or legislation applicable to the product.
2.Identify the type of consumer you want to include in your injury scenario with the hazardous product.U.K.
Start with the intended user and the intended use of the product for your first injury scenario. Take other consumers (See table 1) and uses for further scenarios.
3.Describe an injury scenario in which the product hazard(s) you have selected causes an injury(ies) or adverse health effect(s) to the consumer you selected.U.K.
Describe the steps to the injury(ies) clearly and concisely, without exaggerating the details (‘shortest path to injury’, ‘critical path to injury’). If there are several concurrent injuries in your scenario, include them all in that same scenario.
When you describe the injury scenario, consider the frequency and duration of use, hazard recognition by the consumer, whether the consumer is vulnerable (in particular children), protective equipment, the consumer's behaviour in the case of an accident, the consumer's cultural background, and other factors that you consider important for the risk assessment.
See section 3.3 and table 2 for guidance.
4.Determine the severity of the injury.U.K.
Determine the level of severity (1 to 4) of the injury to the consumer. If the consumer suffers from several injuries in your injury scenario, estimate the severity of all those injuries together.
See table 3 for guidance.
5.Determine the probability of the injury scenario.U.K.
Assign a probability to each step of your injury scenario. Multiply the probabilities to calculate the overall probability of your injury scenario.
See left-hand side of table 4 for guidance.
6.Determine the risk level.U.K.
Combine the severity of the injury and the overall probability of the injury scenario and check the risk level in table 4.
7.Check whether the risk level is plausible.U.K.
If the risk level does not seem plausible, or if you are uncertain about the severity of injury(ies) or about the probability(ies), move them one level up and down and recalculate the risk. This ‘sensitivity analysis’ will show you whether the risk changes when your input changes.
If the risk level remains the same, you can be quite confident of your risk assessment. If it changes easily, you may want to err on the safe side and take the higher risk level as ‘the risk’ of the consumer product.
You could also discuss the plausibility of the risk level with experienced colleagues.
8.Develop several injury scenarios to identify the highest risk of the product.U.K.
If your first injury scenario identifies a risk level below the highest risk level set out in these guidelines, and if you think that the product may pose a higher risk than the one identified,
select other consumers (including vulnerable consumers, in particular children);
identify other uses (including reasonably foreseeable uses),
in order to determine which injury scenario puts the product at its highest risk.
The highest risk is normally ‘the risk’ of the product that allows the most effective risk management measures. In specific cases, a particular hazard may lead to a less-than-highest risk and require specific risk management measures. This has to be taken duly into account.
As a rule of thumb, injury scenarios may lead to the highest risk level set out in these guidelines where:
the injury(ies) considered are at least at levels 3 or 4; —
the overall probability of an injury scenario is at least > 1/100.
See table 4 for guidance.
9.Document and pass on your risk assessment.U.K.
Be transparent and also set out all the uncertainties that you encountered when making your risk assessment.
Examples for reporting risk assessments are provided in section 6 of these guidelines.

6. Examples U.K.
6.1. Folding chair U.K.

A folding chair has a folding mechanism constructed in such a way that the user's fingers can get trapped between the seat and the folding mechanism. This can lead to fractures or even loss of one or more fingers.
Determination of risk(s)
| Injury scenario | Injury type and location | Severity of injury | Probability of injury | Overall probability | Risk | |
|---|---|---|---|---|---|---|
| Person unfolds the chair, grips seat close to the back corner by mistake (Person inattentive/ distracted), finger gets caught between seat and backrest | Minor pinching of finger | 1 | Unfolding the chair | 1 | 1/500 | Low risk |
| Gripping the seat at back corner while unfolding | 1/50 | |||||
| Finger gets caught | 1/10 | > 1/1 000 | ||||
| Minor pinching | 1 | |||||
| Person unfolds the chair, grips seat at the side by mistake (Person inattentive/ distracted), finger gets caught between seat and link | Minor pinching of finger | 1 | Unfolding the chair | 1 | 1/500 | Low risk |
| Gripping the seat at the side while unfolding | 1/50 | |||||
| Finger gets caught | 1/10 | > 1/1 000 | ||||
| Minor pinching | 1 | |||||
| Person unfolds the chair, chair is clamped, person tries to push down the seat and grips seat close to the corner by mistake (Person inattentive/ distracted), finger gets caught between seat and backrest | Fracture of finger | 2 | Unfolding the chair | 1 | 1/500 000 | Low risk |
| Chair clamps | 1/1 000 | |||||
| Gripping the seat at corners while unfolding | 1/50 | |||||
| Finger gets caught | 1/10 | > 1/1 000 000 | ||||
| Fracture of finger | 1 | |||||
| Person unfolds the chair, chair is clamped, person tries to push down the seat and grips seat at the side by mistake (Person inattentive/ distracted), finger gets caught between seat and link | Fracture of finger | 2 | Unfolding the chair | 1 | 1/500 000 | Low risk |
| Chair clamps | 1/1 000 | |||||
| Gripping the seat at the side while unfolding | 1/50 | |||||
| Finger gets caught | 1/10 | > 1/1 000 000 | ||||
| Fracture of finger | 1 | |||||
| Person is sitting on chair, wants to move the chair and tries to lift it by gripping the chair at the rear part of the seat, finger gets caught between seat and backrest | Loss of digit | 3 | Sitting on chair | 1 | 1/6 000 | High risk |
| Moves the chair while sitting | 1/2 | |||||
| Grips chair at rear part while moving | 1/2 | |||||
| Chair partially folds, creating a gap between the backrest and seat | 1/3 | > 1/10 000 | ||||
| Finger is between backrest and seat | 1/5 | |||||
| Finger gets caught | 1/10 | |||||
| Loss of (part of) finger | 1/10 | |||||
| Person is sitting on chair, wants to move the chair and tries to lift it by gripping the chair at the rear part of the seat, finger gets caught between seat and link | Loss of digit | 3 | Sitting on chair | 1 | 1/6 000 | High risk |
| Moves the chair while sitting | 1/2 | |||||
| Grips chair at rear part while moving | 1/2 | |||||
| Chair partially folds, creating a gap between the backrest and seat | 1/3 | > 1/10 000 | ||||
| Finger is between backrest and seat | 1/5 | |||||
| Finger gets caught | 1/10 | |||||
| Loss of (part of) finger | 1/10 | |||||
The overall risk of the folding chair is thus ‘high risk’.
6.2. Socket protectors U.K.

This case deals with socket protectors. These are devices that users (parents) put into the electrical socket outlets to stop small children from accessing live parts by putting a long metal object into one of the holes in the outlet and getting a (fatal) electric shock.
The holes in this particular protector (where the pins of the plug go through) are so narrow that the pins can get stuck. This means that the user may pull the protector off the outlet when the plug is pulled out. The user may not notice this happening.
Determination of risk(s)
| Injury scenario | Injury type and location | Severity of injury | Probability of injury | Overall probability | Risk | |
|---|---|---|---|---|---|---|
| Protector is removed from the socket, which becomes unprotected. Child is playing with thin conductible object, which can be inserted into the socket, accessing high voltage and is electrocuted. | Electrocution | 4 | Removal of protector | 9/10 | 27/160 000 | Serious risk |
| Not noticing the removal of protector | 1/10 | |||||
| Child is playing with thin conductible object | 1/10 | |||||
| Child is unattended when playing | 1/2 | > 1/10 000 | ||||
| Child inserts the object into the socket | 3/10 | |||||
| Access to voltage | 1/2 | |||||
| Electrocution due to voltage (without circuit interrupter) | 1/4 | |||||
| Protector is removed from the socket, which becomes unprotected. Child is playing with thin conductible object, which can be inserted into the socket, accessing high voltage and sustains shock. | Burns 2nd degree | 1 | Removal of protector | 9/10 | 81/160 000 | Low risk |
| Not noticing the removal of protector | 1/10 | |||||
| Child is playing with thin conductible object | 1/10 | |||||
| Child inserts the object into the socket | 3/10 | |||||
| Access to voltage | 1/2 | > 1/10 000 | ||||
| Child is unattended when playing | 1/2 | |||||
| Burn due to electric current (without circuit interrupter) | 3/4 | |||||
| Socket unprotected. Child is playing with thin conductible object, which can be inserted into the socket, accessing high voltage and is electrocuted. | Electrocution | 4 | Child is playing with thin conductible object | 1/10 | 3/80 000 | High risk |
| Child is unattended when playing | 1/100 | |||||
| Child inserts the object into the socket | 3/10 | |||||
| Access to voltage | 1/2 | > 1/100 000 | ||||
| Electrocution due to voltage (without circuit interrupter) | 1/4 | |||||
The overall risk of the socket protectors is thus ‘serious’.
6.3. Sensitivity analysis U.K.
The factors used to calculate the risk of an injury scenario, namely the severity of the injury and the probability, often have to be estimated. This creates uncertainty. Probability in particular can be difficult to estimate, since the behaviour of consumers, for example, can be difficult to predict. Does a person perform a certain action often or only occasionally?
It is therefore important to consider the level of uncertainty of the two factors and to make a sensitivity analysis. The purpose of this analysis is to establish how much the risk level varies when the estimated factors vary. The example provided on the table below only shows the variation of probability, since the severity of the injury is usually predicted with more certainty.
A practical way of performing the sensitivity analysis is to repeat the risk assessment for a certain scenario, but to use a different probability for one or more steps in the scenario. For example, a candle containing seeds could cause a fire, because the seeds can catch fire and generate high flames. Furniture or curtains can catch fire and persons not in the room could inhale toxic fumes and suffer fatal poisoning:
| Injury scenario | Injury type and location | Severity of injury | Probability of injury | Resulting probability | Risk |
|---|---|---|---|---|---|
| Seeds or beans catch fire generating high flames. Furniture or curtains catch fire. Persons are not in room, but inhale toxic fumes. | Fatal poisoning | 4 |
| 0 00675 > 1/1 000 | Serious |
The probability levels for the steps in the scenario were estimated as shown in the table.
The overall probability is 0,00675, which corresponds to > 1/1 000 in table 4. This leads to the conclusion of ‘serious risk’. Note that the exact probability is closer to 1/100 than to 1/1 000, which already gives some confidence in the risk level because it is a little deeper in the serious risk area of table 4 than the > 1/1 000 row suggests.
Suppose we are uncertain about the 5 % probability that persons inhale the toxic fumes. We could put it at a much lower 0,1 % (0 001 = 1 in a thousand). If we recalculate with that assumption, the overall probability is 0,000135, which translates into > 1/10 000. Nevertheless, the risk is still serious. Even if for some reason the probability were to be a factor of 10 lower, the risk would still be high. Therefore, although the probability may vary 10- or 100-fold, we still find a serious or high risk (the latter being quite close to ‘serious’). Thus, this sensitivity analysis lets us confidently assess the risk as serious.
In general, however, risk assessment should be based on ‘reasonable worst cases’: not too pessimistic on every factor, but certainly not too optimistic.
Table 1
Consumers
| Consumers | Description |
|---|---|
| Very vulnerable consumers | Very young children: 0 to 36 months Others: Persons with extensive and complex disabilities |
| Vulnerable consumers | Young children: Children older than 36 months and younger than 8 years. Older children: Children 8 to 14 years Others: Persons with reduced physical, sensory or mental capabilities (e.g. partially disabled, elderly, including those over 65, with some reduction in their physical and mental capabilities), or lack of experience and knowledge |
| Other consumers | Consumers other than very vulnerable or vulnerable consumers |
Table 2
Hazards, typical injury scenarios and typical injuries
| NB: This table is for guidance only; the typical injury scenarios should be adapted when preparing a risk assessment. There is specific risk assessment guidance for chemicals, cosmetics and possibly others. It is highly recommended to use this specific guidance when assessing such products. See section 3.2. | |||
| Hazard group | Hazard(product property) | Typical injury scenario | Typical injury |
|---|---|---|---|
| Size, shape and surface | Product is obstacle | Person trips over product and falls; or person bumps into product | Bruising; fracture, concussion |
| Product is impermeable to air | Product covers mouth and/or nose of a person (typically a child), or covers internal airway | Suffocation | |
| Product is or contains small part | Person (child) swallows small part; the part gets stuck in larynx and blocks airways | Choking, internal airway obstruction | |
| Possible to bite off small part from product | Person (child) swallows small part; the part gets stuck in the digestive tract | Digestive tract obstruction | |
| Sharp corner or point | Person bumps into sharp corner or is hit by moving sharp object; this causes a puncture or penetration injury | Puncture; blinding, foreign body in eye; hearing, foreign body in ear | |
| Sharp edge | Person touches sharp edge; this lacerates the skin or cuts through tissues | Laceration, cut; amputation | |
| Slippery surface | Person walks on surface, slips and falls | Bruising; fracture, concussion | |
| Rough surface | Person slides along rough surface; this causes friction and/or abrasion | Abrasion | |
| Gap or opening between parts | Person puts a limb or body in opening and finger, arm, neck, head, body or clothing is trapped; injury occurs due to gravity or movement | Crushing, fracture, amputation, strangulation | |
| Potential energy | Low mechanical stability | Product tips; person on top of product falls from height, or person near product is hit by the product; electrical product tips, breaks and gives access to live parts, or continues to work heating nearby surfaces | Bruising; dislocation; sprain; fracture, concussion; crushing; electric shock; burns |
| Low mechanical strength | Product collapses by overloading; person on top of product falls from height, or person near product is hit by the product; electrical product tips, breaks and gives access to live parts, or continues to work heating nearby surfaces | Bruising; dislocation; fracture, concussion; crushing; electric shock; burns | |
| High position of user | Person at high position on the product loses balance, has no support to hold on to and falls from height | Bruising; dislocation; fracture, concussion; crushing | |
| Elastic element or spring | Elastic element or spring under tension is suddenly released; person in the line of movement is hit by the product | Bruising; dislocation; fracture, concussion; crushing | |
| Pressurised liquid or gas, or vacuum | Liquid or gas under pressure is suddenly released; person in the vicinity is hit; or implosion of the product produces flying objects | Dislocation; fracture, concussion; crushing; cuts (see also under fire and explosion) | |
| Kinetic Energy | Moving product | Person in the line of movement of the product is hit by the product or run over | Bruising; sprain; fracture, concussion; crushing |
| Parts moving against one another | Person puts a body part between the moving parts while they move together; the body part gets trapped and put under pressure (crushed) | Bruising; dislocation; fracture; crushing | |
| Parts moving past one another | Person puts a body part between the moving parts while they move close by (scissor movement); the body part gets trapped between the moving parts and put under pressure (shearing) | Laceration, cut; amputation | |
| Rotating parts | A body part, hair or clothing of a person is entangled by the rotating part; this causes a pulling force | Bruising; fracture; laceration (skin of the head); strangulation | |
| Rotating parts close to one another | A body part, hair or clothing of a person is drawn in by the rotating parts; this causes a pulling force and pressure on the body part | Crushing, fracture, amputation, strangulation | |
| Acceleration | Person on the accelerating product loses balance, has no support to hold on to and falls with some speed | Dislocation; fracture, concussion; crushing | |
| Flying objects | Person is hit by the flying object and depending on the energy sustains injuries | Bruising; dislocation; fracture, concussion; crushing | |
| Vibration | Person holding the product loses balance and falls; or prolonged contact with vibrating product causes neurological disorders, osteoarticular disorder, trauma of the spine, vascular disorder | Bruising; dislocation; fracture; crushing | |
| Noise | Person is exposed to noise from the product. Tinnitus and hearing loss may occur depending on sound level and distance | Hearing injury | |
| Electrical Energy | High/low voltage | Person touches part of the product that is at high voltage; the person receives an electric shock and may be electrocuted | Electric shock |
| Heat production | Product becomes hot; a person touching it may sustain burns; or the product may emit molten particles, steam, etc., that hits a person | Burn, scald | |
| Live parts too close | Electric arc or sparks occur between the live parts. This may cause a fire and intense radiation | Eye injury; burn, scald | |
| Extreme temperatures | Open flames | A person near the flames may sustain burns, possibly after clothing catches fire | Burn, scald |
| Hot surfaces | Person does not recognise the hot surface and touches it; the person sustains burns | Burn | |
| Hot liquids | Person handling a container of liquid spills some of it; the liquid falls on the skin and causes scalds | Scald | |
| Hot gases | Person breathes in the hot gases emitted from a product; this causes lung burn; or prolonged exposure to hot air causes dehydration | Burn | |
| Cold surfaces | Person does not recognise the cold surface and touches it; the person sustains frostbite | Burn | |
| Radiation | Ultraviolet radiation, laser | Skin or eyes of a person are exposed to radiation emitted by the product | Burn, scald; neurological disorders; eye injury; skin cancer, mutation |
| High intensity electromagnetic field (EMF) source; low frequency or high frequency (microwave) | Person is close to the electromagnetic field (EMF) source, body (central nervous system) is exposed | Neurological (brain) damage, leukaemia (children) | |
| Fire and explosion | Flammable sub stances | Person is near the flammable substance; an ignition source sets the substance on fire; this causes injuries to the person | Burn |
| Explosive mixtures | Person is near the explosive mixture; an ignition source causes an explosion; the person is hit by the shock wave, burning material and/or flames | Burn, scald; eye injury, foreign body in eye; hearing injury, foreign body in ear | |
| Ignition sources | The ignition source causes a fire; a person is injured by flames, or intoxicated by gases from the house fire | Burn; poisoning | |
| Overheating | Product overheats; fire, explosion | Burn, scald; eye injury, foreign body in eye; hearing injury, foreign body in ear | |
| Toxicity | Toxic solid or fluid | Person ingests substance from product, e.g. by putting it in mouth, and/or substance gets on skin | Acute poisoning; irritation, dermatitis |
| Person breathes in solid or fluid, for example vomited material (pulmonary aspiration) | Acute poisoning in lungs (aspiration pneumonia); infection | ||
| Toxic gas, vapour or dust | Person inhales substance from product; and/or substance gets on skin | Acute poisoning in lungs; irritation, dermatitis | |
| Sensitising substance | Person ingests substance from product, e.g. by putting it in mouth; and/or substance gets on skin; and/or person inhales gas, vapour or dust | Sensitisation; allergic reaction | |
| Irritating or corrosive solid or fluid | Person ingests substance from product, e.g. by putting it in mouth, and/or substance gets on skin or in eyes | Irritation, dermatitis; skin burn; eye injury, foreign body in eye | |
| Irritating or corrosive gas or vapour | Person inhales substance from product, and/or substance gets on skin or in eyes | Irritation, dermatitis; skin burn; acute poisoning or corrosive effect in lungs or in eyes | |
| CMR substance | Person ingests substance from product, e.g. by putting it in mouth, and/or substance gets onto skin; and/or person inhales substance as gas, vapour or dust | Cancer, mutation, reproductive toxicity | |
| Microbiological contamination | Microbiological contamination | Person gets into contact with contaminated product by ingestion, inhalation or skin contact | Infection, local or systemic |
| Product operating hazards | Unhealthy posture | Design causes unhealthy posture of person when operating the product | Strain; musculoskeletal disorder |
| Overexertion | Design requires use of considerable force when operating the product | Sprain or strain; musculoskeletal disorder | |
| Anatomical unsuitability | Design is not adapted to human anatomy, which makes it difficult or impossible to operate | Sprain or strain | |
| Ignoring personal protection | Design makes it difficult for a person wearing protection to handle or operate the product | Various injuries | |
| Inadvertent (de)activation | Person can easily (de)activate product, which leads to unwanted operation | Various injuries | |
| Operational inadequacy | Design provokes faulty operation by a person; or product with a protective function does not provide expected protection | Various injuries | |
| Failure to stop | Person wants to stop the product, but it continues to operate in situation where this is unwanted | Various injuries | |
| Unexpected start | Product shuts down during a power failure, but resumes operation in a hazardous way | Various injuries | |
| Inability to stop | In an emergency situation, person is not able to stop operation of the product | Various injuries | |
| Inadequately fitting parts | Person tries to fit a part, needs too much force to fit, product breaks; or part is too loosely fitted and becomes loose during use | Sprain or strain; laceration, cut; bruising; entrapment | |
| Missing or incorrectly fitted protection | Hazardous parts are reachable for a per son | Various injuries | |
| Insufficient warning instructions, signs and symbols | User does not notice warning instructions signs and/or does not understand symbols | Various injuries | |
| Insufficient warning signals | User does not see or hear warning signal (optical or audio), causing dangerous operation | Various injuries | |
Table 3 Severity of injury U.K.
IntroductionU.K.
These risk assessment guidelines distinguish between four levels of injury harm severity. It is important to realise that severity should be assessed completely objectively. The aim is to compare the severity of different scenarios and to set priorities, not to judge the acceptability of a single injury at this stage. Any injury harm that could easily have been avoided will be difficult to accept for a consumer. However, authorities can justifiably invest more effort into avoiding irreversible consequences than into preventing temporary discomfort.
In order to assess the severity of the consequences (acute injury or other damage to health), objective criteria can be found, on the one hand, in the level of medical intervention, and, on the other hand, in the consequences to the further functioning of the victim. Both could be expressed as cost, but the costs of consequences of health damage may be difficult to quantify.
Combining these criteria, the four levels may be defined as follows:
1.
Harm or consequence that after basic treatment (first aid, normally not by a doctor) does not substantially hamper functioning or cause excessive pain; usually the consequences are completely reversible.
2.
Harm or consequence for which a visit to A&E may be necessary, but in general, hospitalisation is not required. Functioning may be affected for a limited period, not more than about 6 months, and recovery is more or less complete.
3.
Harm or consequence that normally requires hospitalisation and will affect functioning for more than 6 months or lead to a permanent loss of function.
4.
Harm or consequence that is or could be fatal, including brain death; consequences that affect reproduction or offspring; severe loss of limbs and/or function, leading to more than approximately 10 % of disability.
The following table, which should be considered as a guide rather than prescriptive or complete, provides examples of injuries at all four levels. National differences may exist, either cultural or caused by different systems of health care and financial arrangements. However, deviating from the proposed classification in the table will affect uniform assessment of risks in the EU; this should be clearly stated and explained in the risk assessment report, and reasons should be given.
| Type of injury | Severity of injury | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Laceration, cut | Superficial | External (deep) (> 10 cm long on body) (> 5 cm long on face) requiring stitches Tendon or into joint White of eye or cornea | Optic nerve Neck artery Trachea Internal organs | Bronchial tube Oesophagus Aorta Spinal cord (low) Deep laceration of internal organs Severed high spinal cord Brain (severe lesion/ dysfunction) |
| Bruising (abrasion/ contusion, swelling, oedema) | Superficial ≤ 25 cm2 on face ≤ 50 cm2 on body | Major > 25 cm2 on face > 50 cm2 on body | Trachea Internal organs (minor) Heart Brain Lung, with blood or air in chest | Brain stem Spinal cord causing paralysis |
| Concussion | — | Very short unconsciousness (minutes) | Prolonged unconsciousness | Coma |
| Entrapment/ pinching | Minor pinching | — | (Use as appropriate the final outcomes of bruising, crushing, fracture, dislocation, amputation, as applicable.) | (Same outcome as for suffocation/ strangulation.) |
| Sprain, strain, musculoskeletal disorder | Extremities Joints Spine (no dislocation or fracture) | Knee ligaments strain | Ligament or tendon rupture/tear Muscle tear Whiplash | — |
| Dislocation | — | Extremities (finger, toe, hand, foot) Elbow Jaw Loosening of tooth | Ankle Wrist Shoulder Hip Knee Spine | Spinal column |
| Fracture | — | Extremities (finger, toe, hand, foot) Wrist Arm Rib Sternum Nose Tooth Jaw Bones around eye | Ankle Leg (femur and lower leg) Hip Thigh Skull Spine (minor compression fracture) Jaw (severe) Larynx Multiple rib fractures Blood or air in chest | Neck Spinal column |
| Crushing | — | — | Extremities (fingers, toe, hand, foot) Elbow Ankle Wrist Forearm Leg Shoulder Trachea Larynx Pelvis | Spinal cord Mid-low neck Chest (massive crushing) Brain stem |
| Amputation | — | — | Finger(s) Toe(s) Hand Foot (Part of) Arm Leg Eye | Both extremities |
| Piercing, puncturing | Limited depth, only skin involved | Deeper than skin Abdominal wall (no organ involvement) | Eye Internal organs Chest wall | Aorta Heart Bronchial tube Deep injuries in organs (liver, kidney, bowel, etc.) |
| Ingestion | — | — | Internal organ injury (Refer also to internal airway obstruction where the ingested object gets stuck high in the oesophagus.) | Permanent damage to internal organ |
| Internal air way obstruction | — | — | Oxygen flow to brain blocked without permanent consequences | Oxygen flow to brain blocked with permanent consequences |
| Suffocation/ Strangulation | — | — | Oxygen flow to brain blocked without permanent consequences | Fatal suffocation/ strangulation |
| Submersion/ Drowning | — | — | — | Fatal drowning |
| Burn/Scald (by heat, cold, or chemical substance) | 1°, up to 100 % of body surface 2°, < 6 % of body surface | 2°, 6-15 % of body surface | 2°, 16-35 % of body surface, or 3°, up to 35 % of body surface Inhalation burn | 2° or 3°, > 35 % of body surface Inhalation burn requiring respiratory assistance |
| Electric shock | (See also under burns as electric current can cause burns.) | Local effects (temporary cramp or muscle paralysis) | — | Electrocution |
| Neurological disorders | — | — | Triggered epileptic seizure | — |
| Eye injury, foreign body in eye | Temporary pain in eye without need for treatment | Temporary loss of sight | Partial loss of sight Permanent loss of sight (one eye) | Permanent loss of sight (both eyes) |
| Hearing injury, foreign body in ear | Temporary pain in ear without need for treatment | Temporary impairment of hearing | Partial loss of hearing Complete loss of hearing (one ear) | Complete loss of hearing (both ears) |
| Poisoning from substances (ingestion, inhalation, dermal) | Diarrhoea, vomiting, local symptoms | Reversible damage to internal organs, e.g. liver, kidney, slight haemolytic anaemia | Irreversible damage to internal organs, e.g. oesophagus, stomach, liver, kidney, haemolytic anaemia, reversible damage to nerve system | Irreversible damage to nerve system Fatality |
| Irritation, dermatitis, inflammation or corrosive effect of substances (inhalation, dermal) | Local slight irritation | Reversible eye damage Reversible systemic effects Inflammatory effects | Lungs, respiratory insufficiency, chemical pneumonia Irreversible systemic effects Partial loss of sight Corrosive effects | Lungs, requiring respiratory assistance Asphyxia |
| Allergic reaction or sensitisation | Mild or local allergic reaction | Allergic reaction, widespread allergic contact dermatitis | Strong sensitisation, provoking allergies to multiple substances | Anaphylactic reaction, shock Fatality |
| Long-term damage from contact with substances or from exposure to radiation | Diarrhoea, vomiting, local symptoms | Reversible damage to internal organs, e.g. liver, kidney, slight haemolytic anaemia | Damage to nervous system, e.g. Organic Psycho Syndrome (OPS; also called Chronic Toxic Encephalopathy, also known as ‘painters' disease’). Irreversible damage to internal organs, e.g. oesophagus, stomach, liver, kidney, haemolytic anaemia, reversible damage to nervous system | Cancer (leukaemia) Effects on reproduction Effects on offspring CNS depression |
| Microbiological infection | Reversible damage | Irreversible effects | Infection requiring prolonged hospitalisation, antibiotics-resistant organisms Fatality | |
Table 4
Risk level from the combination of the severity of injury and probability
| Probability of damage during foreseeable lifetime of the product | Severity of injury | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| High  Low | >50 % | H | S | S | S |
| > 1/10 | M | S | S | S | |
| > 1/100 | M | S | S | S | |
| > 1/1 000 | L | H | S | S | |
| > 1/10 000 | L | M | H | S | |
| > 1/100 000 | L | L | M | H | |
| > 1/1 000 000 | L | L | L | M | |
| < 1/1 000 000 | L | L | L | L | |
S — Serious RiskU.K.
H — High risk
M — Medium risk
L — Low risk
Glossary of termsU.K.
Hazard
:
Source of danger involving the chance of being injured or harmed. A means of quantifying the hazard in a risk assessment is the severity of the possible injury or harm.
Product hazard
:
Hazard created by the properties of a product.
Risk
:
Balanced combination of a hazard and the probability that damage will occur. Risk describes neither the hazard, nor the probability, but both at the same time.
Risk assessment
:
Procedure for identifying and assessing hazards, consisting of three steps:
1.
identification of the seriousness of a hazard;
2.
determination of the probability that a consumer will be injured by that hazard;
3.
combination of the hazard with the probability.
Risk level
:
Degree of risk, which may be ‘serious’, ‘high’, ‘medium’ and ‘low’. When the (highest) level of risk has been identified, the risk assessment is complete.
Risk management
:
Follow-up action, which is separate from risk assessment and aims to reduce or eliminate a risk.
(3)
Commission Decision 2010/15/EU of 16 December 2009 laying down guidelines for the management of the Community Rapid Information System ‘RAPEX’ established under Article 12 and of the notification procedure established under Article 11 of Directive 2001/95/EC (the General Product Safety Directive) (OJ L 22, 26.1.2010, p. 1).
(4)
In other places of these Guidelines, the term ‘Commission’ generally refers to the RAPEX team established in the Commission department responsible for Directive 2001/95/EC and to the relevant Commission services, where appropriate.
(5)
The Information and Communication System on Market Surveillance (‘ICSMS’). This platform is aimed at facilitating communication between market surveillance bodies in the EU and in EFTA countries on non-compliant products.
(6)
In the context of this document, the term ‘Member States’ must be interpreted as not precluding all other actors from being addressed by the provisions contained in these Guidelines.
(7)
See the latest EC Implementing Decision published on https://ec.europa.eu/consumers/consumers_safety/safety_products/rapex/alerts/repository/content/pages/rapex/index_en.htm
(8)
See recital 10 of Directive 2001/95/EC.
(9)
Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety (OJ L 31, 1.2.2002, p. 1).
(10)
Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use (OJ L 311, 28.11.2001, p. 67).
(11)
Directive 2001/82/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to veterinary medicinal products (OJ L 311, 28.11.2001, p. 1).
(12)
Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC (OJ L 117, 5.5.2017, p. 1).
(13)
Council Directive 90/385/EEC of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices (OJ L 189, 20.7.1990, p. 17).
(14)
Directive (EU) 2015/1535 of the European Parliament and of the Council of 9 September 2015 laying down a procedure for the provision of information in the field of technical regulations and of rules on Information Society services (OJ L 241, 17.9.2015, p. 1).
(15)
See EU general risk assessment methodology (Action 5 of Multi-Annual Action Plan for the surveillance of products in the EU (COM(2013)76) providing guidance to authorities with relation to Article 20(2) of Regulation (EC) No 765/2008: http://ec.europa.eu/DocsRoom/documents/17107/attachments/1/translations
(16)
See https://ec.europa.eu/consumers/consumer-safety/rag/#/screen/home
(17)
www.ec.europa.eu/rapex
(18)
See Part I, Chapter 6.2 of these Guidelines.
(19)
See Part II, Chapter 2.2.1 of these Guidelines.
(20)
See Part II, Chapter 2.2.1 of these Guidelines.
(21)
See Part II, Chapter 2.2.2 of these Guidelines.
(22)
See Part II, Chapter 2.2.2 of these Guidelines.
(23)
For more information on follow-up actions, see Part II Chapter 4.4.5 of these Guidelines.
(24)
For more information about notifications where safety aspects are subject to discussions at EU level, see Part II Chapters 3.4.4 and 3.4.7.1.1 of these Guidelines.
(25)
See point 10 of Annex II of Directive 2001/95/EC.
(26)
See point 9 of Annex II of Directive 2001/95/EC.
(27)
For more information on notifications on safety aspects subject to discussions at EU level, see Part II Chapters 3.1.2(d) and 3.4.7.1.1.
(28)
https://ec.europa.eu/consumers/consumers_safety/safety_products/rapex/alerts/?event=main.search
(29)
Practice already agreed at the GPSD Committee of 24 September 2012, of which RAPEX Contact Points were informed at the RAPEX Contact Point meeting of 4 October (agenda point 4) and applied since 2013.
(30)
Paragraph 1 of Article 16(1) of Directive 2001/95/EC and Article 23 paragraph 3 in relation to Article 19(5) of Regulation (EC) No 765/2008.
(31)
Article 16(1) and (2) of Directive 2001/95/EC.
(32)
For more information on the notification criteria, see Part I Chapter 2.
(33)
For more information on notifications sent through the RAPEX application before measures are taken, see Chapter 3.1.2(b).
(34)
For more information on notifications on safety aspects subject to discussions at EU level, see Part II Chapters 3.1.2.d and 3.4.4.
(35)
For more information about deadlines, see Part III Appendix 4 of these Guidelines.
(36)
All deadlines mentioned in these Guidelines are expressed in calendar days.
(37)
See point 10 of Annex II of Directive 2001/95/EC.
(38)
For the purpose of these Guidelines, ‘economic operator’ refers to any natural of legal person defined as ‘economic operator’ in Regulation (EC) No 765/2008 or as ‘producer’ and ‘distributor’ in the GPSD.
(39)
https://webgate.ec.europa.eu/etranslation/translateDocument.html
(40)
There is no need to send notifications via the Permanent Representation of a Member State to the EU.
(41)
See Part I, Chapter 5.3 of these Guidelines.
(42)
The fields contained in the template may be updated following developments agreed between the Commission and Member States.
(43)
The fields contained in the template may be updated following developments agreed between the Commission and Member States.
(44)
If you need more information on the Risk Assessment method for harmonised products (both consumer and professional products) in relation to broader categories of public risks protected under EU harmonisation legislation, please refer to Part I, Chapter 5.3.
(45)
Benis HG (1990): A Product Risk Assessment Nomograph, report prepared for the New Zealand Ministry of Consumer Affairs, dated February 1990. Cited in: European Commission (2005) Establishing a Comparative Inventory of Approaches and Methods Used by Enforcement Authorities for the Assessment of the Safety of Consumer Products Covered by Directive 2001/95/EC on General Product Safety and Identification of Best Practices. Report prepared by Risk & Policy Analysts (RPA), Loddon, Norfolk, UK.
(46)
Method used by the Belgian authorities. Cited in: European Commission (2005) Establishing a Comparative Inventory of Approaches and Methods Used by Enforcement Authorities for the Assessment of the Safety of Consumer Products Covered by Directive 2001/95/EC on General Product Safety and Identification of Best Practices. Report prepared by Risk & Policy Analysts (RPA), Loddon, Norfolk, UK.
(47)
Commission Decision 2004/418/EC of 29 April 2004 laying down guidelines for the management of the EU Rapid Information System (RAPEX) and for notifications presented in accordance with Article 11 of Directive 2001/95/EC (OJ L 151, 30.4.2004, p. 83).
(48)
Directive 2001/95/EC.
(49)
https://webgate.ec.europa.eu/idbpa/.
(50)
NB: uncertainty always has to be taken into account when comparing a test result with a limit. See, for example:
the ‘Report on the relationship between analytical results, measurement uncertainty, recovery factors and the provisions of EU food and feed legislation …’ https://ec.europa.eu/food/safety/chemical_safety/contaminants/catalogue_en
the Summary report on the ‘Preparation of a working document in support of the uniform interpretation of legislative standards and the laboratory quality standards prescribed under Directive 93/99/EEC’. http://ec.europa.eu/food/fs/scoop/9.1_sr_en.pdf
(51)
Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC (OJ L 396, 30.12.2006, p. 1).
(53)
Standard EN 71-1:2005, section 8.2 +A6:2008.
(54)
Article 10 of Regulation (EC) No 1223/2009 (OJ L 342, 22.12.2009, p. 59).
(55)
REACH Regulation and guidance documents on REACH, see http://echa.europa.eu/ European Chemicals Agency (2008). The Guidance on Information Requirements and Chemical Safety Assessment: http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm
(56)
Commission Implementing Decision 2013/674/EU of 25 November 2013 on Guidelines on Annex I to Regulation (EC) No 1223/2009 of the European Parliament and of the Council on cosmetic products (OJ L 315, 26.11.2013, p. 82); SCCS (Scientific Committee on Consumer Safety), SCCS Notes of Guidance for the Testing of Cosmetic Ingredients and their Safety Evaluation 9th revision, 29 September 2015, SCCS/1564/15, revision of 25 April 2016: http://ec.europa.eu/health/scientific_committees/consumer_safety/docs/sccs_o_190.pdf
(57)
See Part I Chapter 1.1, penultimate paragraph.
(58)
This is taken from the definition of ‘safe product’ in Article 2(b) of Directive 2001/95/EC.
Options/Help
Print Options
PrintThe Whole Decision
You have chosen to open the Whole Decision
The Whole Decision you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
Y Rhestrau you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Mae deddfwriaeth ar gael mewn fersiynau gwahanol:
Y Diweddaraf sydd Ar Gael (diwygiedig):Y fersiwn ddiweddaraf sydd ar gael o’r ddeddfwriaeth yn cynnwys newidiadau a wnaed gan ddeddfwriaeth ddilynol ac wedi eu gweithredu gan ein tîm golygyddol. Gellir gweld y newidiadau nad ydym wedi eu gweithredu i’r testun eto yn yr ardal ‘Newidiadau i Ddeddfwriaeth’.
Gwreiddiol (Fel y’i mabwysiadwyd gan yr UE): Mae'r wreiddiol version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Pwynt Penodol mewn Amser: This becomes available after navigating to view revised legislation as it stood at a certain point in time via Advanced Features > Show Timeline of Changes or via a point in time advanced search.
Gweler y wybodaeth ychwanegol ochr yn ochr â’r cynnwys
Rhychwant ddaearyddol: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Dangos Llinell Amser Newidiadau: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Dewisiadau Agor
Dewisiadau gwahanol i agor deddfwriaeth er mwyn gweld rhagor o gynnwys ar y sgrin ar yr un pryd
Rhagor o Adnoddau
Gallwch wneud defnydd o ddogfennau atodol hanfodol a gwybodaeth ar gyfer yr eitem ddeddfwriaeth o’r tab hwn. Yn ddibynnol ar yr eitem ddeddfwriaeth sydd i’w gweld, gallai hyn gynnwys:
- y PDF print gwreiddiol y fel adopted version that was used for the EU Official Journal
- rhestr o newidiadau a wnaed gan a/neu yn effeithio ar yr eitem hon o ddeddfwriaeth
- pob fformat o’r holl ddogfennau cysylltiedig
- slipiau cywiro
- dolenni i ddeddfwriaeth gysylltiedig ac adnoddau gwybodaeth eraill
Llinell Amser Newidiadau
Mae’r llinell amser yma yn dangos y fersiynau gwahanol a gymerwyd o EUR-Lex yn ogystal ag unrhyw fersiynau dilynol a grëwyd ar ôl y diwrnod ymadael o ganlyniad i newidiadau a wnaed gan ddeddfwriaeth y Deyrnas Unedig.
Cymerir dyddiadau fersiynau’r UE o ddyddiadau’r dogfennau ar EUR-Lex ac efallai na fyddant yn cyfateb â’r adeg pan ddaeth y newidiadau i rym ar gyfer y ddogfen.
Ar gyfer unrhyw fersiynau a grëwyd ar ôl y diwrnod ymadael o ganlyniad i newidiadau a wnaed gan ddeddfwriaeth y Deyrnas Unedig, bydd y dyddiad yn cyd-fynd â’r dyddiad cynharaf y daeth y newid (e.e. ychwanegiad, diddymiad neu gyfnewidiad) a weithredwyd i rym. Am ragor o wybodaeth gweler ein canllaw i ddeddfwriaeth ddiwygiedig ar Ddeall Deddfwriaeth.
Rhagor o Adnoddau
Defnyddiwch y ddewislen hon i agor dogfennau hanfodol sy’n cyd-fynd â’r ddeddfwriaeth a gwybodaeth am yr eitem hon o ddeddfwriaeth. Gan ddibynnu ar yr eitem o ddeddfwriaeth sy’n cael ei gweld gall hyn gynnwys:
- y PDF print gwreiddiol y fel adopted fersiwn a ddefnyddiwyd am y copi print
- slipiau cywiro
liciwch ‘Gweld Mwy’ neu ddewis ‘Rhagor o Adnoddau’ am wybodaeth ychwanegol gan gynnwys
- rhestr o newidiadau a wnaed gan a/neu yn effeithio ar yr eitem hon o ddeddfwriaeth
- manylion rhoi grym a newid cyffredinol
- pob fformat o’r holl ddogfennau cysylltiedig
- dolenni i ddeddfwriaeth gysylltiedig ac adnoddau gwybodaeth eraill
The data on this page is available in the alternative data formats listed:
 Mae’r holl gynnwys ar gael dan Drwydded Llywodraeth Agored v3.0 ac eithrio ble nodir yn wahanol. Yn ychwanegol mae’r safle hwn â chynnwys sy’n deillio o EUR-Lex, a ailddefnyddiwyd dan delerau Penderfyniad y Comisiwn 2011/833/EU ar ailddefnyddio dogfennau o sefydliadau’r UE. Am ragor o wybodaeth gweler ddatganiad cyhoeddus Swyddfa Gyhoeddiadau’r UE ar ailddefnyddio.
Mae’r holl gynnwys ar gael dan Drwydded Llywodraeth Agored v3.0 ac eithrio ble nodir yn wahanol. Yn ychwanegol mae’r safle hwn â chynnwys sy’n deillio o EUR-Lex, a ailddefnyddiwyd dan delerau Penderfyniad y Comisiwn 2011/833/EU ar ailddefnyddio dogfennau o sefydliadau’r UE. Am ragor o wybodaeth gweler ddatganiad cyhoeddus Swyddfa Gyhoeddiadau’r UE ar ailddefnyddio.