- Y Diweddaraf sydd Ar Gael (Diwygiedig)
- Pwynt Penodol mewn Amser (12/06/2006)
- Gwreiddiol (Fel y’i mabwysiadwyd gan yr UE)
Commission Directive 2006/56/ECDangos y teitl llawn
Commission Directive 2006/56/EC of 12 June 2006 amending the Annexes to Council Directive 93/85/EEC on the control of potato ring rot
You are here:

Pa Fersiwn
Nodweddion Uwch
- Dangos Graddfa Ddaearyddol(e.e. Lloegr, Cymru, Yr Alban aca Gogledd Iwerddon)
- Dangos Llinell Amser Newidiadau
Rhagor o Adnoddau
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


Mae hon yn eitem o ddeddfwriaeth sy’n deillio o’r UE
Mae unrhyw newidiadau sydd wedi cael eu gwneud yn barod gan y tîm yn ymddangos yn y cynnwys a chyfeirir atynt gydag anodiadau.Ar ôl y diwrnod ymadael bydd tair fersiwn o’r ddeddfwriaeth yma i’w gwirio at ddibenion gwahanol. Y fersiwn legislation.gov.uk yw’r fersiwn sy’n weithredol yn y Deyrnas Unedig. Y Fersiwn UE sydd ar EUR-lex ar hyn o bryd yw’r fersiwn sy’n weithredol yn yr UE h.y. efallai y bydd arnoch angen y fersiwn hon os byddwch yn gweithredu busnes yn yr UE. EUR-Lex Y fersiwn yn yr archif ar y we yw’r fersiwn swyddogol o’r ddeddfwriaeth fel yr oedd ar y diwrnod ymadael cyn cael ei chyhoeddi ar legislation.gov.uk ac unrhyw newidiadau ac effeithiau a weithredwyd yn y Deyrnas Unedig wedyn. Mae’r archif ar y we hefyd yn cynnwys cyfraith achos a ffurfiau mewn ieithoedd eraill o EUR-Lex. The EU Exit Web Archive legislation_originated_from_EU_p3
Changes over time for: ANNEX I
Alternative versions:
Status:
EU Directives are published on this site to aid cross referencing from UK legislation. Since IP completion day (31 December 2020 11.00 p.m.) no amendments have been applied to this version.
ANNEX IU.K.TEST SCHEME FOR DIAGNOSIS, DETECTION AND IDENTIFICATION OF THE RING ROT BACTERIUM, CLAVIBACTER MICHIGANENSIS (Smith) Davis et al. ssp. SEPEDONICUS (Spieckermann et Kotthoff) Davis et al. SCOPE OF THE TEST SCHEME
The presented scheme describes the various procedures involved in:U.K.
(i)
Diagnosis of ring rot in potato tubers and plants;
(ii)
Detection of Clavibacter michiganensis ssp. sepedonicus in samples of potato tubers and plants;
(iii)
Identification of Clavibacter michiganensis ssp. sepedonicus (C. m. subsp. sepedonicus).
GENERAL PRINCIPLESU.K.
Optimized protocols for the various methods, validated reagents and details for the preparation of test and control materials are provided in the Appendices. A list of the laboratories that were included in optimization and validation of protocols is provided in Appendix 1.
Since the protocols involve detection of a quarantine organism and will include the use of viable cultures of C. m. subsp. sepedonicus as control materials, it will be necessary to perform the procedures under suitable quarantined conditions with adequate waste disposal facilities and under the conditions of appropriate licences as issued by the official plant quarantine authorities.
Testing parameters must assure consistent and reproducible detection of levels of C. m. subsp. sepedonicus at the set thresholds of the selected methods.
Precise preparation of positive controls is imperative.
Testing according to the required thresholds also implies correct settings, maintenance and calibration of equipment, careful handling and preservation of reagents and all measures to prevent contamination between samples, e.g. separation of positive controls from test samples. Quality control standards must be applied to avoid administrative and other errors, especially concerning labelling and documentation.
A suspected occurrence, as referred to in article 4(2) in directive 93/85/EEC implies a positive result in diagnostic or screening tests performed on a sample as specified in flow charts.
If the first screening test (IF or PCR/FISH) is positive, then contamination with Cms is suspected and a second screening test must be done. If the second screening test is positive, then the suspicion is confirmed (suspected occurrence) and the testing according to the scheme must be continued. If the second screening test is negative, then the sample is considered not contaminated with Cms.
Therefore a positive IF test as referred to in article 4(2) is defined by a positive IF reading confirmed by a second screening test (PCR/FISH).
Confirmed presence as referred to in article 5(1) in directive 93/85/EEC implies the isolation and identification of a pure culture of C. m. subsp. sepedonicus with confirmation of pathogenicity.
1.FLOW CHART DIAGRAM PRESENTATIONU.K.
1.1.Detection scheme for the diagnosis of Ring Rot in potato tubers and potato plants with symptoms of ring rotU.K.
The testing procedure is intended for potato tubers and plants with symptoms typical or suspect of ring rot. It involves a rapid screening test, isolation of the pathogen from infected vascular tissue on diagnostic media and, in case of a positive result, identification of the culture as C. m. subsp. sepedonicus.

1.2.Scheme for detection and identification of Clavibacter michiganensis ssp. sepedonicus in samples of asymptomatic potato tubersU.K.
PrincipleU.K.
The testing procedure is intended for detection of latent infections in potato tubers. A positive result from at least two screening tests, based on different biological principles, must be complemented by the isolation of the pathogen; followed by, in case of isolation of typical colonies, confirmation of a pure culture as C. m. subsp. sepedonicus. A positive result from only one of the screening tests is not sufficient to consider the sample suspect.
Screening tests and isolation tests must permit a detection threshold of 103 to 104 cells/ml resuspended pellet, included as positive controls in each series of tests.
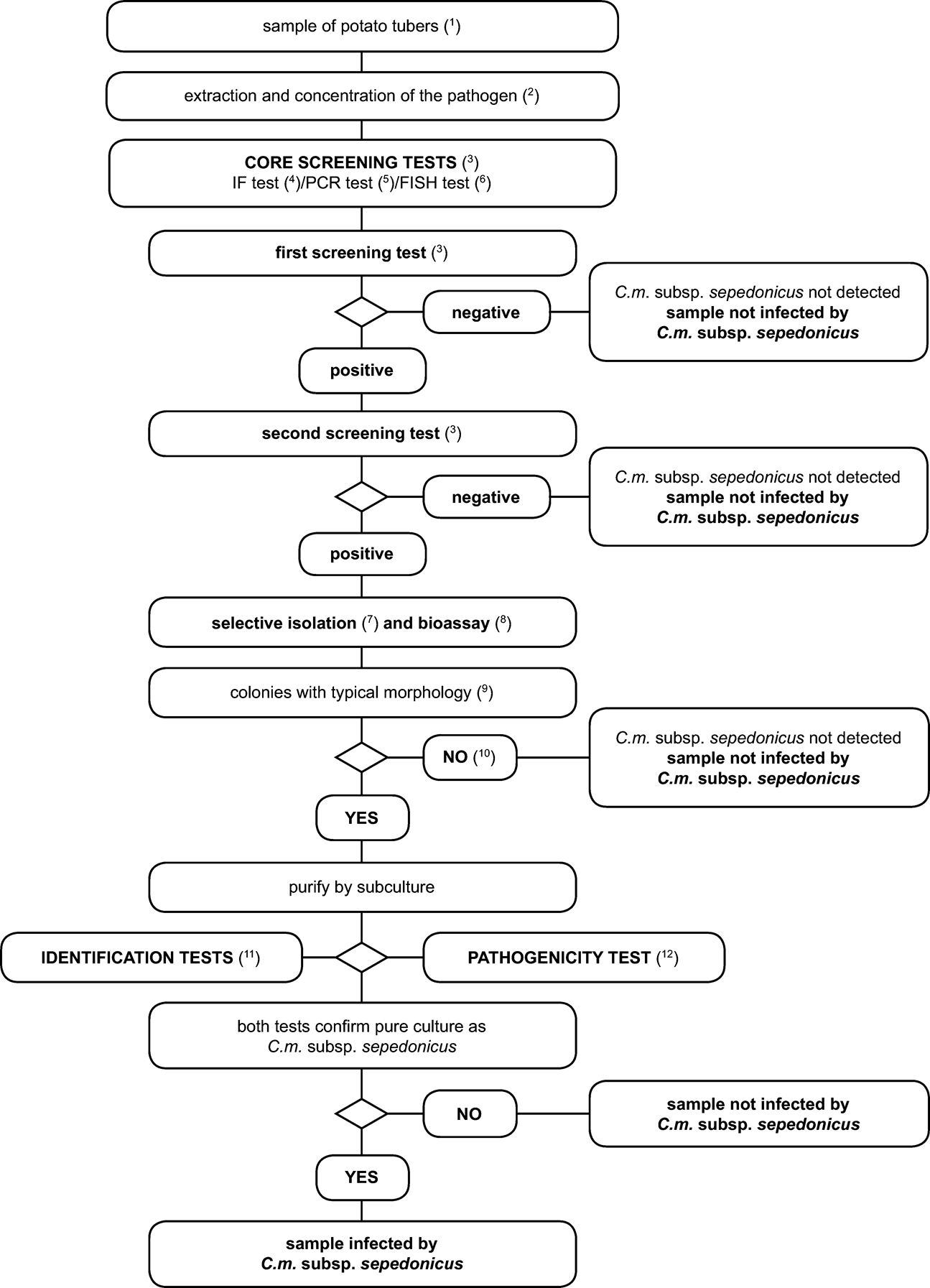

1.3.Scheme for detection and identification of Clavibacter michiganensis ssp. sepedonicus in samples of asymptomatic potato plantsU.K.


2.VISUAL EXAMINATION FOR RING ROT SYMPTOMSU.K.
2.1.Potato plantsU.K.
Under European climatic conditions symptoms are rarely found in the field and often only at the end of the season. Moreover the symptoms are frequently masked or confused by/with other diseases, senescence or mechanical damages. Therefore it may be easy to miss symptoms in field inspections. Wilting symptoms are very different from those of brown rot; wilting is usually slow and initially limited to the leaf margins. Young infected leaves often continue to expand although, less so in the infected zones. This creates odd shaped leaves. Leaves affected by blocking of the vascular tissues further down the stem often develop chlorotic, yellow to orange, intercostal areas. Infected leaflets, leaves and even stems may eventually die. Often leaves and tubers are simply reduced in size. Occasionally plants are stunted. Coloured pictures of a range of symptoms can be found on the web site http://forum.europa.eu.int/Public/irc/sanco/Home/main
2.2.Potato tubersU.K.
The earliest symptoms are a slight glassiness or translucence of the tissue without softening around the vascular system, particularly near the heel end. The vascular ring at the heel end may be slightly darker in colour than normal. The first readily identifiable symptom is one whereby the vascular ring has a yellowish coloration and when the tuber is gently squeezed, pillars of cheese-like material emerge from the vessels. This exudation contains millions of bacteria. Browning of the vascular tissue may develop and tuber symptoms at this stage are similar to those of brown rot caused by Ralstonia solanacearum. At first, these symptoms may be restricted to one part of the ring, not necessarily close to the heel end and may gradually extend to the whole ring. As the infection progresses, destruction of the vascular tissue occurs; the outer cortex may become separated from the inner cortex. In advanced stages of infection, cracks appear on the surface of the tuber, which are often reddish-brown at their margins. Recently in Europe several cases have occurred where the central cortex rots at the same time as the vascular ring resulting in secondary invasion with internal hollowing and necrosis. Secondary fungal or bacterial invasion may mask the symptoms and it may be difficult, if not impossible, to distinguish advanced ring rot symptoms from other tuber rots. Atypical symptoms may be possible. Coloured pictures of a range of symptoms can be found on the web site http://forum.europa.eu.int/Public/irc/sanco/Home/main
3.SAMPLE PREPARATIONU.K.
3.1.Potato tubersU.K.
Note:U.K.
The standard sample size is 200 tubers per test. More intensive sampling requires more tests on samples of this size. Larger numbers of tubers in the sample will lead to inhibition or difficult interpretation of the results. However, the procedure can be conveniently applied for samples with less than 200 tubers where fewer tubers are available.
Validation of all detection methods described below is based on testing of samples of 200 tubers.
The potato extract described below can also be used for detection of the potato brown rot bacterium, Ralstonia solanacearum.
Optional pre-treatment in advance to sample preparation:U.K.
Wash the tubers. Use appropriate disinfectants (chlorine compounds when PCR-test is to be used in order to remove eventual pathogen DNA) and detergents between each sample. Air dry the tubers. This washing procedure is particularly useful (but not required) for samples with excess soil and if a PCR-test or direct isolation procedure is to be performed.
3.1.1.Remove with a clean and disinfected scalpel or vegetable knife the skin at the heel end of each tuber so that the vascular tissue becomes visible. Carefully cut out a small core of vascular tissue at the heel end and keep the amount of non-vascular tissue to a minimum (see web site: http://forum.europa.eu.int/Public/irc/sanco/Home/main)U.K.
Note:U.K.
Set aside any tubers with suspected ring rot symptoms and test separately.
If during removal of the heel end core suspect symptoms of ring rot are observed, the tuber should be visually inspected after cutting near the heel end. Any cut tuber with suspected symptoms should be suberised at room temperature for two days and stored under quarantine (at 4 to 10 °C) until all tests have been completed. All tubers in the sample (including those with suspicious symptoms) should be kept according to Annex II.
3.1.2.Collect the heel end cores in unused disposable containers which can be closed and/or sealed (in case containers are reused they should be thoroughly cleaned and disinfected using chlorine compounds). Preferably, the heel end cores should be processed immediately. If this is not possible, store them in the container, without addition of buffer, refrigerated for not longer than 72 hours or for not longer than 24 hours at room temperature. Drying and suberisation of cores, and growth of saprophytes during storage may hinder detection of the ring rot bacterium.U.K.
3.1.3.Process the heel end cores by one of the following procedures: either,U.K.
(a)
cover the cores with sufficient volume (approximately 40 ml) of extraction buffer (Appendix 3) and agitate on a rotary shaker (50 to 100 rpm) for four hours below 24 °C or for 16 to 24 hours refrigerated,
or
(b)
homogenise the cores with sufficient volume (approximately 40 ml) of extraction buffer (Appendix 3), either in a blender (e.g. Waring or Ultra Thurax) or by crushing in a sealed disposable maceration bag (e.g. Stomacher or Bioreba strong guage polythene, 150 mm × 250 mm; radiation sterilised) using a rubber mallet or suitable grinding apparatus (e.g. Homex).
Note:U.K.
The risk of cross-contamination of samples is high when samples are homogenized using a blender. Take precautions to avoid aerosol generation or spillage during the extraction process. Ensure that freshly sterilised blender blades and vessels are used for each sample. If the PCR test is to be used, avoid carry-over of DNA on containers or grinding apparatus. Crushing in disposable bags and use of disposable tubes is recommended where PCR is to be used.
3.1.4.Decant the supernatant. If excessively cloudy, clarify either by slow speed centrifugation (at not more than 180 g for 10 minutes at a temperature between 4 to 10 °C) or by vacuum filtration (40 to 100 µm), washing the filter with additional (10 ml) extraction buffer (Appendix 3).U.K.
3.1.5.Concentrate the bacterial fraction by centrifugation at 7 000 g for 15 minutes (or 10 000 g for 10 minutes) at a temperature between 4 to 10 °C and discard the supernatant without disturbing the pellet.U.K.
3.1.6.Resuspend the pellet in 1,5 ml pellet buffer (Appendix 3). Use 500 µl to test for C. m. subsp. sepedonicus, 500 µl for Ralstonia solanacearum and 500 µl for reference purposes. Add sterile glycerol to final concentration of 10 to 25 % (v/v) to the 500 µl of the reference aliquot and to the remaining test aliquot, vortex and store at -16 to -24 °C (weeks) or at 68 to -86 °C (months). Preserve the test aliquots at 4 to 10 °C during testing.U.K.
Repeated freezing and thawing is not advisable.
If transport of the extract is required, ensure delivery in a cool box within 24 to 48 hours.
3.1.7.It is imperative that all C. m. subsp. sepedonicus positive controls and samples are treated separately to avoid contamination. This applies to IF slides and to all tests.U.K.
3.2.Potato plantsU.K.
Note:U.K.
For detection of latent C. m. subsp. sepedonicus populations it is advised to test composite samples. The procedure can be conveniently applied for composite samples of up to 200 stem parts. (Where surveys are performed they should be based on a statistically representative sample of the plant population under investigation.)
3.2.1.With a clean disinfected knife or pruning shears, remove a 1 to 2 cm segment from the base of each stem, just above the soil level.U.K.
Disinfect stem segments briefly with ethanol 70 % and immediately blot dry on tissue paper.
Collect stem segments in a closed sterile container according to the following sampling procedures:
3.2.2.Process the stem segments by one of the following procedures: either,U.K.
(a)
cover the segments with sufficient volume (approximately 40 ml) of extraction buffer (Appendix 3) and agitate on a rotary shaker (50 to 100 rpm) for four hours below 24 °C or for 16 to 24 hours refrigerated,
or
(b)
process immediately. By crushing the segments in a strong maceration bag (e.g. Stomacher or Bioreba) with an appropriate volume of extraction buffer (Appendix 3) using a rubber mallet or appropriate grinding apparatus (e.g. Homex). If this is not possible, store the stem segments refrigerated for not longer than 72 hours or for not longer than 24 hours at room temperature.
3.2.3.Decant the supernatant after settling for 15 minutes.U.K.
3.2.4.Further clarification of the extract or concentration of the bacterial fraction are not usually required but may be achieved by filtration and/or centrifugation as described in section 3.1.4 to 3.1.6.U.K.
3.2.5.Divide the neat or concentrated sample extract into 2 equal parts. Maintain one half at 4 to 10 °C during testing and store the other half with 10 to 25 % (v/v) sterile glycerol at -16 to -24 °C (weeks) or at -68 to -86 °C (months) in case further testing is required.U.K.
4.IF TESTU.K.
PrincipleU.K.
The use of the IF test as the principal screening test is recommended because of its proven robustness to achieve the required thresholds.
When the IF test is used as the principal screening test and the IF reading is positive, the PCR or FISH test must be performed as a second screening test. When the IF test is used as the second screening test and the IF reading is positive, further testing according to the flow scheme is required to complete the analysis.
Note:U.K.
Always use a polyclonal antibody, when the IF test is used as the principal screening test. In case of a positive IF reading with a polyclonal antibody further screening of the sample with a monoclonal antibody may provide more specificity but can be less sensitive.
Use antibodies to a reference strain of C. m. subsp. sepedonicus. It is recommended that the titre is determined for each new batch of antibodies. The titre is defined as the highest dilution at which optimum reaction occurs when testing a suspension containing 105 to 106 cells per ml of the homologous strain of C. m. subsp. sepedonicus and using an appropriate fluorescein isothiocyanate (FITC) conjugate according to the manufacturer's recommendations. The crude polyclonal or monoclonal antibodies should have an IF titre of at least 1:2000. During testing, the antibodies should be used at a working dilution(s) (WD) close to or at the titre. Use validated antibodies (see web site http://forum.europa.eu.int/Public/irc/sanco/Home/main).
The test should be performed on freshly-prepared sample extracts. If necessary, it can be successfully performed on extracts stored at -68 to -86 °C under glycerol. Glycerol can be removed from the sample by addition of 1 ml pellet buffer (Appendix 4), re-centrifugation for 15 minutes at 7 000 g and resuspension in an equal volume of pellet buffer. This is often not necessary, especially if slides samples are fixed to the slides by flaming (see 2.2).
Prepare separate positive control slides of the homologous strain or any other reference strain of C. m. subsp. sepedonicus, suspended in potato extract, as specified in Appendix 2, and optionally in buffer.
Naturally infected tissue (maintained by lyophilization or freezing at -16 to -24 °C) should be used where possible as a similar control on the same slide.
As negative controls, use aliquots of sample extract which previously tested negative.
Use multiwell microscope slides with preferably 10 windows of at least 6 mm diameter.
Test control material in an identical manner as the sample(s).
4.1.Prepare the test slides by one of the following procedures:U.K.
(i)
For pellets with relatively little starch sediment:
Pipette a measured standard volume (15 µl is appropriate for 6 mm window diameter-scale up volume for larger windows) of a 1/100 dilution of the resuspended potato pellet onto the first window. Subsequently pipette a similar volume of undiluted pellet (1/1) onto the remaining windows on the row. The second row can be used as duplicate or for a second sample as presented in Figure 1.
(ii)
For other pellets:
Prepare decimal dilutions (1/10 and 1/100) of the resuspended pellet in pellet buffer. Pipette a measured standard volume (15 µl is appropriate for 6 mm window diameter-scale up volume for larger windows) of the resuspended pellet and each dilution on a row of windows. The second row can be used as duplicate or for a second sample as presented in Figure 2.
4.2.Dry the droplets at ambient temperatures or by warming at temperature of 40 to 45 °C. Fix the bacterial cells to the slide either by heating (15 minutes at 60 °C), flaming, with 95 % ethanol or according to specific instructions from the suppliers of the antibodies.U.K.
If necessary, fixed slides may then be stored frozen in a desiccated box for as little time as necessary (up to a maximum of 3 months) prior to further testing.
4.3.
IF procedure:U.K.
(i)
According to test slide preparation in 4.1(i):
Prepare a set of twofold dilutions of the antibody in IF buffer. The first well should have 1/2 of the titre (T/2), the others 1/4 of the titre (T/4), 1/2 of the titre (T/2), the titre (T) and twice the titre (2T).
(ii)
According to test slide preparation in 4.1(ii):
Prepare the working dilution (WD) of the antibody in IF buffer. The working dilution affects the specificity.
Figure 1.
Preparation of the test slide according to 4.1(i) and 4.3(i)
| Dilutions of resuspended pellet | |||||||
| 1/100 | 1/1 | 1/1 | 1/1 | 1/1 | | Dilution of resuspended pellet | |
| (T = titre) | T/2 | T/4 | T/2 | T | 2T | | Twofold dilutions of antiserum/antibody |
| Sample 1 |  |  |  |  |  | ||
| 1 | 2 | 3 | 4 | 5 | |||
| Duplicate of sample 1 or sample 2 |  |  |  |  |  | ||
| 6 | 7 | 8 | 9 | 10 | |||
Figure 2.
Preparation of the test slide according to 4.1(ii) and 4.3(ii)
| Working dilution of antiserum/antibody | |||||||
| 1/1 | 1/10 | 1/100 | empty | empty | | Decimal dilution of resuspended pellet | |
| Sample 1 |  |  |  |  |  | ||
| 1 | 2 | 3 | 4 | 5 | |||
| Duplicate of sample 1 or sample 2 |  |  |  |  |  | ||
| 6 | 7 | 8 | 9 | 10 | |||
4.3.1.Arrange the slides on moist paper. Cover each test window completely with the antibody dilution(s). The volume of antibody applied on each window must be at least the volume of extract applied.U.K.
The following procedure should be carried out in the absence of specific instructions from the suppliers of the antibodies:
4.3.2.Incubate the slides on moist paper under a cover for 30 minutes at ambient temperature (18 to 25 °C).U.K.
4.3.3.Shake the droplets off each slide and rinse carefully with IF buffer. Wash by submerging for 5 minutes in IF buffer-Tween (Appendix 3) and subsequently for 5 minutes in IF buffer. Avoid causing aerosols or droplet transfer that could result in cross-contamination. Carefully remove excess moisture by blotting gently.U.K.
4.3.4.Arrange the slides on moist paper. Cover the test windows with the dilution of FITC conjugate used to determine the titre. The volume of conjugate applied on the windows must be identical to the volume of antibody applied.U.K.
4.3.5.Incubate the slides on moist paper under a cover for 30 minutes at ambient temperature (18 to 25 °C).U.K.
4.3.6.Shake the droplets of conjugate off the slide. Rinse and wash as before (4.3.3).U.K.
Carefully remove excess moisture.
4.3.7.Pipette 5 to 10 µl of 0,1M phosphate-buffered glycerol (Appendix 3) or a commercially antifading mountant on each window and apply a coverslip.U.K.
4.4.
Reading the IF test:U.K.
4.4.1.Examine test slides on an epifluorescence microscope with filters suitable for excitation of FITC, under oil or water immersion at a magnification of 500 to 1 000. Scan windows across two diameters at right angles and around the perimeter. For samples showing no or low number of cells observe at least 40 microscope fields.U.K.
Check the positive control slide first. Cells must be bright fluorescent and completely stained at the determined antibody titre or working dilution. The IF test (section 4) must be repeated if the staining is aberrant.
4.4.2.Observe for bright fluorescing cells with characteristic morphology of C. m. subsp. sepedonicus in the test windows of the test slides (see web site http://forum.europa.eu.int/Public/irc/sanco/Home/main). The fluorescence intensity must be equivalent or better to the positive control strain at the same antibody dilution. Cells with incomplete staining or with weak fluorescence must be disregarded.U.K.
If any contamination is suspected the test must be repeated. This may be the case when all slides in a batch show positive cells due to the contamination of buffer or if positive cells are found (outside of the slide windows) on the slide coating.
4.4.3.There are several problems inherent to the specificity of the immunofluorescence test. Background populations of fluorescing cells with atypical morphology and cross reacting saprophytic bacteria with size and morphology similar to C. m. sepedonicus are likely to occur in potato heel end core and stem segment pellets.U.K.
4.4.4.Consider only fluorescing cells with typical size and morphology at the titre or working dilution of the antibodies as in 4.3.U.K.
4.4.5.Interpretation of the IF reading:U.K.
(i)
If bright fluorescing cells with characteristic morphology are found, estimate the average number of typical cells per microscope field and calculate the number of typical cells per ml of resuspended pellet (Appendix 4).
The IF reading is positive for samples with at least 5 × 103 typical cells per ml of resuspended pellet. The sample is considered potentially contaminated. and further testing is required.
(ii)
The IF reading is negative for samples with less than 5 × 103 cells per ml resuspended pellet and the sample is considered negative. Further testing is not required.
5.FISH TESTU.K.
PrincipleU.K.
When the FISH test is used as the first screening test and found to be positive, the IF test must be performed as a second compulsory screening test. When the FISH test is used as the second screening test and found to be positive, further testing according to the flow scheme is required to complete the diagnosis.
Note:U.K.
Use validated C. m. subsp. sepedonicus-specific oligo-probes (Appendix 7). Preliminary testing with this method should permit reproducible detection of at least 103 to 104 cells of C. m. subsp. sepedonicus per ml added to sample extracts which previously tested negative.
The following procedure should preferably be performed on freshly prepared sample extract but can also be successfully performed on sample extract that has been stored under glycerol at -16 to -24 °C or -68 to -86 °C.
As negative controls, use aliquots of sample extract that previously tested negative for C. m. subsp. sepedonicus.
As positive controls prepare suspensions containing 105 to 106 cells per ml of C. m. subsp. sepedonicus (e.g. strain NCPPB 4053, or PD 406) in 0,01M phosphate buffer (PB) from a three to five day culture (preparation see Appendix 2). Prepare separate positive control slides of the homologous strain or any other reference strain of C. m. subsp. sepedonicus, suspended in potato extract, as specified in Appendix 2.
The use of the FITC-labelled eubacterial oligo-probe offers a control for the hybridisation process, since it will stain all eubacteria that are present in the sample.
Test control material in an identical manner as the sample(s).
5.1.Potato extract fixationU.K.
The following protocol is based upon Wullings et al., (1998):U.K.
5.1.1.Prepare fixative solution (see Appendix 7).U.K.
5.1.2.Pipette 100 µl of each sample extract into an Eppendorf tube and centrifuge for eight min at 7 000 g.U.K.
5.1.3.Remove the supernatant and dissolve the pellet in 500 µl of fixative prepared < 24 hours previously. Vortex and incubate overnight at 4 °C.U.K.
An alternative fixative is 96 % ethanol. To use this dissolve the pellet from step 5.1.2 in 50 µl 0,01M PB and 50 µl 96 % ethanol. Vortex mix and incubate at 4 °C for 30 to 60 minutes.
5.1.4.Centrifuge for 8 min. at 7 000 g, remove the supernatant and resuspend the pellet in 75 µl 0,01M PB (see Appendix 3).U.K.
5.1.5.Spot 16 µl of the fixed suspensions onto a clean multitest slide as shown in Fig. 3. Apply two different samples per slide, undiluted and use 10 µl to make a 1:100 dilution (in 0,01M PB). The remaining sample solution (49 µl) can be stored at -20 °C after addition of 1 volume of 96 % ethanol. In case the FISH assay requires repeating, remove the ethanol by centrifugation and add an equal volume of 0,01M PB (mix by vortexing).U.K.
Figure 3.
Layout for FISH slide
| Sample 1 | Blank | Blank | Blank | Sample 2 |
 |  |  |  |  |
| window 1 | window 2 | window 3 | window 4 | window 5 |
| Sample 1 | Blank | Blank | Blank | Sample 2 |
 |  |  |  |  |
| window 6 | window 7 | window 8 | window 9 | window 10 |
| Coverslip 1 | Coverslip 2 | |||
5.1.6.Air-dry the slides (or on slide dryer at 37 °C) and fix them by flaming.U.K.
At this stage the procedure may be interrupted and the hybridisation continued the following day. Slides should be stored dust-free and dry at room temperature.
5.2.Pre-hybridisation and hybridisationU.K.
5.2.1.Prepare a lysozyme solution containing 10 mg lysozyme (Sigma L–6876) in 10 ml buffer (100 mM Tris-HCl, 50 mM EDTA, pH8.0). This solution can be stored but it should only be freeze-thawed once. Cover each sample well with approximately 50 µl of lysozyme solution and incubate for 10 minutes at room temperature. Then dip the slides in demineralised water, once only and dry with filter paper.U.K.
Alternatively, instead of lysozyme add 50 µl of 40 to 400µg ml–1 proteinase K in buffer (20 mM Tris-HCl, 2 mM CaCl2, pH 7,4) to each well and incubate at 37 °C for 30 minutes.
5.2.2.Dehydrate the cells in a graded ethanol series of 50 %, 80 % and 96 % for one minute each. Air dry the slides in a slide-holder.U.K.
5.2.3.Prepare a moist incubation chamber by covering the bottom of an air-tight box with tissue or filter paper soaked in 1x hybmix (Appendix 7). Pre-incubate the box in the hybridisation oven at 55 °C for at least 10 minutes.U.K.
5.2.4.Prepare the hybridisation solution (Appendix 7) allowing 45 µl per slide, and preincubate for five minutes at 55 °C.U.K.
5.2.5.Place slides on a hot plate at 45 °C and apply 10 µl of hybridization solution to each of the four wells on the slide(s).U.K.
5.2.6.Apply two coverslips (24 × 24 mm) to each slide without trapping air. Place the slides in the pre-warmed moist chamber and hybridise overnight in the oven at 55 °C in the dark.U.K.
5.2.7.Prepare three beakers containing 1 l of Ultra pure water (UPW), 1 l of 1x hybmix (334 ml 3x hybmix and 666 ml UPW) and 1 l of 1/2x hybmix (167 ml 3x hybmix and 833 ml UPW). Pre-incubate each in a waterbath at 55 °C.U.K.
5.2.8.Remove the coverslips from the slides and place the slides in a slide holder.U.K.
5.2.9.Wash away excess probe by incubation for 15 mins. in the beaker with 1x hybmix at 55 °C.U.K.
5.2.10.Transfer the slide holder to 1/2 hybmix washing solution and incubate for a further 15 mins.U.K.
5.2.11.Dip the slides briefly in UPW and place them on filter paper. Remove excess moisture by covering the surface gently with filter paper. Pipette 5 to 10 μl of anti-fading mountant solution (e.g. Vectashield, Vecta Laboratories, CA, USA or equivalent) on each window and apply a large coverslip (24 × 60 mm) over the whole slide.U.K.
5.3.Reading the FISH testU.K.
5.3.1.Observe the slides immediately with a microscope fitted for epifluorescence microscopy at 630 or 1000x magnification under immersions oil. With a filter suitable for fluorescein isothiocyanate (FITC) eubacterial cells (including most gram negative cells) in the sample are stained fluorescent green. Using a filter for tetramethylrhodamine-5-isothiocyanate, Cy3-stained cells of C. m. subsp. sepedonicus appear fluorescent red. Compare cell morphology with that of the positive controls. Cells must be bright fluorescent and completely stained The FISH test (section 9.4) must be repeated if the staining is aberrant. Scan windows across two diameters at right angles and around the perimeter. For samples showing no or low number of cells observe at least 40 microscope fields.U.K.
5.3.2.Observe for bright fluorescing cells with characteristic morphology of C. m. subsp. sepedonicus in the test windows of the test slides (see web site http://forum.europa.eu.int/Public/irc/sanco/Home/main). The fluorescence intensity must be equivalent or better than that of the positive control strain. Cells with incomplete staining or with weak fluorescence must be disregarded.U.K.
5.3.3.If any contamination is suspected the test must be repeated. This may be the case when all slides in a batch show positive cells due to the contamination of buffer or if positive cells are found (outside of the slide windows) on the slide coating.U.K.
5.3.4.There are several problems inherent to the specificity of the FISH test. Background populations of fluorescing cells with atypical morphology and cross reacting saprophytic bacteria with size and morphology similar to C. m. subsp. sepedonicus may occur, although much less frequent than in the IF test, in potato heel end core and stem segment pellets.U.K.
5.3.5.Consider only fluorescing cells with typical size and morphology, see in 5.3.2.U.K.
5.3.6.Interpretation of the FISH test result:U.K.
(i)
Valid FISH test results are obtained if bright green fluorescent cells of size and morphology typical of C. m. subsp. sepedonicus are observed using the FITC filter and if bright red fluorescent cells using the rhodamine filter in all positive controls and not in any of the negative controls. If bright fluorescing cells with characteristic morphology are found, estimate the average number of typical cells per microscope field and calculate the number of typical cells per ml of resuspended pellet (Appendix 4). Samples with at least 5 × 103 typical cells per ml of resuspended pellet are considered potentially contaminated. Further testing is required. Samples with less than 5 × 103 typical cells per ml of resuspended pellet are considered negative.
(ii)
The FISH test is negative if bright red fluorescent cells with size and morphology typical of C. m. subsp. sepedonicus are not observed using the rhodamine filter, provided that typical bright red fluorescent cells are observed in the positive control preparations when using the rhodamine filter.
6.PCR TESTU.K.
PrinciplesU.K.
When the PCR test is used as the principal screening test and found to be positive, the IF test must be performed as a second compulsory screening test. When the PCR test is used as the second screening test and found to be positive, further testing according to the flow scheme is required to complete the diagnosis.
Full exploitation of this method as principal screening test is only recommended when specialised expertise has been acquired.
Note:U.K.
Preliminary testing with this method should permit reproducible detection of 103 to 104 cells of C. m. subsp. sepedonicus per ml added to sample extracts which previously tested negative. Optimisation experiments may be required to achieve maximum levels of sensitivity and specificity in all laboratories.
Use validated PCR reagents and protocols. Preferably select a method with an internal control.
Use appropriate precautions to avoid contamination of sample with target DNA. The PCR test should be performed by experienced technicians, in dedicated molecular biology laboratories, in order to minimise the possibility of contamination with target DNA.
Negative controls (for DNA extraction and PCR procedures) should always be handled as final samples in the procedure, to make evident whether any carry over of DNA has occurred.
The following negative controls should be included in the PCR test:
sample extract that previously tested negative for C. m. subsp. Sepedonicus,
buffer controls used for extracting the bacterium and the DNA from the sample,
PCR-reaction mix.
The following positive controls should be included:
aliquots of resuspended pellets to which C. m. subsp. sepedonicus has been added (preparation see Appendix 2),
a suspension of 106 cells per ml of C. m. subsp. sepedonicus in water from a virulent isolate (e.g. NCPPB 2140 or NCPPB 4053),
if possible also use DNA extracted from positive control samples in the PCR test.
To avoid potential contamination prepare positive controls in a separate environment from samples to be tested.
Sample extracts should be as free as possible from soil. It could therefore in certain cases advisible to prepare extractions from washed potatoes if PCR protocols are to be used.
6.1.DNA purification methodsU.K.
Use positive and negative control samples as described above.U.K.
Prepare control material in an identical manner as the sample(s).
A variety of methods are available for purification of target DNA from complex sample substrates, thus removing inhibitors of PCR and other enzymatic reactions and concentrating target DNA in the sample extract.
The following method has been optimised for use with the validated PCR method shown in Appendix 6.
6.1.(a)Method according to Pastrik (2000)U.K.
1.
Pipette 220 µl of lysis buffer (100 mM NaCl, 10 mM Tris-HCl [pH 8,0], 1 mM EDTA [pH 8,0]) into a 1,5 ml Eppendorf tube.
2.
Add 100 µl sample extract and place in a heating block or waterbath at 95 °C for 10 minutes.
3.
Put tube on ice for five minutes.
4.
Add 80 µl Lysozyme stock solution (50 mg lysozyme per ml in 10 mM Tris HCl, pH 8,0) and incubate at 37 °C for 30 minutes.
5.
Add 220 µl of Easy DNA® solution A (Invitrogen), mix well by vortexing and incubate at 65 °C for 30 minutes.
6.
Add 100 µl of Easy DNA® solution B (Invitrogen), vortex vigorously until the precipitate runs freely in the tube and the sample is uniformly viscous.
7.
Add 500 µl of chloroform and vortex until the viscosity decreases and the mixture is homogeneous.
8.
Centrifuge at 15 000 g for 20 minutes at 4 °C to separate phases and form the interphase.
9.
Transfer the upper phase into a fresh Eppendorf tube.
10.
Add 1 ml of 100 % ethanol (-20 °C) vortex briefly and incubate on ice for 10 minutes.
11.
Centrifuge at 15 000 g for 20 minutes at 4 °C and remove ethanol from pellet.
12.
Add 500 µl 80 % ethanol (-20 °C) and mix by inverting the tube.
13.
Centrifuge at 15 000 g for 10 minutes at 4 °C, save the pellet and remove ethanol.
14.
Allow the pellet to dry in air or in a DNA speed vac.
15.
Resuspend the pellet in 100 µl sterile UPW and leave at room temperature for at least 20 minutes.
16.
Store at -20 °C until required for PCR.
17.
Spin down any white precipitate by centrifugation and use 5 µl of the supernatant containing DNA for the PCR.
6.1.(b)Other methodsU.K.
Other DNA extraction methods (e.g. Qiagen DNeasy Plant Kit) could be applied providing that they are proven to be equally as effective in purifying DNA from control samples containing 103 to 104 pathogen cells per ml.
6.2.PCRU.K.
6.2.1.Prepare test and control templates for PCR according to the validated protocol (Appendix 6). Prepare one decimal dilution of sample DNA extract (1:10 in UPW).U.K.
6.2.2.Prepare the appropriate PCR reaction mix in a contamination-free environment according to the published protocol (Appendix 6). The validated PCR protocol is a multiplex reaction that also incorporates an internal PCR control.U.K.
6.2.3.Add 5 µl of DNA extract per 25 µl PCR reaction in sterile PCR tubes.U.K.
6.2.4.Incorporate a negative control sample containing only PCR reaction mix and add the same source of UPW as used in the PCR mix in place of sample.U.K.
6.2.5.Place tubes in the same thermal cycler which was used in preliminary testing and run the appropriately optimised PCR programme (Appendix 6).U.K.
6.3.Analysis of the PCR productU.K.
6.3.1.Resolve PCR amplicons by agarose gel electrophoresis. Run at least 12 µl of amplified DNA reaction mixture from each sample mixed with 3 µl loading buffer (Appendix 6) in 2,0 % (w/v) agarose gels in tris-acetate-EDTA (TAE) buffer (Appendix 6) at 5 to 8 V per cm. Use an appropriate DNA marker, e.g. 100 bp ladder.U.K.
6.3.2.Reveal DNA bands by staining in ethidium bromide (0,5 mg per L) for 30 to 45 min taking appropriate precautions for handling this mutagen.U.K.
6.3.3.Observe stained gel under short wave UV transillumination (e.g. λ = 302 nm) for amplified PCR products of the expected size (Appendix 6) and document.U.K.
6.3.4.For all new findings/cases verify authenticity of the PCR amplicon by performing restriction enzyme analysis on a sample of the remaining amplified DNA by incubating at the optimum temperature and time with an appropriate enzyme and buffer (see Appendix 6). Resolve the digested fragments by agarose gel electrophoresis as before and observe characteristic restriction fragment pattern under UV transillumination after ethidium bromide staining and compare with the undigested and digested positive control.U.K.
Interpretation of the PCR test result:
The PCR test is negative if the C. m. subsp. sepedonicus-specific PCR amplicon of expected size is not detected for the sample in question but is detected for all positive control samples (in case of multiplex PCR with plant specific internal control primers: a second PCR-product of expected size must be amplified with the sample in question).
The PCR test is positive if the C. m. subsp. sepedonicus-specific PCR amplicon of expected size and restriction pattern (when required) is detected, providing that it is not amplified from any of the negative control samples. Reliable confirmation of a positive result can also be obtained by repeating the test with a second set of PCR primers (section 9.3).
Note:U.K.
Inhibition of the PCR may be suspected if the expected amplicon is obtained from the positive control sample containing C. m. subsp. sepedonicus in water but negative results are obtained from positive controls with C. m. subsp. sepedonicus in potato extract. In multiplex PCR protocols with internal PCR controls, inhibition of the reaction is indicated when neither of the two amplicons are obtained.
Contamination may be suspected if the expected amplicon is obtained from one or more of the negative controls.
7.BIOASSAY TESTU.K.
Note:U.K.
Preliminary testing with this method should permit reproducible detection of 103 to 104 colony-forming units of C. m. subsp. sepedonicus per ml added to sample extracts that previously tested negative (preparation see Appendix 2).
Highest sensitivity of detection can be expected when using freshly prepared sample extract and optimal growth conditions. However, the method can be successfully applied to extracts that have been stored under glycerol at -68 to -86 °C.U.K.
Some varieties of eggplant provide an excellent selective enrichment medium for the growth of C. m. subsp. sepedonicus even in the absence of symptoms and also provide an excellent confirmatory host test.
Growth conditions should be optimal to reduce the risk of false negative test results.
For cultural details, see Appendix 8.
7.1.Distribute the whole of the remaining test aliquot of the resuspended pellet from section 3.1.6 or 3.2.5 between eggplants by one of the methods given below (7.3 or 7.4). Use only plants at leaf stage two to three up to full expansion of the third true leaf. In order to ensure complete utilisation of the resuspended pellet as well as effective inoculation the procedures outlined below will require 15 to 25 eggplants per sample.U.K.
7.2.Do not water eggplants for one to two days prior to inoculation to reduce turgor pressure.U.K.
7.3.
Slit inoculationU.K.
7.3.1.Holding the plant between two fingers, pipette a drop (approximately 5 to 10 µl) of the suspended pellet on the stem between the cotyledons and the first leaf.U.K.
7.3.2.Using a sterile scalpel, make a diagonal slit, about 1,0 cm long and approximately 2/3 of the stem thickness deep, starting the cut from the pellet drop.U.K.
7.3.3.Seal the cut with sterile vaseline from a syringe.U.K.
7.4.Syringe inoculationU.K.
Inoculate the eggplant stems just above the cotyledons using a syringe fitted with a hypodermic needle (not less than 23 G). Distribute the sample between the eggplants.
7.5.As the positive controls, inoculate 5 plants with an aqueous suspension of 105 to 106 cells per ml of a known culture of C. m. subsp. sepedonicus and, where possible, with naturally infected tuber tissue (see section 4) by the same inoculation method (7.3 or 7.4).U.K.
7.6.As the negative control, inoculate 5 plants with sterile pellet buffer by the same inoculation method (7.3 or 7.4).U.K.
7.7.Incubate plants in quarantine facilities for up to four weeks at 18 to 24 °C. Incubate plants with sufficient light and high humidity (70 to 80 %) and water to prevent water logging or wilting through water deficiency. C. m. sepedonicus cells are killed at temperatures above 30 °C and the optimum temperature is 21 °C. To avoid contamination incubate positive control and negative control plants on clearly separated benches in a glasshouse or growth chamber or, in case space is limited, ensure strict separation between treatments. If plants for different samples must be incubated close together, separate them with appropriate screens. When fertilising, watering, inspecting and any other manipulations take great care to avoid cross-contamination. It is essential to keep glasshouses and growth chambers free of all insect pests since they may transmit the bacterium from sample to sample.U.K.
7.8.Examine regularly for symptoms starting after a week. Count the number of plants showing symptoms. C. m. subsp. sepedonicus causes leaf wilting in eggplants which may commence as a marginal or interveinal flaccidity. Wilted tissue may initially appear dark green or mottled but turns paler before becoming necrotic. Interveinal wilts often have a greasy water-soaked appearance. Necrotic tissue often has a bright yellow margin. Plants are not necessarily killed; the longer the period before symptoms develop, the greater the chance of survival. Plants may outgrow the infection. Young eggplants are much more susceptible to low populations of C. m. subsp. sepedonicus than are older plants, hence the necessity to use plants at or just before leaf stage 3.U.K.
Wilts may also be induced by populations of other bacteria or fungi present in the tuber tissue pellet. These include Ralstonia solanacearum, Erwinia carotovora subsp. carotovora and E. carotovora subsp. atroseptica, Erwinia chrysanthemi, Phoma exigua var. foveata, as well as large populations of saprophytic bacteria. In particular Erwinia chrysanthemi can cause leaf symptoms and wilt that is very similar to symptoms of C. m. sepedonicus. The only difference is blackening of the stems in case in Erwinia chrysanthemi infections. Other wilts can be distinguished from those caused by C. m. subsp. sepedonicus since whole leaves or whole plants wilt rapidly. Also a Gram stain can be prepared: this test will differentiate C. m. subsp. sepedonicus from Erwinia spp.
7.9.As soon as symptoms in eggplants are observed reisolation should be performed, using sections of wilted leaf tissue or stem tissue from plants (see 3.1. 3 for tissue maceration). Surface disinfect the eggplant leaves and stems by wiping with 70 % ethanol. Perform an IF test or PCR on the eggplant sap and isolate on to suitable (selective) media (see section 8). A Gram stain (Appendix 9) may also be prepared. Identify purified cultures of presumptive C. m. subsp. sepedonicus and confirm pathogenicity (see section 9 and 10).U.K.
7.10.Under certain circumstances, in particular where growing conditions are not optimal, it may be possible for C. m. subsp. sepedonicus to exist as a latent infection within eggplants even after incubation periods up to 4 weeks. If no symptoms are observed after 4 weeks perform IF/PCR on a composite sample of 1 cm stem sections of each test plant taken above the inoculation site. If the test is positive reisolation on suitable (selective) media should be performed following the procedure under section 8. Identify purified cultures of presumptive C. m. subsp. sepedonicus and confirm pathogenicity (section 9 and 10).U.K.
Interpretation of the bioassay test result.
Valid Bioassay test results are obtained when plants of the positive control show typical symptoms, the bacteria can be reisolated from these plants and no symptoms are found on the negative controls.
The bioassay test is negative if test plants are not infected by C. m. subsp. sepedonicus, and provided that C. m. subsp. sepedonicus is detected in positive controls.
The bioassay test is positive if the test plants are infected by C. m. subsp. sepedonicus.
8.ISOLATION OF C. M. SUBSP. SEPEDONICUS U.K.
Note:U.K.
Diagnosis is only completed if C. m. subsp. sepedonicus is isolated, subsequently identified (see section 9). and confirmed by a pathogenicity test (section 10). Although C. m. subsp. sepedonicus is a fastidious organism, it can be isolated from symptomatic tissue.
However, it may be outgrown by rapidly growing saprophytic bacteria and, therefore, isolations directly from the tuber or stem tissue pellet (section 3.1.6 or 3.2.5) is difficult. With selective medium and appropriate dilution of the resuspended pellet from the heel end cores or stems of potatoes direct isolation of C. m. subsp. sepedonicus may be possible.U.K.
Isolations shall be made from all symptomatic potato tubers or stem segments and from eggplants were no symptoms are observed but IF/PCR test from the composite sample was positive (see section 7.10). Maceration of eggplants stems when necessary should be carried out as described in section 3.1.3.
As positive controls, prepare decimal dilutions from a suspension of 106 cfu per ml of C. m. subsp. sepedonicus (e.g. NCPPB 4053 or PD 406). To avoid any possibility of contamination, prepare positive controls totally separated from samples to be tested.
For each newly prepared batch of a selective medium its suitability for growth of the pathogen should be tested before it is used to test routine samples.
Test control material in an identical manner as the sample(s).
8.1.Selective platingU.K.
8.1.1.From a 100 µl aliquot from a resuspended potato pellet sample or eggplant sap make 10-fold dilutions in pellet buffer (Appendix 3).U.K.
8.1.2.Isolation from undiluted potato pellet usually fails due to the fastidious growth habit of Cms and competition by saprophytes. Since the bacterium is usually present in high populations in infected tissues, the saprophytes can usually be diluted out, whilst the pathogen remains. It is therefore recommended to spread 100 µl from each of the samples, 1/100 up to 1/10 000 dilutions onto MTNA medium or NCP-88 medium (Appendix 5) (if using 90 mm diameter petri dishes- adjust volume for alternative dish sizes), using spreaders (hockey sticks) and the spread plate technique.U.K.
Note:U.K.
An alternative strategy is to spread out the initial 100 µl potato pellet aliquot onto a first agar plate with a spreader and then remove the spreader to a second agar plate, streaking out any residue left on the spreader; finally repeat this with a third plate, thus giving a dilution plating effect via the spreader.
8.1.3.Incubate plates in the dark at 21 to 23 °C.U.K.
8.1.4.Initial examinations of the plates including, by reference to the control plates, counts of any C. m. subsp. sepedonicus like colonies are made after 3 days, with further counts after 5, 7, eventually 10 days.U.K.
8.2.Purification of suspicious coloniesU.K.
Note:U.K.
Subculturing of C. m. subsp. sepedonicus-like colonies should be carried out onto YGM media for eggplant inoculation and/or subsequent identification; this should be done before the plates become too overgrown i.e. preferably after three to five days.
8.2.1.Streak C. m. subsp. sepedonicus –like colonies on to one of the following media: (formulae are given in Appendix 5):U.K.
nutrient dextrose agar (for use in subculturing only),
yeast peptone glucose agar,
yeast extract mineral salts agar.
Incubate at 21 °C to 24 °C for up to 10 days.
C. m. subsp. sepedonicus is slow-growing, usually producing pin-point, cream, domed colonies within 10 days. Photos of typical colonies of C. m. subsp. sepedonicus (see web site http://forum.europa.eu.int/Public/irc/sanco/Home/main).
8.2.2.Re-streak to establish purity.U.K.
Growth rates are improved with subculture. Typical colonies are creamy-white or ivory, occasionally yellow, rounded, smooth, raised, convex-domed, mucoid-fluidal, with entire edges and usually 1 to 3 mm in diameter.
A simple Gram stain (Appendix 9) may help to select colonies for further testing.
8.2.3.Identify presumptive cultures (see section 9) and perform a pathogenicity test (see section 10).U.K.
9.IDENTIFICATIONU.K.
Identify pure cultures of presumptive C. m. subsp. sepedonicus isolates using at least two of the following tests based on different biological principles.U.K.
Include known reference strains where appropriate for each test performed.
9.1.Nutritional and enzymatic identification testsU.K.
Determine the following phenotypic properties which are universally present or absent in C. m. subsp. sepedonicus, according to the methods of Lelliott and Stead (1987), Klement et al. (1990), Schaad (2001), Anonymous (1987).
All media should be incubated at 21 °C and examined after six days. If no growth has occurred, incubate for up to 20 days.
All tests must include a known C. m. subsp.sepedonicus control. Nutritional and physiological tests must be made using inocula from nutrient agar subcultures. Morphological comparisons must be made from nutrient dextrose agar cultures.
| Tests | Expected result |
|---|---|
| Oxidation/Fermentation (O/F) test | Inert or weakly oxidative |
| Oxidase activity | – |
| Growth at 37 °C | – |
| Urease activity | – |
| Aesculin hydrolysis | + |
| Starch hydrolysis | – or weak |
| Tolerance of 7 % NaCl | – |
| Indole production | – |
| Catalase activity | + |
| H2S production | – |
| Citrate utilization | – |
| Gelatin liquefaction | – |
| Acid glycerol | – |
| Acid from lactose | – or weak |
| Acid from rhamnose | – |
| Acid from salicin | – |
| Gram stain (Appendix 9) | + |
9.2.IF-testU.K.
(a)
Prepare a suspension of approximately 106 cells per ml in IF buffer (Appendix 3).
(b)
Prepare a 2-fold dilution series of an appropriate antiserum.
(c)
Apply the IF procedure (section 4).
(d)
A positive IF test is achieved if the IF titre of the culture is equivalent to that of the positive control.
9.3.PCR testU.K.
(a)
Prepare a suspension of approximately 106 cells per ml in ultra pure water (UPW).
(b)
Heat 100 µl of the cell suspension in closed tubes in a heating block or boiling waterbath at 100 °C for four minutes. If required, addition of freshly-prepared NaOH to a final concentration of 0,05M may assist cell lysis. The samples may then be stored at -16 to -24 °C until required.
(c)
Apply appropriate PCR procedures to amplify C. m. subsp. sepedonicus specific amplicons (e. g. Pastrik, 2000; see Appendix 4; Li and de Boer, 1995; Mills et al., 1997; Pastrik and Rainey, 1999; Schaad et al., 1999.
(d)
A positive identification of C. m. subsp.sepedonicus is achieved if the PCR amplicons are the same size and have the same restriction fragment length polymorphisms as for the positive control strain.
9.4.FISH testU.K.
(a)
Prepare a suspension of approximately 106 cells per ml in UPW.
(b)
Apply the FISH procedure (section 5).
(c)
A positive FISH test is achieved if the same reactions are achieved from the culture and the positive control.
9.5.Fatty acid profiling (FAP)U.K.
(a)
Grow the culture on trypticase soy agar (Oxoid) for 72 hours at 21 °C (+/– 1°).
(b)
Apply an appropriate FAP procedure (Janse, 1991; Stead, 1992).
(c)
A positive FAP test is achieved if the profile of the presumptive culture is identical to that of the positive control. The presence of characteristic fatty acids are 15:1 Anteiso A, 15:0 Iso, 15:0 Anteiso, 16:0 Iso, 16:0 and 17:0 Anteiso is highly indicative of C. m. sepedonicus. Other genera such as Curtobacterium, Arthrobacter and Micrococcus also have some of these acids but 15:1 Anteiso A is a rare acid in these bacteria but occurs in all Clavibacter spp. at between 1 to 5 %. In C. m. sepedonicus the value is usually around 5 %.
9.6.BOX-PCRU.K.
(a)
Prepare a suspension of approximately 106 cells per ml in UPW.
(b)
Apply the test according the procedure (Smith et al., 2001).
10.CONFIRMATION TESTU.K.
The pathogenicity test must be performed as final confirmation of a diagnosis of C. m. subsp. sepedonicus and for assessment of virulence of cultures identified as C. m. subsp. sepedonicus:U.K.
10.1.Prepare an inoculum of approximately 106 cells per ml from three day cultures of the isolate to be tested and an appropriate positive control strain of C. m. subsp. sepedonicus.U.K.
10.2.Inoculate 5 to 10 eggplant stems of young seedlings at leaf stage 3 (section 7.3 or 7.4).U.K.
10.3.Incubate at 18 to 24 °C with sufficient light and high relative humidity with appropriate watering to avoid waterlogging or drought stress (section 7.7). With pure cultures, typical wilting should be obtained within two weeks but plants not showing symptoms (see section 7.8) after this time should be incubated up to three weeks at temperatures conducive to eggplant growth but not exceeding 25 °C (Appendix 8). If after three weeks symptoms are not present, the culture cannot be confirmed as being a pathogenic form of C. m. subsp. sepedonicus.U.K.
10.4.Isolate from symptomatic plants by removing a section of stem 2 cm above the inoculation point. Comminute and suspend in a small volume of sterile distilled water or 50 mM phosphate buffer (Appendix 3). Isolate from the suspension by dilution spreading or streaking onto MTNA and YPGA (Appendix 5), incubate for three to five days at 21 to 23 °C and observe the formation of colonies typical of C. m. subsp. sepedonicus.U.K.
Appendix 1Laboratories involved in optimisation and validation of protocols
| a Contact scientists: see website http://forum.europa.eu.int/Public/irc/sanco/Home/main | ||
| Laboratorya | Location | Country |
|---|---|---|
| Agentur für Gesundheit und Ernährungssicherheit | Vienna and Linz | Austria |
| Departement Gewasbescherming | Merelbeke | Belgium |
| Plantedirektoratet | Lyngby | Denmark |
| Central Science Laboratory | York | England |
| Scottish Agricultural Science Agency | Edinburgh | Scotland |
| Laboratoire National de la Protection des Végétaux, Unité de Bactériologie | Angers | France |
| Laboratoire National de la Protection des Végétaux, Station de Quarantaine de la Pomme de Terre | Le Rheu | France |
| Biologische Bundesanstalt | Kleinmachnow | Germany |
| Pflanzenschutzamt Hannover | Hannover | Germany |
| State Laboratory | Dublin | Ireland |
| Plantenziektenkundige Dienst | Wageningen | Netherlands |
| Norwegian Crop Research Institute, Plant Protection Centre | Aas | Norway |
| Direcção-Geral de Protecção das Culturas | Lisbon | Portugal |
| Nacionalni institut za biologijo | Ljubljana | Slovenia |
| Centro de Diagnostico de Aldearrubia | Salamanca | Spain |
Appendix 2Preparation of positive and negative controls for the core screening tests PCR/IF and FISH
Produce a 72 hour culture of a virulent strain of C. m. subsp. sepedonicus (NCPPB 4053 or PD 406) on MTNA basal medium and suspend in 10 mM phosphate buffer to obtain a cell density of approximately 1 to 2 × 108 cfu per ml. This is usually obtained by a faintly turbid suspension equivalent to an optical density of 0,20 at 600 nm.
Remove the heel end cores of 200 tubers taken from a white skin variety production known to be free from C. m. subsp. sepedonicus.
Process the heel ends as usual and resuspend the pellet in 10 ml.
Prepare 10 sterile 1,5 ml microvials with 900 µl of the resuspended pellet.
Transfer 100 µl of the suspension of C. m. subsp. sepedonicus to the first microvial. Vortex.
Establish decimal levels of contamination by further diluting in the next five microvials.
The six contaminated microvials will be used as positive controls. The four non-contaminated microvials will be used as negative controls. Label the microvials accordingly.
Prepare aliquots of 100 µl in sterile 1,5 ml microvials thus obtaining nine replicas of each control sample. Store at -16 to -24 °C until use.
The presence and quantification of C. m. subsp. sepedonicus in the control samples should be first confirmed by IF.
For the PCR test perform DNA extraction from positive and negative control samples with each series of test samples.
For IF and FISH tests perform assays on positive and negative control samples with each series of test samples.
For IF, FISH and PCR assays C. m. subsp. sepedonicus must be detected in at least the 106 and 104 cells/ml of the positive controls and not in any of the negative controls.
Appendix 3Buffers for test procedures
GENERAL: Unopened sterilized buffers can be stored for up to one year.U.K.
1.Buffers for extraction procedureU.K.
1.1.Extraction buffer (50 mM phosphate buffer, pH 7,0)U.K.
This buffer is used for extraction of the bacterium from plant tissues by homogenisation or shaking.
| Na2HPO4 (anhydrous) | 4,26 g |
| KH2PO4 | 2,72 g |
| Distilled water | 1.00 L |
Dissolve ingredients, check pH and sterilise by autoclaving at 121 °C for 15 min.
Additional components may be useful as follows:
1.2.Pellet buffer (10 mM phosphate buffer, pH 7,2)U.K.
This buffer is used for resuspension and dilution of potato tuber heel-end core extracts following concentration to a pellet by centrifugation.
| Na2HPO4.12H2O | 2,7 g |
| NaH2PO4.2H2O | 0,4 g |
| Distilled water | 1,00 L |
Dissolve ingredients, check pH and sterilise by autoclaving at 121 °C for 15 min.
2.Buffers for the IF testU.K.
2.1.IF-Buffer (10 mM phosphate buffered saline (PBS), pH 7,2)U.K.
This buffer is used for dilution of antibodies.
| Na2HPO4.12H2O | 2,7 g |
| NaH2PO4.2H2O | 0,4 g |
| NaCl | 8,0 g |
| Distilled water | 1.0 L |
Dissolve ingredients, check pH and sterilise by autoclaving at 121 °C for 15 min.
2.2.IF-buffer-TweenU.K.
This buffer is used to wash slides.
Add 0,1 % Tween 20 to the IF buffer.
2.3.Phosphate buffered glycerol, pH 7,6U.K.
This buffer is used as a mountant fluid on the windows of IF slides to enhance fluorescence.
| Na2HPO4.12H2O | 3,2 g |
| NaH2PO4.2H2O | 0,15 g |
| Glycerol | 50 ml |
| Distilled water | 100 ml |
Anti-fading mountant solutions are commercially available e.g. Vectashield® (Vector Laboratories) or Citifluor® (Leica).
Appendix 4Determination of contamination level in IF and FISH tests
1.
Count the mean number of typical fluorescent cells per field of view (c)
2.
Calculate the number of typical fluorescent cells per microscope slide window (C)
C = c × S/s
| where | S | = | surface area of window of multispot slide, and |
| s | = | surface area of objective field. |
| s = πi2/4G2K2 | where | i | = | field coefficient (varies from 8 to 24 depending upon ocular type) |
| K | = | tube coefficient (1 or 1,25) | ||
| G | = | magnification of objective (100x, 40x etc.). |
3.
Calculate the number of typical fluorescent cells per ml of resuspended pellet (N)
N = C × 1 000/y × F
| where | y | = | volume of resuspended pellet on each window, and |
| F | = | dilution factor of resuspended pellet. |
Appendix 5Media for isolation and culture of C. m. subsp. sepedonicus
(a)General growth mediaU.K.
Nutrient agar (NA)U.K.
| Nutrient Agar (Difco) | 23,0 g |
| Distilled water | 1,0 L |
Dissolve ingredients and sterilise by autoclaving at 121 °C for 15 min.
Nutrient dextrose agar (NDA)U.K.
Difco bacto nutrient agar containing 1 % D(+) glucose (monohydrate). Sterilize by autoclaving at 115 °C for 20 minutes.
Yeast peptone glucose agar (YPGA)U.K.
| Yeast Extract (Difco) | 5,0 g |
| Bacto-Peptone (Difco) | 5,0 g |
| D(+) Glucose (monohydrate) | 10,0 g |
| Bacto-Agar (Difco) | 15,0 g |
| Distilled water | 1,0 L |
Dissolve ingredients and sterilise by autoclaving at 121 °C for 15 min.
Yeast extract mineral salts medium (YGM)U.K.
| Bacto-Yeast-Extract (Difco) | 2,0 g |
| D(+) Glucose (monohydrate) | 2,5 g |
| K2HPO4 | 0,25 g |
| KH2PO4 | 0,25 g |
| MgSO4.7H2O | 0,1 g |
| MnSO4.H2O | 0,015 g |
| NaCl | 0,05 g |
| FeSO4.7H2O | 0,005 g |
| Bacto-Agar (Difco) | 18 g |
| Distilled water | 1,0 L |
Dissolve ingredients and sterilize 0,5 litre volumes of medium by autoclaving at 115 °C for 20 minutes.
(b)Validated selective growth mediaU.K.
MTNA mediumU.K.
Unless otherwise stated all media components are from BDH.
| Yeast extract (Difco) | 2,0 g |
| Mannitol | 2,5 g |
| K2HPO4 | 0,25 g |
| KH2PO4 | 0,25 g |
| NaCl | 0,05 g |
| MgSO4.7H2O | 0,1 g |
| MnSO4.H2O | 0,015 g |
| FeSO4.7H2O | 0,005 g |
| Agar (Oxoid no. 1) | 16,0 g |
| Distilled water | 1,0 L |
Dissolve ingredients, adjust pH to 7,2. After autoclaving (at 121 °C for 15 minutes) and cooling down to 50 °C, add the antibiotics: trimethoprim 0,06 g, nalidixic acid 0,002 g, amphotericin B 0,01 g.
Stock antibiotic solutions: trimethoprim (Sigma) and nalidixic acid (Sigma) (both at 5 mg/ml), in 96 % methanol, amphotericin B (Sigma) (1 mg/ml) in dimethyl sulfoxide. Stock solutions are filter-sterilized.
Note:U.K.
Durability of basal medium is three months. After antibiotics are added durability is one month when stored refrigerated.
NCP-88 mediumU.K.
| Nutrient agar (Difco) | 23 g |
| Yeast extract (Difco) | 2 g |
| D-mannitol | 5 g |
| K2HPO4 | 2 g |
| KH2PO4 | 0,5 g |
| MgSO4.7H2O | 0,25 g |
| Distilled water | 1,0 L |
Dissolve ingredients, adjust pH to 7,2. After autoclaving and cooling down to 50 °C, add the following antibiotics: Polymyxin B sulphate (Sigma) 0,003 g, nalidixic acid (Sigma) 0,008 g, Cycloheximide (Sigma) 0,2 g.
Dissolve antibiotics in stock solutions as follows: nalidixic acid in 0,01 M NaOH, cycloheximide in 50 % ethanol, polymyxin B sulphate in distilled water. Stock solutions are filter-sterilized.
Note:U.K.
Durability of basal medium is three months. After antibiotics are added durability is one month when stored refrigerated.
Appendix 6Validated PCR protocol and reagents
Note:U.K.
Preliminary testing should permit reproducible detection of at least 103 to 104 cells of C. m. sepedonicus per ml of sample extract.
Preliminary testing should also show no false positive results with a panel of selected bacterial strains.
1.Multiplex PCR protocol with internal PCR control (Pastrik, 2000)U.K.
1.1.Oligonucleotide primersU.K.
| Forward primer PSA-1 | 5′- ctc ctt gtg ggg tgg gaa aa -3′ |
| Reverse primer PSA-R | 5′- tac tga gat gtt tca ctt ccc c -3′ |
| Forward primer NS-7-F | 5′- gag gca ata aca ggt ctg tga tgc -3′ |
| Reverse Primer NS-8-R | 5’- tcc gca ggt tca cct acg ga -3’ |
Expected amplicon size from C. m. subsp. sepedonicus template DNA = 502 bp (PSA-primer set).
Expected amplicon size from the 18S rRNA internal PCR control = 377 bp (NS-primer set).
1.2.PCR reaction mixU.K.
| a Methods were validated using Taq polymerase from Perkin Elmer (AmpliTaq or Gold) and Gibco BRL. | ||
| b Concentration of primers NS-7 F and NS-8-R were optimised for potato heel end core extraction using the homogenisation method and DNA purification according to Pastrik (2000) (see section 6.1.a) and 6.2). Re-optimisation of reagent concentrations will be required if extraction by shaking or other DNA isolation methods are used. | ||
| Reagent | Quantity per reaction | Final concentration |
|---|---|---|
| Sterile UPW | 15,725 µl | |
| 10x PCR buffera (15 mM MgCl2) | 2,5 µl | 1x (1,5 mM MgCl2) |
| BSA (fraction V) (10 %) | 0,25 µl | 0,1 % |
| d-nTP mix (20 mM) | 0,125 µl | 0,1 mM |
| Primer PSA-1 (10 µM) | 0,5 µl | 0,2 µM |
| Primer PSA-R (10 µM) | 0,5 µl | 0,2 µM |
| Primer NS-7-F (10 µM)b | 0,1 µl | 0,04 µM |
| Primer NS-8-R (10 µM)b | 0,1 µl | 0,04 µM |
| Taq polymerase (5 U/µl)a | 0,2 µl | 1,0 U |
| Sample volume | 5,0 µl | |
| Total volume | 25,0 µl | |
1.3.PCR reaction conditionsU.K.
Run the following programme:
| 1 cycle of: | (i) | 3 minutes at 95 °C (denaturation of template DNA) |
| 10 cycles of: | (ii) | 1 minute at 95 °C (denaturation of template DNA) |
| (iii) | 1 minute at 64 °C (annealing of primers) | |
| (iv) | 1 minute at 72 °C (extension of copy) | |
| 25 cycles of: | (v) | 30 seconds at 95 °C (denaturation of template DNA) |
| (vi) | 30 seconds at 62 °C (annealing of primers) | |
| (vii) | 1 minute at 72 °C (extension of copy) | |
| 1 cycle of: | (viii) | 5 minutes at 72 °C (final extension) |
| (ix) | hold at 4 °C. |
Note:U.K.
This programme is optimised for use with an MJ Research PTC 200 thermal cycler. Modification of the duration steps of cycles (ii), (iii) (iv), (v), (vi) and (vii) may be required for use with other models.
1.4.Restriction enzyme analysis of amplicon.U.K.
PCR products amplified from C. m. subsp. sepedonicus DNA produce a distinctive restriction fragment length polymorphism with enzyme Bgl II after incubation at 37 °C for 30 minutes. The restriction fragments obtained from C. m. subsp. sepedonicus-specific fragment are 282 bp and 220 bp in size.
2.Preparation of the Loading bufferU.K.
2.1.Bromphenol blue (10 %-stock solution)U.K.
| Bromphenol blue | 5 g |
| Distilled Water (bidest) | 50 ml |
2.2.Loading bufferU.K.
| Glycerol (86 %) | 3,5 ml |
| Bromphenol blue (5.1) | 300 µl |
| Distilled Water (bidest) | 6,2 ml |
3.10x Tris Acetate EDTA (TAE) buffer, pH 8,0U.K.
| Tris buffer | 48,4 g |
| Glacial acetic acid | 11,42 ml |
| EDTA (disodium salt) | 3,72 g |
| Distilled water | 1,00 L |
Dilute to 1x before use.
Also commercially available (e.g. Invitrogen or equivalent).
Appendix 7Validated reagents for FISH test
1.Oligo-probesU.K.
| Cms-specific probe CMS-CY3-01: | 5′- ttg cgg ggc gca cat ctc tgc acg -3′ |
| Non-specific eubacterial probe EUB-338-FITC: | 5′- gct gcc tcc cgt agg agt -3′ |
2.Fixative solutionU.K.
[WARNING! THE FIXATIVE CONTAINS PARAFORMALDEHYDE WHICH IS TOXIC. WEAR GLOVES AND DO NOT INHALE. IT IS ADVISABLE TO WORK IN A FUME CUPBOARD]
(i)
Heat nine ml molecular grade water (e.g. Ultra pure water (UPW)) to about 60 °C and add 0,4 g paraformaldehyde. Paraformaldehyde dissolves after adding five drops of 1N NaOH and stirring with a magnetic stirrer.
(ii)
Adjust pH to 7,0 by addition of 1ml of 0,1 M phosphate buffer (PB; pH 7,0) and five drops of 1 N HCl. Check pH with indicator strips and adjust if necessary with HCl or NaOH.
[WARNING! DO NOT USE A PH METER IN SOLUTIONS WITH PARAFORMALEDHYDE]
(iii)
Filter the solution through a 0,22 µm membrane filter and keep dust-free at 4 °C until further use.
(iv)
Note:
Alternative fixative solution: 96 % ethanol.
3.3x HybmixU.K.
| NaCl | 2,7 M |
| Tris-HCl | 60 mM (pH 7,4) |
| EDTA (filter sterilised and autoclaved) | 15 mM |
Dilute to 1x as required.
4.Hybridisation solutionU.K.
1x Hybmix
| Sodium dodecyl sulphate (SDS) | 0,01 % |
| probe EUB 338 | 5 ng/μl |
| probe CMSCY301 | 5 ng/μl |
Prepare quantities of hybridisation solution according to the calculations in Table. For each slide (containing two different samples in duplicate) 90 μl hybridisation solution is required.
Table:
Suggested quantities for preparation of hybridisation mix
| 2 slides | 8 slides | |
|---|---|---|
| Sterile UPW | 50,1 | 200,4 |
| 3x hybmix | 30,0 | 120,0 |
| 1 % SDS | 0,9 | 3,6 |
| Probe EUB 338 (100 ng/μl) | 4,5 | 18,0 |
| Probe CMSCY301 (100 ng/μl) | 4,5 | 18,0 |
| Total volume (μl) | 90,0 | 360,0 |
NB. Store all solutions containing light sensitive oligo-probes in the dark at -20 °C. Protect from direct sunlight or electric light during use.U.K.
5.0,1M Phosphate buffer, pH 7,0U.K.
| Na2HPO4 | 8,52 g |
| KH2PO4 | 5,44 g |
| Distilled water | 1,00 L |
Dissolve ingredients, check pH and sterilise by autoclaving at 121 °C for 15 min.
Appendix 8Eggplant culture
Sow seeds of eggplant (Solanum melongena) in pasteurized seed compost. Transplant seedlings with fully expanded cotyledons (10 to 14 days) into pasteurized potting compost.
Eggplants should be grown in a glasshouse with the following environmental conditions:
| Day length: | 14 hours or natural day length if greater; | |
| Temperature: | day: | 21 to 24 °C, |
| night: | 15 °C. |
| Susceptible varieties of eggplant: | ‘Black Beauty’, |
| ‘Long Tom’, | |
| ‘Rima’, | |
| ‘Balsas’ |
Supplier: see website http://forum.europa.eu.int/Public/irc/sanco/Home/main
Appendix 9Gram stain procedure (Hucker's modification) (Doetsch, 1981)(1)
Crystal violet solutionU.K.
Dissolve 2 g crystal violet in 20 ml 95 % ethanol.
Dissolve 0,8 g ammonium oxalate in 80 ml distilled water.
Mix the two solutions.
Lugol's iodineU.K.
| Iodine | 1 g |
| Potassium iodide | 2 g |
| Distilled water | 300 ml |
Grind the solids together in a pestle and mortar. Add to the water and stir to dissolve in a closed container.
Safranin counterstain solutionU.K.
Stock solution:
| Safranin O | 2,5 g |
| 95 % ethanol | 100 ml |
Mix and store.
Dilute: 1:10 to obtain a working solution.
Staining procedureU.K.
1.
Prepare smears, air dry and heat fix.
2.
Flood slide with crystal violet solution for one minute.
3.
Wash briefly with tap water.
4.
Flood with Lugol's iodine for one minute.
5.
Wash with tap water and blot dry.
6.
Decolourise with 95 % ethanol, added dropwise, until no further colour is removed or immerse with gentle agitation for 30 seconds.
7.
Wash in tap water and blot dry.
8.
Flood with safranin solution for 10 seconds.
9.
Wash with tap water and blot dry.
Gram positive bacteria stain violet-blue; Gram negative bacteria stain pink-red.
REFERENCESU.K.
1.Anonymous, 1987. Scheme of the detection and diagnosis of the ring rot bacterium Corynebacterium sepedonicum in batches of potato tubers. Commission of the European Communities, Luxembourg. Publ EUR 11288 EN, 21 pp.U.K.
2.Bradbury, J. F., 1970. Isolation and preliminary study of bacteria from plants. Rev. Pl. Path., 49, 213-218.U.K.
3.Dinesen, I. G., 1984. The extraction and diagnosis of Corynebacterium sepedonicum from diseased potato tubers. EPPO Bull. 14 (2), 147-152.U.K.
4.Doetsch, R. N., 1981. Determinative methods of light microscopy. In: Manual of methods for general bacteriology, American Society for Microbiology, Washington, 21-23.U.K.
5.Hugh, R. and Leifson, F., 1953. The taxonomic significance of fermentative versus oxidative metabolism of carbohydrates by various gram-negative bacteria. J. Bact., 66, 24-26.U.K.
6.Janse, J. D., 1991. Infra- and intra-specific classification of Pseudomonas solanacearum strains using whole cell fatty-acid analysis. Systematic and Applied Microbiology 14; 335-345.U.K.
7.Janse, J. D. and J. Van Vaerenbergh. The interpretation of the EC method for the detection of latent ring rot infections (Corynebacterium sepedonicum) in potato. EPPO Bull., No 17, 1987, pp. 1-10.U.K.
8.Jansing, H. and K. Rudolph, 1998. Physiological capabilities of Clavibacter michiganensis ssp. sepedonicus and development of a semi-selective medium. Journal of Plant Diseases and Protection, 105, 590-601.U.K.
9.Kovacs, N., 1956. Identification of Pseudomonas pyocyanea by the oxidase reaction. Nature, Lond., 178, 703.U.K.
10.Klement Z.; Rudolph, K and D. C. Sands, 1990. Methods in Phytobacteriology. Akadémiai Kiadó, Budapest, 568 pp.U.K.
11.Lelliott, R. A., 1966. The plant pathogenic coryneform bacteria. J. appl. Bact., 29, 114-118.U.K.
12.Lelliott, R. A., E. Billing and A. C. Hayward, 1966. A determinative scheme for the fluorescent plant pathogenic pseudomonads J. appl. Bact., 29, 470-489.U.K.
13.Lelliott, R. A. and P. W., Sellar, 1976. The detection of latent ring rot (Corynebacterium sepedonicum (Spiek. et Kotth.) Skapt. et Burkh.) in potato stocks. EPPO Bull., 6 (2), 101-106.U.K.
14.Li, X. and S.H. de Boer, 1995. Selection of Polymerase Chain Reaction primers from RNA intergenic spacer region for specific detection of Clavibacter michiganensis ssp. sepedonicus. Phytopathology, 85, 837-842.U.K.
15.Mills, D., Russell, B., W. and J., W. Hanus, 1997. Specific detection of Clavibacter michiganensis ssp. sepedonicus by amplification of three unique DNA sequences isolated by subtraction hybridization. Phytopathology, 87, 8, 853-861.U.K.
16.Pastrik, K.-H. and R.A. Rainey. 1999. Identification and differentiation of Clavibacter michiganensis subspecies by polymerase chain reaction-based techniques. J. Phytopathology 147; 687-693.U.K.
17.Pastrik, K.-H., 2000. Detection of Clavibacter michiganensis ssp. sepedonicus in potato tubers by multiplex PCR with coamplification of host DNA. European Journal of Plant Pathology, 106, 155-165.U.K.
18.Ramamurthi, C. S., 1959. Comparative studies on some Gram-positive phytopathogenic bacteria and their relationship to the Corynebacteria. Mem. Cornell agric. Exp. Sta., 366, 52 pp.U.K.
19.Schaad, W., Berthier-Schaad, Y., Sechler, A. and Knorr, D. (1999) Detection of Clavibacter michiganensis subsp. sepedonicus in potato tubers by BIO-PCR and an automated real-time fluorescence detection system. Plant Disease 83; 1095–1100.U.K.
20.Schaad, W. 2001. Laboratory guide for identification of plant pathogenic bacteria. Schaad [Hrsg.]. — 3. ed.; St. Paul, Minnesota:, 373 pp.U.K.
21.Skerman, V. B. D., 1967. A guide to the identification of the genera of bacteria. 2nd ed., William and Wilkins Company, Baltimore.U.K.
22.Smith, N. C.; Hennesy, J; Stead, D.E., 2001. Repetetive sequence-derived PCR profiling using the BOX-A1Ralstonia solanacearum primer for rapid identification of plant pathogen Clavibacter michiganensis ssp. sepedonicus. European Journal of Plant Pathology, 107 (7), 739-748.U.K.
23.Sneath, P. H. A. and V. G. Collins, 1974. A study in test reproductibility between laboratories: report of Pseudomonas working party. Antonie van Leeuwenhoek, 40, 481-527.U.K.
24.Stead, D.E. 1992. Grouping of plant pathogenic and some other Pseudomonas spp. using cellular fatty-acid profiles. International Journal of Systematic Bacteriology 42; 281-295.U.K.
25.Wullings, B. A.; van Beuningen, A. R.; Janse, J. D. and A. D. L. Akkermans, 1998. Detection of Ralstonia solanacearum, which causes brown rot of potato, by fluorescent in situ hybridization with 23s rRNA-targeted probes. Appl. Environ. Microbiol. 64, 4546–4554.U.K.
(1)
Commercially available solutions and staining kits can also be used.
Options/Help
Print Options
PrintThe Whole Directive
PrintThis Annex only
You have chosen to open the Whole Directive
The Whole Directive you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
Y Rhestrau you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Mae deddfwriaeth ar gael mewn fersiynau gwahanol:
Y Diweddaraf sydd Ar Gael (diwygiedig):Y fersiwn ddiweddaraf sydd ar gael o’r ddeddfwriaeth yn cynnwys newidiadau a wnaed gan ddeddfwriaeth ddilynol ac wedi eu gweithredu gan ein tîm golygyddol. Gellir gweld y newidiadau nad ydym wedi eu gweithredu i’r testun eto yn yr ardal ‘Newidiadau i Ddeddfwriaeth’.
Gwreiddiol (Fel y’i mabwysiadwyd gan yr UE): Mae'r wreiddiol version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Pwynt Penodol mewn Amser: This becomes available after navigating to view revised legislation as it stood at a certain point in time via Advanced Features > Show Timeline of Changes or via a point in time advanced search.
Gweler y wybodaeth ychwanegol ochr yn ochr â’r cynnwys
Rhychwant ddaearyddol: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Dangos Llinell Amser Newidiadau: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Dewisiadau Agor
Dewisiadau gwahanol i agor deddfwriaeth er mwyn gweld rhagor o gynnwys ar y sgrin ar yr un pryd
Rhagor o Adnoddau
Gallwch wneud defnydd o ddogfennau atodol hanfodol a gwybodaeth ar gyfer yr eitem ddeddfwriaeth o’r tab hwn. Yn ddibynnol ar yr eitem ddeddfwriaeth sydd i’w gweld, gallai hyn gynnwys:
- y PDF print gwreiddiol y fel adopted version that was used for the EU Official Journal
- rhestr o newidiadau a wnaed gan a/neu yn effeithio ar yr eitem hon o ddeddfwriaeth
- pob fformat o’r holl ddogfennau cysylltiedig
- slipiau cywiro
- dolenni i ddeddfwriaeth gysylltiedig ac adnoddau gwybodaeth eraill
Llinell Amser Newidiadau
Mae’r llinell amser yma yn dangos y fersiynau gwahanol a gymerwyd o EUR-Lex yn ogystal ag unrhyw fersiynau dilynol a grëwyd ar ôl y diwrnod ymadael o ganlyniad i newidiadau a wnaed gan ddeddfwriaeth y Deyrnas Unedig.
Cymerir dyddiadau fersiynau’r UE o ddyddiadau’r dogfennau ar EUR-Lex ac efallai na fyddant yn cyfateb â’r adeg pan ddaeth y newidiadau i rym ar gyfer y ddogfen.
Ar gyfer unrhyw fersiynau a grëwyd ar ôl y diwrnod ymadael o ganlyniad i newidiadau a wnaed gan ddeddfwriaeth y Deyrnas Unedig, bydd y dyddiad yn cyd-fynd â’r dyddiad cynharaf y daeth y newid (e.e. ychwanegiad, diddymiad neu gyfnewidiad) a weithredwyd i rym. Am ragor o wybodaeth gweler ein canllaw i ddeddfwriaeth ddiwygiedig ar Ddeall Deddfwriaeth.
Rhagor o Adnoddau
Defnyddiwch y ddewislen hon i agor dogfennau hanfodol sy’n cyd-fynd â’r ddeddfwriaeth a gwybodaeth am yr eitem hon o ddeddfwriaeth. Gan ddibynnu ar yr eitem o ddeddfwriaeth sy’n cael ei gweld gall hyn gynnwys:
- y PDF print gwreiddiol y fel adopted fersiwn a ddefnyddiwyd am y copi print
- slipiau cywiro
liciwch ‘Gweld Mwy’ neu ddewis ‘Rhagor o Adnoddau’ am wybodaeth ychwanegol gan gynnwys
- rhestr o newidiadau a wnaed gan a/neu yn effeithio ar yr eitem hon o ddeddfwriaeth
- manylion rhoi grym a newid cyffredinol
- pob fformat o’r holl ddogfennau cysylltiedig
- dolenni i ddeddfwriaeth gysylltiedig ac adnoddau gwybodaeth eraill
 Mae’r holl gynnwys ar gael dan Drwydded Llywodraeth Agored v3.0 ac eithrio ble nodir yn wahanol. Yn ychwanegol mae’r safle hwn â chynnwys sy’n deillio o EUR-Lex, a ailddefnyddiwyd dan delerau Penderfyniad y Comisiwn 2011/833/EU ar ailddefnyddio dogfennau o sefydliadau’r UE. Am ragor o wybodaeth gweler ddatganiad cyhoeddus Swyddfa Gyhoeddiadau’r UE ar ailddefnyddio.
Mae’r holl gynnwys ar gael dan Drwydded Llywodraeth Agored v3.0 ac eithrio ble nodir yn wahanol. Yn ychwanegol mae’r safle hwn â chynnwys sy’n deillio o EUR-Lex, a ailddefnyddiwyd dan delerau Penderfyniad y Comisiwn 2011/833/EU ar ailddefnyddio dogfennau o sefydliadau’r UE. Am ragor o wybodaeth gweler ddatganiad cyhoeddus Swyddfa Gyhoeddiadau’r UE ar ailddefnyddio.