
Statutory Instruments
2013 No. 2033
Medicines
The Veterinary Medicines Regulations 2013
Made
6th August 2013
Laid before Parliament
20th August 2013
Coming into force
1st October 2013
The Secretary of State is a Minister designated(1) for the purposes of making Regulations under section 2(2) of the European Communities Act 1972(2) in relation to measures in the veterinary and phytosanitary fields for the protection of public health.
The Secretary of State has carried out the consultation required by Article 9 of Regulation (EC) No 178/2002 of the European Parliament and of the Council laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety(3).
In accordance with section 56(1) of the Finance Act 1973(4), the Treasury consent to the making of these Regulations.
The Secretary of State makes these Regulations in exercise of the powers conferred by section 2(2) of the European Communities Act 1972 and by section 56(1) of the Finance Act 1973.
PART 1U.K.Introduction
Title and commencementU.K.
1. These Regulations may be cited as the Veterinary Medicines Regulations 2013 and come into force on 1st October 2013.
Definition of “veterinary medicinal product”, interpretation and scopeE+W+S
2.—(1) In these Regulations “veterinary medicinal product” means—
(a)any substance or combination of substances presented as having properties for treating or preventing disease in animals; or
(b)any substance or combination of substances that may be used in, or administered to, animals with a view either to restoring, correcting or modifying physiological functions by exerting a pharmacological, immunological or metabolic action, or to making a medical diagnosis.
(2) In these Regulations—
“adverse reaction” means a reaction to a veterinary medicinal product that is harmful and unintended and that occurs at doses normally used in animals for the prophylaxis, diagnosis or treatment of disease or to restore, correct or modify a physiological function;
F1...
“animal” means all animals other than man and includes birds, reptiles, fish, molluscs, crustacea and bees;
“the cascade” has the meaning given in paragraph 1 of Schedule 4;
[F2“Regulation (EC) No 178/2002” means Regulation (EC) No 178/2002 of the European Parliament and of the Council laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety(6);]
[F3“Regulation (EC) No 1831/2003” means Regulation (EC) No 1831/2003 of the European Parliament and of the Council on additives for use in animal nutrition(7);]
[F4“Regulation (EU) 2017/625” means Regulation (EU) 2017/625 of the European Parliament and of the Council on official controls and other official activities performed to ensure the application of food and feed law, rules on animal health and welfare, plant health and plant protection products;]
F5...
[F6“Regulation (EC) No 183/2005” means Regulation (EC) No 183/2005 of the European Parliament and of the Council laying down requirements for feed hygiene(9);]
“Commission Regulation (EC) No 1234/2008” means Commission Regulation (EC) No 1234/2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products(6) [F7, as last amended by Regulation (EU) No 712/2012](5);
F1...
“extension variation” has the same meaning as “Extension of a marketing authorisation” in Article 2 of Commission Regulation EC No 1234/2008;
“horse passport” means [F8a passport issued in accordance with the provisions of Commission Regulation (EC) No 504/2008 implementing Council Directives 90/426/EEC and 90/427/EEC as regards] [F8an identification document which complies with Commission Implementing Regulation (EU) 2015/262 laying down rules pursuant to Council Directives 90/427/EEC and 2009/156/EC as regards the] methods for the identification of equidae(8);
“immunological veterinary medicinal product” means a veterinary medicinal product administered to animals in order to produce active or passive immunity or to diagnose the state of immunity;
“Regulation (EC) No 470/2009 of the European Parliament and of the Council” means Regulation (EC) No 470/2009 of the European Parliament and of the Council laying down Community procedures for the establishment of residue limits of pharmacologically active substances in foodstuffs of animal origin(9);
“Regulation (EC) No 767/2009 of the European Parliament and of the Council” means Regulation (EC) No 767/2009 of the European Parliament and of the Council on the placing on the market and use of feed in relation to feedingstuffs containing specified feed additives(10) [F9, as last amended by Regulation (EU) No 2017/2279];
“risk-benefit balance” means an evaluation of the positive therapeutic effects of the veterinary medicinal product in relation to—
any risk to human or animal health relating to the quality, safety or efficacy of the veterinary medicinal product; or
any risk of undesirable effects on the environment;
“strength” means the amount of active substances in a dosage unit or unit of volume or weight.
(3) In these Regulations references to types of variation are to those specified in Commission Regulation (EC) No 1234/2008;
F10(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(5) For the avoidance of doubt, these Regulations apply to veterinary medicinal products irrespective of whether or not there is other legislation controlling a product.
Extent Information
E1This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F1Words in reg. 2(2) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(2)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F2Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(b)
F3Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(c)
F4Words in reg. 2(2) inserted (E.) (14.12.2019) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) Regulations 2019 (S.I. 2019/1488), regs. 1(1), 27(a)(ii); and words inserted (W.) (31.1.2020) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) (Wales) Regulations 2020 (S.I. 2020/44), regs. 1(2), 24(1)(a)(ii); and words inserted (S.N.I.) (31.12.2020) by The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(2)(b)
F5Words in reg. 2(2) omitted (E.) (14.12.2019) by virtue of The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) Regulations 2019 (S.I. 2019/1488), regs. 1(1), 27(a)(i); and words omitted (W.) (31.1.2020) by virtue of The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) (Wales) Regulations 2020 (S.I. 2020/44), regs. 1(2), 24(1)(a)(i); and words omitted (S.N.I.) (31.12.2020) by virtue of The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(2)(a)
F6Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(e)
F7Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(a)
F8Words in reg. 2 substituted (E.) (1.10.2018) by The Equine Identification (England) Regulations 2018 (S.I. 2018/761), regs. 1(1), 49
F9Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(f)
F10Reg. 2(4) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(2)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Definition of “veterinary medicinal product”, interpretation and scopeN.I.
2.—(1) In these Regulations “veterinary medicinal product” means—
(a)any substance or combination of substances presented as having properties for treating or preventing disease in animals; or
(b)any substance or combination of substances that may be used in, or administered to, animals with a view either to restoring, correcting or modifying physiological functions by exerting a pharmacological, immunological or metabolic action, or to making a medical diagnosis.
(2) In these Regulations—
“adverse reaction” means a reaction to a veterinary medicinal product that is harmful and unintended and that occurs at doses normally used in animals for the prophylaxis, diagnosis or treatment of disease or to restore, correct or modify a physiological function;
“the Agency” means the European Medicines Agency established by Regulation (EC) No 726/2004 of the European Parliament and of the Council laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency(5);
“animal” means all animals other than man and includes birds, reptiles, fish, molluscs, crustacea and bees;
“the cascade” has the meaning given in paragraph 1 of Schedule 4;
“Commission Regulation (EC) No 1234/2008” means Commission Regulation (EC) No 1234/2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products(6) [F166, as last amended by Regulation (EU) No 712/2012](5);
“Commission Regulation (EU) No 37/2010” means Commission Regulation (EU) No 37/2010 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin(7);
“extension variation” has the same meaning as “Extension of a marketing authorisation” in Article 2 of Commission Regulation EC No 1234/2008;
“horse passport” means a passport issued in accordance with the provisions of Commission Regulation (EC) No 504/2008 implementing Council Directives 90/426/EEC and 90/427/EEC as regards methods for the identification of equidae(8);
“immunological veterinary medicinal product” means a veterinary medicinal product administered to animals in order to produce active or passive immunity or to diagnose the state of immunity;
[F167“Regulation (EC) No 178/2002” means Regulation (EC) No 178/2002 of the European Parliament and of the Council laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety(6);]
[F168“Regulation (EC) No 1831/2003” means Regulation (EC) No 1831/2003 of the European Parliament and of the Council on additives for use in animal nutrition(7);]
F169...
[F170“Regulation (EC) No 183/2005” means Regulation (EC) No 183/2005 of the European Parliament and of the Council laying down requirements for feed hygiene(9);]
“Regulation (EC) No 470/2009 of the European Parliament and of the Council” means Regulation (EC) No 470/2009 of the European Parliament and of the Council laying down Community procedures for the establishment of residue limits of pharmacologically active substances in foodstuffs of animal origin(9);
“Regulation (EC) No 767/2009 of the European Parliament and of the Council” means Regulation (EC) No 767/2009 of the European Parliament and of the Council on the placing on the market and use of feed in relation to feedingstuffs containing specified feed additives(10) [F171, as last amended by Regulation (EU) No 2017/2279];
[F172“Regulation (EU) 2017/625” means Regulation (EU) 2017/625 of the European Parliament and of the Council on official controls and other official activities performed to ensure the application of food and feed law, rules on animal health and welfare, plant health and plant protection products;]
“risk-benefit balance” means an evaluation of the positive therapeutic effects of the veterinary medicinal product in relation to—
any risk to human or animal health relating to the quality, safety or efficacy of the veterinary medicinal product; or
any risk of undesirable effects on the environment;
“strength” means the amount of active substances in a dosage unit or unit of volume or weight.
(3) In these Regulations references to types of variation are to those specified in Commission Regulation (EC) No 1234/2008;
(4) In these Regulations any reference to a member State is a reference to a member State of the European Union and Norway, Iceland and Liechtenstein.
(5) For the avoidance of doubt, these Regulations apply to veterinary medicinal products irrespective of whether or not there is other legislation controlling a product.
Extent Information
E72This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F166Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(a)
F167Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(b)
F168Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(c)
F169Words in reg. 2(2) omitted (S.N.I.) (31.12.2020) by virtue of The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(2)(a)
F170Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(e)
F171Words in reg. 2(2) inserted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(2)(f)
F172Words in reg. 2(2) inserted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(2); and also inserted (S.N.I.) (31.12.2020) by The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(2)(b)
Products to which these Regulations do not applyU.K.
3.—(1) These Regulations do not apply to a veterinary medicinal product based on radio-active isotopes.
(2) They do not apply in relation to a product intended for administration in the course of a procedure licensed under the Animals (Scientific Procedures) Act 1986(11), except that, if the animals are to be put into the human food chain, the only products that may be administered to the animals are—
(a)authorised veterinary medicinal products administered in accordance with their marketing authorisation; or
(b)products administered in accordance with an animal test certificate granted under paragraph 9 of Schedule 4.
PART 2U.K.Authorised veterinary medicinal products
Placing a veterinary medicinal product on the marketE+W+S
4.—[F11(1) No person may place a veterinary medicinal product on the market unless the Secretary of State has—
(a)as regards a product to which Schedule 1B applies, issued a QNIG certificate in respect of that product;
(b)otherwise, granted a marketing authorisation in respect of that product.]
(2) No person may certify data in relation to an application for a marketing authorisation or in relation to an existing marketing authorisation if they know that those data are false, or do not believe that they are accurate.
(3) Schedule 1 (marketing authorisations) has effect.
[F12(4) Schedule 1A (converted EU marketing authorisations) has effect.]
[F13(5) Schedule 1B (Northern Ireland qualifying good marketing authorisations) has effect. ]
Extent Information
E2This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F11Reg. 4(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1, 3(3) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(a)); 2020 c. 1, Sch. 5 para. 1(1)
F12Reg. 4(4) inserted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(2); 2020 c. 1, Sch. 5 para. 1(1)
F13Reg. 4(5) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(2)
Placing a veterinary medicinal product on the marketN.I.
4.—(1) No person may place a veterinary medicinal product on the market unless that product has been granted a marketing authorisation by the Secretary of State or the Agency.
(2) No person may certify data in relation to an application for a marketing authorisation or in relation to an existing marketing authorisation if they know that those data are false, or do not believe that they are accurate.
(3) Schedule 1 (marketing authorisations) has effect.
Extent Information
E73This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Manufacture of veterinary medicinal productsU.K.
5.—(1) The holder of a marketing authorisation must ensure that every stage in the manufacture of the veterinary medicinal product is carried out by the manufacturer specified in the marketing authorisation(12).
(2) Schedule 2 (the manufacture of veterinary medicinal products) has effect.
(3) “Manufacture” includes any part of the manufacture of a veterinary medicinal product until the finished product is ready for sale in its final form as specified in the marketing authorisation but does not include the manufacture of an ingredient or breaking open the package of a veterinary medicinal product(13).
Marketing of products not in accordance with a marketing authorisationU.K.
6. The holder of a marketing authorisation for a veterinary medicinal product is guilty of an offence if either the holder or the manufacturer supplies a product that is not completely in accordance with the marketing authorisation.
Classification, supply and possession of the productU.K.
7.—(1) Schedule 3 (classification and supply, wholesale dealers and sheep dip) has effect.
(2) No person may supply a veterinary medicinal product that has passed its expiry date.
(3) No person may open the package (including the outer package) of a veterinary medicinal product before it has been supplied to the final user, other than as permitted under Schedule 3.
(4) No person may supply an authorised human medicinal product for administration to an animal (other than a product supplied by a veterinary surgeon or in accordance with a written prescription from a veterinary surgeon that includes all the information specified in paragraph 6 of Schedule 3).
(5) No person may be in possession of a veterinary medicinal product that was supplied to that person other than in accordance with Schedule 3.
Administration of the productE+W+S
8. No person may administer a veterinary medicinal product to an animal unless—
(a)the product has a marketing authorisation authorising its administration in the United Kingdom, and the administration is in accordance with that marketing authorisation; or
(b)it is administered in accordance with Schedule 4 (administration of a veterinary medicinal product outside the terms of a marketing authorisation) or Schedule 6 (exemptions for small pet animals).
Extent Information
E3This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Administration of the productN.I.
8. No person may administer a veterinary medicinal product to an animal unless—
(a)the product has a marketing authorisation authorising its administration in [F173Northern Ireland], and the administration is in accordance with that marketing authorisation; or
(b)it is administered in accordance with Schedule 4 (administration of a veterinary medicinal product outside the terms of a marketing authorisation) or Schedule 6 (exemptions for small pet animals).
Extent Information
E74This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F173Words in reg. 8(a) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(3)
Importation of authorised veterinary medicinal productsE+W+S
9.—[F14(1) No person may import, or move into Great Britain from Northern Ireland, a veterinary medicinal product authorised for use in Great Britain except in accordance with this regulation.]
(2) A holder of a marketing authorisation for a veterinary medicinal product may import that veterinary medicinal product.
(3) A holder of a manufacturing authorisation may import a veterinary medicinal product to which that authorisation relates.
(4) An authorised wholesale dealer may import a veterinary medicinal product if—
(a)the authorisation covers the product; and
(b)the dealer has notified the holder of the marketing authorisation in writing before importation.
(5) A veterinary surgeon or a pharmacist may import any authorised veterinary medicinal product.
(6) A suitably qualified person (registered in accordance with paragraph 14 of Schedule 3) may import any authorised veterinary medicinal product that that person is permitted to supply.
(7) There are no restrictions on the importation of an authorised veterinary medicinal product in category AVM-GSL.
Extent Information
E4This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F14Reg. 9(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(3)
Importation of authorised veterinary medicinal productsN.I.
9.—(1) No person may import a veterinary medicinal product authorised for use in [F174Northern Ireland] except in accordance with this regulation.
(2) A holder of a marketing authorisation for a veterinary medicinal product may import that veterinary medicinal product.
(3) A holder of a manufacturing authorisation may import a veterinary medicinal product to which that authorisation relates.
(4) An authorised wholesale dealer may import a veterinary medicinal product if—
(a)the authorisation covers the product; and
(b)the dealer has notified the holder of the marketing authorisation in writing before importation.
(5) A veterinary surgeon or a pharmacist may import any authorised veterinary medicinal product.
(6) A suitably qualified person (registered in accordance with paragraph 14 of Schedule 3) may import any authorised veterinary medicinal product that that person is permitted to supply.
(7) There are no restrictions on the importation of an authorised veterinary medicinal product in category AVM-GSL.
Extent Information
E75This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F174Words in reg. 9(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(4)
Advertising the productU.K.
10.—(1) No person may advertise a veterinary medicinal product if the advertisement is misleading or contains any medicinal claim that is not in the summary of product characteristics.
(2) No person may advertise an authorised human medicinal product for administration to animals (including sending a price list of, or including, authorised human medicinal products to a veterinary surgeon or veterinary practice).
(3) Paragraph (2) does not apply to the holder of a wholesale dealer’s authorisation who supplies a list of authorised human medicinal products, together with prices, to a veterinary surgeon for use under the cascade provided that—
(a)the list is sent following a request from the veterinary surgeon to whom it is sent; and
(b)the list states clearly that the product does not have a marketing authorisation as a veterinary medicinal product, and may only be prescribed and administered under the cascade.
Advertising of prescription products and products containing psychotropic drugs or narcoticsU.K.
11.—(1) No person may advertise a veterinary medicinal product that—
(a)is available on veterinary prescription only; or
(b)contains psychotropic drugs or narcotics.
(2) In the case of a product containing psychotropic drugs or narcotics, paragraph (1) does not apply to advertisements aimed at veterinary surgeons or pharmacists.
(3) Subject to paragraph (4) in the case of POM-V medicines, paragraph (1) does not apply to price lists, or to advertisements aimed at—
(a)veterinary surgeons;
(b)veterinary nurses;
(c)pharmacists; or
(d)professional keepers of animals.
(4) No person may advertise anti-microbials to professional keepers of animals.
(5) In the case of POM-VPS medicines, paragraph (1) does not apply to price lists, or to advertisements aimed at—
(a)veterinary surgeons;
(b)pharmacists;
(c)suitably qualified persons registered in accordance with paragraph 14 of Schedule 3;
(d)other veterinary health care professionals; or
(e)professional keepers of animals.
Defence of publication in the course of businessU.K.
12. In proceedings for an offence under these Regulation 43(g), it is a defence for the person charged to prove—
(a)that that person’s business is to publish or arrange for the publication of advertisements, and
(b)that the advertisement was received in the ordinary course of business and the person charged did not know and had no reason to suspect that its publication would amount to an offence under these Regulations.
Wholesale dealingU.K.
13. No person may buy a veterinary medicinal product, other than by retail or for the purposes of retail supply in accordance with Schedule 3, unless the buyer has a wholesale dealer’s authorisation granted by the Secretary of State under this regulation and Schedule 3.
FeedingstuffsU.K.
14. Schedule 5 (medicated feedingstuffs and specified feed additives) has effect.
ExemptionsU.K.
15.—(1) These Regulations do not apply to an inactivated autogenous vaccine that is manufactured, on the instructions of a veterinary surgeon, from pathogens or antigens obtained from an animal and used for the treatment of that animal.
(2) Schedule 1 and Part 1 of Schedule 2 do not apply in relation to an inactivated autogenous vaccine that is—
(a)manufactured by a person and in premises authorised in accordance with Part 2 of Schedule 2, on the instructions of a veterinary surgeon, from pathogens or antigens obtained from an animal; and
(b)used for the treatment of—
(i)other animals on the same site;
(ii)animals intended to be sent to those premises; or
(iii)animals on a site that receives animals from those premises.
(3) Schedule 1 and Part 1 of Schedule 2 do not apply in relation to—
(a)blood or blood constituents from a blood bank authorised in accordance with Part 3 of Schedule 2;
(b)a product manufactured for administration under the cascade by a person and in premises authorised in accordance with Part 4 of Schedule 2; or
(c)equine stem cell products for use as an autologous treatment for horses from an equine collection centre authorised in accordance with Part 5 of Schedule 2.
(4) Schedule 6 (exemptions for small pet animals) has effect.
FeesU.K.
16. Schedule 7 (fees) has effect.
PART 3U.K.Records
Food-producing animals: proof of purchase of veterinary medicinal productsU.K.
17. The keeper of a food-producing animal must keep proof of purchase of all veterinary medicinal products acquired for the animal (or, if they were not bought, documentary evidence of how they were acquired).
Food-producing animals: records of administration by a veterinary surgeonU.K.
18. A veterinary surgeon who administers a veterinary medicinal product to a food-producing animal must either enter the following information personally in the keeper’s records or give it to the keeper in writing (in which case the keeper must enter the following into those records)—
(a)the name of the veterinary surgeon;
(b)the name of the product and the batch number;
(c)the date of administration of the product;
(d)the amount of product administered;
(e)the identification of the animals treated; and
(f)the withdrawal period.
Food-producing animals: records of acquisition and administrationU.K.
19.—(1) When a veterinary medicinal product is bought or otherwise acquired for a food-producing animal the keeper must, at the time, record—
(a)the name of the product and the batch number;
(b)the date of acquisition;
(c)the quantity acquired; and
(d)the name and address of the supplier.
(2) At the time of administration (unless the administration is by a veterinary surgeon in which case the record must be in accordance with regulation 18) the keeper must record—
(a)the name of the product;
(b)the date of administration;
(c)the quantity administered;
(d)the withdrawal period; and
(e)the identification of the animals treated.
(3) A keeper who disposes of any or all of the veterinary medicinal product other than by treating an animal must record—
(a)the date of disposal;
(b)the quantity of product involved; and
(c)how and where it was disposed of.
Food-producing animals: retention of recordsU.K.
20. The keeper of a food-producing animal must keep the documentation on the acquisition of a veterinary medicinal product and the records relating to the product for at least five years following the administration or other disposal of the product, irrespective of whether or not the animal concerned is no longer in that keeper’s possession or has been slaughtered or has died during that period.
Records by a holder of a manufacturing authorisationU.K.
21.—(1) A holder of a manufacturing authorisation must, as soon as is reasonably practicable, make a record of each batch of veterinary medicinal product manufactured, assembled or supplied, which must include—
(a)the name of the product;
(b)the quantity manufactured, assembled or supplied;
(c)the date of manufacture, assembly or supply;
(d)the batch number and expiry date; and
(e)in the case of supply, the name and address of the recipient.
(2) The holder must keep with the record all certification provided by the qualified person (manufacture) in relation to that batch.
(3) The holder must keep all records and certificates for at least five years from the date the veterinary medicinal product is placed on the market.
Records by a holder of a wholesale dealer’s authorisationU.K.
22. A holder of a wholesale dealer’s authorisation must record the following as soon as is reasonably practicable after each incoming or outgoing transaction (including disposal) relating to a veterinary medicinal product—
(a)the date and nature of the transaction,
(b)the name of the veterinary medicinal product,
(c)the manufacturer’s batch number,
(d)the expiry date,
(e)the quantity, and
(f)the name and address of the supplier or recipient,
and must keep the records for at least three years.
Records of the receipt or supply of prescription productsU.K.
23.—(1) Any person permitted under these Regulations to supply a veterinary medicinal product classified as POM-V or POM-VPS who receives or supplies any such veterinary medicinal product must keep all documents relating to the transaction that show—
(a)the date;
(b)the name of the veterinary medicinal product;
(c)the batch number (except that, in the case of a product for a non-food-producing animal, this need only be recorded either on the date of receipt of the batch or the date a veterinary medicinal product from the batch is first supplied);
(d)the quantity;
(e)the name and address of the supplier or recipient; and
(f)if there is a written prescription, the name and address of the person who wrote the prescription and a copy of the prescription.
(2) If the documents do not include this information that person must make a record of the missing information as soon as is reasonably practicable following the transaction.
(3) As an alternative to paragraphs (1) and (2) that person may make a record of all the information required there provided that this is done as soon as is reasonably practicable following the transaction.
(4) The documentation and records must be kept for at least five years.
Records of products administered to a food-producing animal under the cascadeU.K.
24. A veterinary surgeon administering a veterinary medicinal product to food-producing animals under the cascade, or permitting another person to administer it under that veterinary surgeon’s responsibility, must, as soon as is reasonably practicable, record—
(a)the date of examination of the animals;
(b)the name and address of the owner;
(c)the identification and number of animals treated;
(d)the result of the veterinary surgeon’s clinical assessment;
(e)the trade name of the product if there is one;
(f)the manufacturer’s batch number shown on the product if there is one;
(g)the name and quantity of the active substances;
(h)the doses administered or supplied;
(i)the duration of treatment; and
(j)the withdrawal period,
and must keep the record for at least five years.
PART 4U.K.Unauthorised veterinary medicinal products
Importation of an unauthorised veterinary medicinal productE+W+S
25.—(1) No person may import or be concerned in the importation of an unauthorised veterinary medicinal product except in accordance with this regulation.
(2) A holder of a marketing authorisation may import an unauthorised veterinary medicinal product if it is for the purpose of the manufacture of a veterinary medicinal product for which the importer holds the marketing authorisation.
(3) A holder of a manufacturing authorisation may import an unauthorised veterinary medicinal product if it is for the manufacture of a veterinary medicinal product that the importer is permitted to manufacture.
(4) A holder of a wholesale dealer’s authorisation may import an unauthorised veterinary medicinal product for the purposes of re-export.
(5) A veterinary surgeon may import an unauthorised veterinary medicinal product that is authorised in another [F15country] if it is for the purpose of administration by that veterinary surgeon or under that veterinary surgeon’s responsibility under the cascade or administration in exceptional circumstances in accordance with Schedule 4; the import must be in accordance with the appropriate certificate granted by the Secretary of State, and the product may be imported by the veterinary surgeon personally or by using a wholesale dealer or pharmacist as an agent.
(6) A wholesale dealer or a pharmacist may import an unauthorised veterinary medicinal product for the purpose of storing it pending administration by a veterinary surgeon under the cascade or administration in exceptional circumstances in accordance with Schedule 4 if—
(a)the veterinary medicinal product is authorised in another F16... country;
(b)the Secretary of State has issued a certificate certifying that—
(i)the disease or condition is such that the veterinary medicinal product is likely to be needed as a matter of urgency for the treatment of an animal;
(ii)delay in administering the product will seriously affect the health or welfare of the animal; and
(iii)there is no suitable veterinary medicinal product authorised in [F17Great Britain]; and
(c)in the case of a wholesale dealer, the product is within the terms of the authorisation.
(7) The holder of an animal test certificate granted under paragraph 9 of Schedule 4 may import anything specified in the animal test certificate in accordance with the conditions in that certificate.
(8) The Secretary of State may authorise in writing the importation of any product or substance for use under a licence granted under the Animals (Scientific Procedures) Act 1986.
[F18(9) For the purposes of this regulation, references to the import or importation of an unauthorised veterinary medical product include the movement of such a product into Great Britain from Northern Ireland.]
Extent Information
E5This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F15Word in reg. 25(5) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(4)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F16Words in reg. 25(6)(a) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(4)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F17Words in reg. 25(6)(b)(iii) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(4)(a)
F18Reg. 25(9) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(4)(b)
Importation of an unauthorised veterinary medicinal productN.I.
25.—(1) No person may import or be concerned in the importation of an unauthorised veterinary medicinal product except in accordance with this regulation.
(2) A holder of a marketing authorisation may import an unauthorised veterinary medicinal product if it is for the purpose of the manufacture of a veterinary medicinal product for which the importer holds the marketing authorisation.
(3) A holder of a manufacturing authorisation may import an unauthorised veterinary medicinal product if it is for the manufacture of a veterinary medicinal product that the importer is permitted to manufacture.
(4) A holder of a wholesale dealer’s authorisation may import an unauthorised veterinary medicinal product for the purposes of re-export.
(5) A veterinary surgeon may import an unauthorised veterinary medicinal product that is authorised in [F175a] member State if it is for the purpose of administration by that veterinary surgeon or under that veterinary surgeon’s responsibility under the cascade or administration in exceptional circumstances in accordance with Schedule 4; the import must be in accordance with the appropriate certificate granted by the Secretary of State, and the product may be imported by the veterinary surgeon personally or by using a wholesale dealer or pharmacist as an agent.
(6) A wholesale dealer or a pharmacist may import an unauthorised veterinary medicinal product for the purpose of storing it pending administration by a veterinary surgeon under the cascade or administration in exceptional circumstances in accordance with Schedule 4 if—
(a)the veterinary medicinal product is authorised in [F176a] member State or a third country;
(b)the Secretary of State has issued a certificate certifying that—
(i)the disease or condition is such that the veterinary medicinal product is likely to be needed as a matter of urgency for the treatment of an animal;
(ii)delay in administering the product will seriously affect the health or welfare of the animal; and
(iii)there is no suitable veterinary medicinal product authorised in [F177Northern Ireland]; and
(c)in the case of a wholesale dealer, the product is within the terms of the authorisation.
(7) The holder of an animal test certificate granted under paragraph 9 of Schedule 4 may import anything specified in the animal test certificate in accordance with the conditions in that certificate.
(8) The Secretary of State may authorise in writing the importation of any product or substance for use under a licence granted under the Animals (Scientific Procedures) Act 1986.
[F178(9) For the purpose of this regulation, references to import or importation of an unauthorised veterinary medicinal product include the movement of such a product from Great Britain to Northern Ireland.]
Extent Information
E76This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F175Word in reg. 25(5) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(5)(a)
F176Word in reg. 25(6)(a) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(5)(a)
F177Words in reg. 25(6)(b)(iii) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(5)(b)
F178Reg. 25(9) inserted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(5)(c)
Possession of an unauthorised veterinary medicinal productE+W+S
26.—(1) No person may be in possession of an unauthorised veterinary medicinal product.
(2) No person may be in possession of an unauthorised veterinary medicinal product with the intention of supplying that product to another person.
(3) This regulation does not apply to—
(a)a veterinary medicinal product imported in accordance with a certificate granted by the Secretary of State under these Regulations;
(b)a product prescribed by a veterinary surgeon under the cascade;
(c)a holder of a manufacturing authorisation if the possession is for export;
(d)a holder of a wholesale dealer’s authorisation if the possession is for export or re-export; or
(e)a holder of a manufacturer’s authorisation or marketing authorisation if the intention is to manufacture a veterinary medicinal product.
(4) A veterinary surgeon who practises in both [F19Great Britain] and another [F20country] may hold veterinary medicinal products authorised in the other [F20country] provided that the amount held does not exceed the amount expected to be used in that [F20country].
(5) It is a defence for a person charged with failing to comply with paragraph (1) to prove that the product was for the purposes of research or development of a veterinary medicinal product.
(6) A veterinary surgeon may have possession of an authorised human medicinal product intended for administration to animals under the cascade, provided that the amount held does not exceed the amount expected to be used under the cascade.
Extent Information
E6This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F19Words in reg. 26(4) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(5)
F20Word in reg. 26(4) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(5) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Possession of an unauthorised veterinary medicinal productN.I.
26.—(1) No person may be in possession of an unauthorised veterinary medicinal product.
(2) No person may be in possession of an unauthorised veterinary medicinal product with the intention of supplying that product to another person.
(3) This regulation does not apply to—
(a)a veterinary medicinal product imported in accordance with a certificate granted by the Secretary of State under these Regulations;
(b)a product prescribed by a veterinary surgeon under the cascade;
(c)a holder of a manufacturing authorisation if the possession is for export;
(d)a holder of a wholesale dealer’s authorisation if the possession is for export or re-export; or
(e)a holder of a manufacturer’s authorisation or marketing authorisation if the intention is to manufacture a veterinary medicinal product.
[F179(4) A veterinary surgeon who practices in Northern Ireland and a member State may hold veterinary medicinal products authorised in the member State provided that the amount held does not exceed the amount expected to be used in that member State.]
(5) It is a defence for a person charged with failing to comply with paragraph (1) to prove that the product was for the purposes of research or development of a veterinary medicinal product.
(6) A veterinary surgeon may have possession of an authorised human medicinal product intended for administration to animals under the cascade, provided that the amount held does not exceed the amount expected to be used under the cascade.
Extent Information
E77This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
Supply of an unauthorised veterinary medicinal productU.K.
27.—(1) No person may supply an unauthorised veterinary medicinal product.
(2) This regulation does not apply to—
(a)a veterinary medicinal product prescribed by a veterinary surgeon under the cascade; or
(b)a product supplied in accordance with a certificate granted by the Secretary of State under these Regulations.
(3) It is a defence for a person charged with failing to comply with paragraph (1) to prove that the supply was for the purposes of research or development of a veterinary medicinal product.
PART 5U.K.Miscellaneous provisions, enforcement and offences
The Veterinary Products CommitteeU.K.
28.—(1) There shall continue to be a Veterinary Products Committee.
(2) The Secretary of State may appoint members of the Committee from professional people who are eminent in their field, and any lay members as the Secretary of State sees fit.
(3) The function of the Committee is to provide scientific advice on any aspect of veterinary medicinal products asked for by the Secretary of State and to carry out any functions specified in these Regulations.
(4) The Secretary of State may pay members of the Committee such amounts as the Secretary of State may decide.
(5) The Secretary of State may consult the Committee at any time.
Veterinary Products Committee appeals procedureU.K.
29.—(1) The following procedure applies when any person receives a notification from the Secretary of State informing that person (the appellant) of a right to an appeal to the Veterinary Products Committee.
(2) The appellant must inform the Secretary of State of an intention to appeal within 28 days of the notification which is the subject of the appeal.
(3) The appeal may be written or oral, or both, at the choice of the appellant.
(4) The appellant may not present to the Committee any new data not available to the Secretary of State at the time of the original decision.
(5) The Committee must consider the appeal and any representations made by the Secretary of State, and report its findings in writing to the Secretary of State together with its recommendations.
(6) The Secretary of State must send a copy of the report to the appellant on request.
(7) The Secretary of State must consider the report and then form a provisional decision.
(8) The Secretary of State must then notify the provisional decision to the appellant, together with the reasons for it.
Appeals to an appointed personU.K.
30.—(1) A person aggrieved by a provisional decision of the Secretary of State under regulation 29 may appeal against the decision to a person appointed for the purpose by the Secretary of State in accordance with this regulation.
(2) So may an applicant for—
(a)a manufacturing authorisation;
(b)appointment as a Qualified Person for the purposes of a manufacturing authorisation;
(c)authorisation for a person or premises to manufacture autogenous vaccines;
(d)an authorisation of a blood bank;
(e)authorisation of a person and premises to manufacture an unauthorised veterinary medicinal product for administration under the cascade;
(f)authorisation of an equine stem cell centre;
(g)a wholesale dealer’s authorisation;
(h)the approval of premises for the supply of POM-VPS or NFA-VPS veterinary medicinal products by a suitably qualified person,
if such an application is refused.
(3) A holder of any of the above authorisations, appointment or approvals may appeal against a suspension or compulsory variation in the same way.
(4) The appointed person must consider the appeal (but may not consider any new data not available to the Secretary of State at the time of the original decision) and any representations made by the Secretary of State and report in writing, with a recommended course of action, to the Secretary of State.
(5) The Secretary of State must then reach a final decision and notify the appellant, together with the reasons for it.
ExportsE+W+S
31.—(1) No person may export a veterinary medicinal product for use in another [F21country] unless the veterinary medicinal product may be lawfully supplied or administered in that [F21country].
(2) If a veterinary medicinal product has been manufactured in accordance with a marketing authorisation, or if a product without a marketing authorisation has been manufactured under a manufacturing authorisation, and the product is intended for export F22..., the Secretary of State must, at the request of the exporter or the competent authorities of the country to which it is being exported, provide a certificate to that effect.
(3) When issuing the certificate the Secretary of State must take account of the model certificates issued by the World Health Organization(14).
(4) If the veterinary medicinal product is authorised in the United Kingdom the Secretary of State must ensure that the exporter or the competent authorities of the [F23importing] country has access to the summary of product characteristics.
Extent Information
E7This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F21Word in reg. 31(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(6)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F22Word in reg. 31(2) omitted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(6)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F23Word in reg. 31(4) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(6)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
ExportsN.I.
31.—(1) No person may export a veterinary medicinal product for use in another [F180country] unless the veterinary medicinal product may be lawfully supplied or administered in that [F180country].
(2) If a veterinary medicinal product has been manufactured in accordance with a marketing authorisation, or if a product without a marketing authorisation has been manufactured under a manufacturing authorisation, and the product is intended for export outside the European Union, the Secretary of State must, at the request of the exporter or the competent authorities of the country to which it is being exported, provide a certificate to that effect.
(3) When issuing the certificate the Secretary of State must take account of the model certificates issued by the World Health Organization(14).
(4) If the veterinary medicinal product is authorised in the United Kingdom the Secretary of State must ensure that the exporter or the competent authorities of the third country has access to the summary of product characteristics.
Extent Information
E78This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F180Word in reg. 31(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(7)
Time limitsU.K.
32.—(1) In any provision in these Regulations requiring the Secretary of State to issue an authorisation within a set time, the clock does not start to run until the Secretary of State has checked that the application dossier is in accordance with these Regulations and has validated the application.
(2) In calculating the period during which the Secretary of State must issue any authorisation requires the clock is stopped when the Secretary of State requires an applicant to provide further data until all the further data required have been provided.
(3) The clock is also stopped during any period that the applicant is given to provide oral or written explanations.
(4) The Secretary of State may stop the clock pending payment of outstanding fees.
Appointment of inspectorsU.K.
33. The Secretary of State must appoint inspectors for the purposes of the enforcement of these Regulations and in these Regulations “inspector” means an inspector appointed under this regulation or a veterinary inspector appointed under the Animal Health Act 1981(15).
Powers of entryE+W+S
34.—(1) An inspector may, on giving reasonable notice, and on producing a duly authenticated authorisation if required, enter any premises at any reasonable hour for the purpose of ensuring that the provisions of these Regulations are being complied with; and in this regulation “premises” includes any place, vehicle, trailer, container, stall, moveable structure, ship or aircraft.
(2) The requirement to give notice does not apply—
(a)where the entry is pursuant to any provision of an [F24enactment] which requires inspection without notice;
(b)where the requirement has been waived;
(c)where reasonable efforts to agree an appointment have failed;
(d)where an inspector has reasonable suspicion of a failure to comply with these Regulations; or
(e)in an emergency.
(3) Paragraph (1) does not apply in relation to any premises which are used wholly or mainly as a private dwelling, unless those premises, or any part of them, are approved, registered or authorised for the sale of veterinary medicines under paragraph 8, 10, 14(4) or 18 of Schedule 3 or for use as a feed business under paragraph 5(2)(e) or 7(2) of Schedule 5.
(4) Paragraphs (1) and (3) do not affect any right of entry conferred by a warrant issued by a justice of the peace.
[F25(5) An inspector may be accompanied by such other persons as the inspector considers necessary.]
(6) If a justice of the peace, on sworn information in writing, is satisfied that there are reasonable grounds for entry into any premises for the purposes of the enforcement of these Regulations, and either—
(a)admission has been refused, or a refusal is expected, and (in either case) that notice to apply for a warrant has been given to the occupier;
(b)asking for admission, or the giving of such a notice, would defeat the object of the entry;
(c)the case is one of urgency; or
(d)the premises are unoccupied or the occupier is temporarily absent,
the justice may by signed warrant authorise an inspector to enter the premises, if need be by reasonable force.
(7) A warrant under this regulation is valid for one month.
(8) An inspector who enters any unoccupied premises must leave them as effectively secured against unauthorised entry as they were before entry.
(9) An inspector may enter the premises of manufacturers of active substances used as starting materials for veterinary medicinal products, and the premises of the marketing authorisation holder.
F26(10) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(11) In this regulation, a reference to a justice of the peace —
(a)in Scotland includes a reference to the sheriff and to a magistrate; and
(b)in Northern Ireland, is a reference to a lay magistrate.
Extent Information
E8This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F24Word in reg. 34(2)(a) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(7)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F25Reg. 34(5) substituted (E.W.S) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(7)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F26Reg. 34(10) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(7)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Powers of entryN.I.
34.—(1) An inspector may, on giving reasonable notice, and on producing a duly authenticated authorisation if required, enter any premises at any reasonable hour for the purpose of ensuring that the provisions of these Regulations are being complied with; and in this regulation “premises” includes any place, vehicle, trailer, container, stall, moveable structure, ship or aircraft.
(2) The requirement to give notice does not apply—
(a)where the entry is pursuant to any provision of an EU instrument which requires inspection without notice;
(b)where the requirement has been waived;
(c)where reasonable efforts to agree an appointment have failed;
(d)where an inspector has reasonable suspicion of a failure to comply with these Regulations; or
(e)in an emergency.
(3) Paragraph (1) does not apply in relation to any premises which are used wholly or mainly as a private dwelling, unless those premises, or any part of them, are approved, registered or authorised for the sale of veterinary medicines under paragraph 8, 10, 14(4) or 18 of Schedule 3 or for use as a feed business under paragraph 5(2)(e) or 7(2) of Schedule 5.
(4) Paragraphs (1) and (3) do not affect any right of entry conferred by a warrant issued by a justice of the peace.
(5) An inspector may be accompanied by—
(a)such other persons as the inspector considers necessary; F181...
F181(b). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(6) If a justice of the peace, on sworn information in writing, is satisfied that there are reasonable grounds for entry into any premises for the purposes of the enforcement of these Regulations, and either—
(a)admission has been refused, or a refusal is expected, and (in either case) that notice to apply for a warrant has been given to the occupier;
(b)asking for admission, or the giving of such a notice, would defeat the object of the entry;
(c)the case is one of urgency; or
(d)the premises are unoccupied or the occupier is temporarily absent,
the justice may by signed warrant authorise an inspector to enter the premises, if need be by reasonable force.
(7) A warrant under this regulation is valid for one month.
(8) An inspector who enters any unoccupied premises must leave them as effectively secured against unauthorised entry as they were before entry.
(9) An inspector may enter the premises of manufacturers of active substances used as starting materials for veterinary medicinal products, and the premises of the marketing authorisation holder.
(10) An inspector may carry out an inspection at the request of [F182a] member State, the European Commission or the Agency.
[F183(11) In this regulation, a reference to justice of the peace is a reference to a lay magistrate.]
Extent Information
E79This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F181Reg. 34(5)(b) and word omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs., 1(3), 10(8(a)
F182Word in reg. 34(10) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(8)(b)
Powers of an inspectorU.K.
35.—(1) An inspector entering premises under the previous regulation may—
(a)inspect the premises, and any plant, machinery or equipment;
(b)search the premises;
(c)take samples;
(d)seize any computers and associated equipment;
(e)seize any veterinary medicinal product or any additive to which Schedule 5 applies, if it is not authorised in the United Kingdom;
(f)seize any premixture or feedingstuff that contains a veterinary medicinal product or additive to which Schedule 5 applies that is not authorised in the United Kingdom;
(g)seize any veterinary medicinal product, any additive to which Schedule 5 applies, any premixture or feedingstuff if—
(i)it has not been lawfully manufactured, incorporated or supplied in accordance with these Regulations;
(ii)it has been stored in a way that affects its safety, quality or efficacy; or
(iii)it is sold or offered for sale by a person not permitted to supply it under these Regulations;
(h)carry out any inquiries, examinations and tests;
(i)have access to, and inspect and copy or seize any documents or records (in whatever form they are held) relating to these Regulations; and
(j)have access to, inspect and check the operation of any computer and any associated apparatus or material that is or has been in use in connection with the records; and for this purpose may require any person having charge of, or otherwise concerned with the operation of, the computer, apparatus or material to afford such assistance as may reasonably be required and, where a record is kept by means of a computer, may require the records to be produced in a form in which they may be taken away.
(2) The powers of seizure under sub-paragraph (1)(e), (f) and (g) include a power to seize anything which purports to be, or which an inspector reasonably believes to be, something the inspector is entitled to seize under these powers.
(3) An officer of any local authority who has entered premises exercising any statutory power of entry for the purposes of enforcing any legislation relating to food hygiene, feed hygiene or animal health, may inspect any records made under these Regulations (in whatever form they are held) relating to food-producing animals, and may remove them to enable them to be copied.
(4) Where an inspector has entered any premises and it is not reasonably practicable to determine at the time whether documents on those premises are relevant to these Regulations, the inspector may seize them to ascertain whether or not they are relevant.
Inspection of pharmaciesU.K.
36. In relation to a pharmacy, all the powers of an inspector to enforce these Regulations may also be exercised by an officer of the General Pharmaceutical Council appointed for the purpose.
ObstructionU.K.
37. No person may—
(a)intentionally obstruct any person acting in the execution of these Regulations;
(b)without reasonable cause, fail to give to any person acting in the execution of these Regulations any assistance or information that that person may reasonably require under these Regulations;
(c)furnish to any person acting in the execution of these Regulations any information knowing it to be false or misleading; or
(d)fail to produce a record when required to do so to any person acting in the execution of these Regulations.
Improvement noticesU.K.
38.—(1) An inspector who has reasonable grounds for believing that any person is failing to comply with these Regulations may serve a notice on that person (in these Regulations referred to as an “improvement notice”) that—
(a)states the inspector’s grounds for believing this;
(b)specifies the matters that constitute the failure to comply;
(c)specifies the measures that, in the inspector’s opinion, the person must take in order to secure compliance; and
(d)requires the person to take those measures, or measures at least equivalent to them, within the period (being not less than 14 days) specified in the notice.
(2) An improvement notice must state—
(a)the right of appeal to a magistrates’ court or to the sheriff; and
(b)the period within which such an appeal may be brought.
Appeals against improvement noticesE+W+S
39.—(1) Any person who is aggrieved by an improvement notice may appeal to a magistrates’ court or, in Scotland, to the sheriff.
(2) The procedure on an appeal to a magistrates’ court under paragraph (1) is by way of complaint, and the Magistrates’ Courts Act 1980(16) applies to the proceedings.
(3) An appeal to the sheriff under paragraph (1) is by summary application.
(4) The period within which an appeal may be brought is 28 days or the period specified in the improvement notice, whichever ends the earlier.
(5) A court may suspend an improvement notice pending an appeal.
Extent Information
E9This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Appeals against improvement noticesN.I.
39.—(1) Any person who is aggrieved by an improvement notice may appeal to a magistrates’ court F184....
(2) The procedure on an appeal to a magistrates’ court under paragraph (1) is by way of complaint, and the Magistrates’ Courts Act 1980(16) applies to the proceedings.
F185(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(4) The period within which an appeal may be brought is 28 days or the period specified in the improvement notice, whichever ends the earlier.
(5) A court may suspend an improvement notice pending an appeal.
Extent Information
E80This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F184Words in reg. 39(1) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(9)(a)
F185Reg. 39(3) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(9)(b)
Powers of a court on appealU.K.
40. On an appeal against an improvement notice, the court may either cancel the notice or confirm it, with or without modification.
Seizure noticesE+W+S
41.—(1) An inspector must follow the procedures set out in this regulation when seizing anything under these Regulations.
(2) The inspector must serve on the person appearing to be in charge of the seized product a notice (referred to in these Regulations as a “seizure notice”)—
(a)giving the grounds for seizing the product; and
(b)informing that person of the rights under this regulation to make a claim, and the address for the service of the claim.
(3) An inspector who is not able to remove products seized immediately may mark the products in any way, and serve a notice on the person in charge of the products identifying them, and prohibiting the removal of the products from the premises until they are collected by an inspector, and no person other than an inspector may remove products identified under this paragraph from the premises.
(4) The person on whom the seizure notice was served or the owner of the seized product may, within 28 days of seizure, notify any claim that the product was not liable to seizure to the Secretary of State at the address specified in the seizure notice, setting out the grounds in full.
(5) If a notification of a claim is not received within 28 days, the Secretary of State may destroy the product.
(6) If a notification of a claim is received within 28 days, then, unless the product seized is being held for the purposes of pending or contemplated criminal proceedings, or for a criminal investigation, the Secretary of State must either return the product or take proceedings for an order for the confirmation of the seizure notice and the destruction of the veterinary medicinal product in a magistrates’ court (or, in Scotland, the sheriff court), and if the court confirms the notice it must order its destruction.
(7) The procedure in a magistrates’ court under this regulation is by way of complaint, and the Magistrates’ Courts Act 1980 applies to the proceedings.
(8) The procedure before the sheriff is by summary application.
(9) The person on whom the seizure notice was served is liable for the costs of transport, storage for up to 28 days and destruction of the product seized unless a claim is made to a court and the court directs otherwise.
Extent Information
E10This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Seizure noticesN.I.
41.—(1) An inspector must follow the procedures set out in this regulation when seizing anything under these Regulations.
(2) The inspector must serve on the person appearing to be in charge of the seized product a notice (referred to in these Regulations as a “seizure notice”)—
(a)giving the grounds for seizing the product; and
(b)informing that person of the rights under this regulation to make a claim, and the address for the service of the claim.
(3) An inspector who is not able to remove products seized immediately may mark the products in any way, and serve a notice on the person in charge of the products identifying them, and prohibiting the removal of the products from the premises until they are collected by an inspector, and no person other than an inspector may remove products identified under this paragraph from the premises.
(4) The person on whom the seizure notice was served or the owner of the seized product may, within 28 days of seizure, notify any claim that the product was not liable to seizure to the Secretary of State at the address specified in the seizure notice, setting out the grounds in full.
(5) If a notification of a claim is not received within 28 days, the Secretary of State may destroy the product.
(6) If a notification of a claim is received within 28 days, then, unless the product seized is being held for the purposes of pending or contemplated criminal proceedings, or for a criminal investigation, the Secretary of State must either return the product or take proceedings for an order for the confirmation of the seizure notice and the destruction of the veterinary medicinal product in a magistrates’ court F186..., and if the court confirms the notice it must order its destruction.
(7) The procedure in a magistrates’ court under this regulation is by way of complaint, and the Magistrates’ Courts Act 1980 applies to the proceedings.
F187(8) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(9) The person on whom the seizure notice was served is liable for the costs of transport, storage for up to 28 days and destruction of the product seized unless a claim is made to a court and the court directs otherwise.
Extent Information
E81This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F186Words in reg. 41(6) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(10)(a)
F187Reg. 41(8) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(10)(b)
PublicationU.K.
42.—(1) The Secretary of State must publicise all improvement notices and seizure notices issued under these Regulations and the suspension or revocation of anything issued under these Regulations, and may do so in such manner as the Secretary of State sees fit.
(2) This does not apply in relation to a seizure notice issued to a common carrier who does not own the seized goods.
OffenceU.K.
43. It is an offence(17) to fail to comply with—
(a)regulation 4(1) or (2);
(b)regulation 5(1);
(c)regulation 7(2), (3), (4) or (5);
(d)regulation 8;
(e)regulation 9(1);
(f)regulation 10(1) or (2);
(g)regulation 11(1);
(h)regulation 13;
(i)regulation 17;
(j)regulation 18
(k)regulation 19
(l)regulation 20
(m)regulation 21
(n)regulation 22
(o)regulation 23
(p)regulation 24;
(q)regulation 25(1);
(r)regulation 26(1), (2) or (6);
(s)regulation 27(1);
(t)regulation 31(1);
(u)regulation 37;
(v)an improvement notice issued under regulation 38; or
(w)regulation 41(3).
PenaltiesU.K.
44.—(1) A person guilty of an offence under these Regulations is liable—
(a)on summary conviction, to a fine not exceeding the statutory maximum or to imprisonment for a term not exceeding six months or both, or
(b)on conviction on indictment, to a fine or to imprisonment for a term not exceeding two years or both.
(2) Where a body corporate is guilty of an offence under these Regulations, and that offence is proved to have been committed with the consent or connivance of, or to have been attributable to any neglect on the part of—
(a)a qualified person appointed as such for the purposes of these Regulations;
(b)any director, manager, secretary or other similar person of the body corporate; or
(c)any person who was purporting to act in any such capacity,
that person is guilty of the offence as well as the body corporate.
(3) If an offence under these Regulations committed by a partnership is shown—
(a)to have been committed with the consent or connivance of a partner; or
(b)to be attributable to any neglect on their part,
the partner as well as the partnership is guilty of the offence and liable to be proceeded against and punished accordingly.
(4) For the purposes of paragraph (2) above, “director”, in relation to a body corporate whose affairs are managed by its members, means a member of the body corporate.
(5) Where an offence that has been committed by a Scottish partnership is proved to have been committed with the consent or connivance of, or to be attributable to any neglect on the part of, a partner, the partner as well as the partnership is guilty of the offence.
Northern IrelandE+W+S
F2745. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Textual Amendments
F27Reg. 45 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(6)
Northern IrelandN.I.
45.—(1) This regulation has effect in relation to the enforcement of these Regulations in Northern Ireland.
(2) The Department of Agriculture and Rural Development or the Department of Health, Social Services and Public Safety (or both Departments acting jointly) instead of the Secretary of State exercise the powers of the Secretary of State in—
(a)regulation 33 (appointment of inspectors);
(b)regulation 41 (seizure notices);
(c)regulation 42 (publication); and
(d)sub-paragraph (4) of paragraph 14 of Schedule 3 (approval of premises for suitably qualified persons).
(3) The Department of Agriculture and Rural Development is the competent authority for—
[F188(a)Regulation (EC) No 178/2002;]
[F189(b)Regulation (EC) No 1831/2003;]
[F190(c)Regulation (EU) 2017/625; and]
[F191(d)Regulation (EC) No 183/2005.]
(4) In relation to pharmacies, an officer of the Pharmaceutical Society of Northern Ireland appointed by the Society for the purpose has all the powers of an inspector to enforce these Regulations.
(5) In proceedings in a magistrates’ court relating to an improvement notice under regulation 38 or a seizure notice under regulation 41, the Magistrates’ Courts (Northern Ireland) Order 1981(18) applies.
Extent Information
E82This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F188Reg. 45(3)(a) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(3)(a)
F189Reg. 45(3)(b) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(3)(b)
F190Reg. 45(3)(c) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(11); and substituted (N.I.) (31.12.2020) by The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(3)
ReviewE+W+S
46.—(1) Before the end of each review period, the Secretary of State must—
(a)carry out a review of these Regulations other than the fees provisions;
(b)set out the conclusions of the review in a report; and
(c)publish the report.
(2) In carrying out the review the Secretary of State must, so far as is reasonable, have regard to how the EU instruments, or provisions of EU instruments, to which this regulation applies are implemented in other member States.
(3) The EU instruments, and provisions of EU instruments, to which this regulation applies are—
(a)Council Directive 90/167/EEC laying down conditions governing the preparation, placing on the market and use of medicated feedingstuffs in the Community, so far it is not superseded by Regulation (EC) No 183/2005(19);
(b)Commission Directive 91/412/EEC laying down the principles and guidelines of good manufacturing practice for veterinary medicinal products(20);
(c)Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products(21);
(d)Regulation (EC) No 178/2002 of the European Parliament and of the Council laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety, in so far as it applies to veterinary medicinal products used in feedingstuffs;
(e)Regulation (EC) No 1831/2003 of the European Parliament and of the Council on additives for use in animal nutrition, in so far as it applies to veterinary medicinal products used in feedingstuffs;
(f)[F28Regulation (EC) No 882/2004 of the European Parliament and of the Council on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules, in so far as it applies to veterinary medicinal products used in feedingstuffs]
[F28Regulation (EU) 2017/625 of the European Parliament and of the Council on official controls and other official activities performed to ensure the application of food and feed law, rules on animal health and welfare, plant health and plant protection products;]
(g)Regulation (EC) No 183/2005 of the European Parliament and of the Council laying down requirements for feed hygiene, in so far as it applies to veterinary medicinal products used in feedingstuffs;
(h)Commission Regulation (EC) No 1234/2008(22);
(i)Regulation (EC) No 470/2009 of the European Parliament and of the Council(23);
(j)Article 8 of Regulation (EC) No 767/2009 of the European Parliament and of the Council, and Articles 15 and 17 of that Regulation as they refer to the labelling requirements for feedingstuffs containing specified feed additives(24); and
(k)Commission Regulation (EU) No 37/2010(25).
(4) The report must in particular—
(a)set out the objectives intended to be achieved by the regulatory system established by these Regulations, other than the fees provisions;
(b)assess the extent to which those objectives are achieved; and
(c)assess whether those objectives remain appropriate and, if so, the extent to which they could be achieved with a system that imposes less regulation.
(5) In this regulation—
(a)“review period” means the period of five years beginning with the day on which these Regulations come into force, and, subject to paragraph (6), each successive period of five years thereafter; and
(b)“the fees provisions” means regulation 16 and Schedule 7.
(6) If a report under this regulation is published before the last day of the review period to which it relates, the following review period is to begin with the day on which that report is published.
Extent Information
E11This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F28Reg. 46(3)(f) substituted (E.) (14.12.2019) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) Regulations 2019 (S.I. 2019/1488), regs. 1(1), 27(c); and substituted (W.) (31.1.2020) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) (Wales) Regulations 2020 (S.I. 2020/44), regs. 1(2), 24(1)(c)
ReviewN.I.
46.—(1) Before the end of each review period, the Secretary of State must—
(a)carry out a review of these Regulations other than the fees provisions;
(b)set out the conclusions of the review in a report; and
(c)publish the report.
(2) In carrying out the review the Secretary of State must, so far as is reasonable, have regard to how the EU instruments, or provisions of EU instruments, to which this regulation applies are implemented in F192... member States.
(3) The EU instruments, and provisions of EU instruments, to which this regulation applies are—
(a)Council Directive 90/167/EEC laying down conditions governing the preparation, placing on the market and use of medicated feedingstuffs in the Community, so far it is not superseded by Regulation (EC) No 183/2005(19);
(b)Commission Directive 91/412/EEC laying down the principles and guidelines of good manufacturing practice for veterinary medicinal products(20);
(c)Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products(21);
(d)Regulation (EC) No 178/2002 of the European Parliament and of the Council laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety, in so far as it applies to veterinary medicinal products used in feedingstuffs;
(e)Regulation (EC) No 1831/2003 of the European Parliament and of the Council on additives for use in animal nutrition, in so far as it applies to veterinary medicinal products used in feedingstuffs;
[F193(f)Regulation (EU) 2017/625;]
(g)Regulation (EC) No 183/2005 of the European Parliament and of the Council laying down requirements for feed hygiene, in so far as it applies to veterinary medicinal products used in feedingstuffs;
(h)Commission Regulation (EC) No 1234/2008(22);
(i)Regulation (EC) No 470/2009 of the European Parliament and of the Council(23);
(j)Article 8 of Regulation (EC) No 767/2009 of the European Parliament and of the Council, and Articles 15 and 17 of that Regulation as they refer to the labelling requirements for feedingstuffs containing specified feed additives(24); and
(k)Commission Regulation (EU) No 37/2010(25).
(4) The report must in particular—
(a)set out the objectives intended to be achieved by the regulatory system established by these Regulations, other than the fees provisions;
(b)assess the extent to which those objectives are achieved; and
(c)assess whether those objectives remain appropriate and, if so, the extent to which they could be achieved with a system that imposes less regulation.
(5) In this regulation—
(a)“review period” means the period of five years beginning with the day on which these Regulations come into force, and, subject to paragraph (6), each successive period of five years thereafter; and
(b)“the fees provisions” means regulation 16 and Schedule 7.
(6) If a report under this regulation is published before the last day of the review period to which it relates, the following review period is to begin with the day on which that report is published.
Extent Information
E83This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F192Word in reg. 46(2) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(12)(a)
RevocationsU.K.
47. The following Regulations are revoked—
(a)the Veterinary Medicines Regulations 2011(26); and
(b)the Veterinary Medicines (Amendment) Regulations 2012(27).
David Heath
Minister of State for Agriculture and Food
Department for Environment, Food and Rural Affairs
We consent
Anne Milton
Mark Lancaster
Two of the Lords Commissioners of Her Majesty’s Treasury
Regulation 4(3)
SCHEDULE 1U.K.Marketing authorisations [F29in Great Britain] [F30in Northern Ireland]
Textual Amendments
F29Words in Sch. 1 heading inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(7)(a)
F30Words in Sch. 1 heading inserted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(a)
PART 1U.K.Application for a marketing authorisation
Application for a marketing authorisationU.K.
1. An application under these Regulations for a marketing authorisation for a veterinary medicinal product must be made to the Secretary of State.
Information with the applicationE+W+S
2.—(1) An application must include all necessary administrative information, and all scientific documentation necessary for demonstrating the safety, quality and efficacy of the product.
(2) In particular, the applicant must provide all the data required in Annex I to Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products(28), generated in accordance with that Annex.
[F31(2A) The reference in paragraph 2(2) to Annex 1 to Directive 2001/82/EC is to be read subject to the following modifications—
(a)a reference to a member State is to be read as a reference to Great Britain;
(b)a reference to the national pharmacopoeia of a member State is to be read as a reference to the British Pharmacopoeia;
(c)a reference to an application for a marketing authorisation pursuant to Article 12 or 13 is to be read as a reference to an application for a marketing authorisation pursuant to this Schedule;
(d)a reference to the Note for Guidance on minimising the risk of transmitting animal spongiform encephalopathy agents via veterinary medicinal products is to be read as a reference to that document as it had effect immediately before IP completion day;
(e)a reference to Council Directive 87/18/EEC is to be read as a reference to Directive 2004/10/EC of the European Parliament and of the Council on the harmonisation of laws, regulations and administrative provisions relating to the application of the principles of good laboratory practice and the verification of their applications for tests on chemical substances;
(f)a reference to Annex 5 of Council Directive 67/548/EEC is to be read as a reference to Regulation (EC) No 1272/2008 of the European Parliament and of the Council on classification, labelling and packaging of substances and mixtures;
(e)the following provisions are to be ignored—
(i)in Title 1—
(aa)in Part 1, in Chapter A, the fourth paragraph;
(bb)in Part 2, in Chapter A, paragraph 3.3;
(ii)in Title 2, in Part 5, in Chapter A, the fifth paragraph.]
(3) The application must contain the following information—
(a)the name of the person who will hold the marketing authorisation, that person’s address and, if different, the name and address of all the manufacturers involved in each stage of the manufacture, and the sites where the manufacture will take place;
(b)the name of the veterinary medicinal product, which may be either—
(i)an invented name provided that this is not liable to be confused with the common name of the product or the international non-proprietary name (INN) recommended by the World Health Organization; or
(ii)a common or scientific name accompanied by a trademark or the name of the marketing authorisation holder;
(c)the qualitative and quantitative particulars of all the constituents of the veterinary medicinal product, including its INN recommended by the World Health Organization, where an INN exists, or its chemical name;
(d)a description of the method of manufacture;
(e)all therapeutic indications, contra-indications and adverse reactions;
(f)the dosage for each species of animal for which the veterinary medicinal product is intended, its pharmaceutical form, method and route of administration and proposed shelf life;
(g)any proposed precautionary and safety measures to be taken when storing the veterinary medicinal product, administering it to animals or disposing of waste, together with an indication of potential risks that the veterinary medicinal product might pose to the environment, to human or animal health or to plants, together with the reasons;
(h)in the case of medicinal products intended for food‑producing species, the proposed withdrawal period necessary to ensure that the maximum residue limits [F32established by an appropriate authority under] Regulation (EC) No 470/2009 of the European Parliament and of the Council are not exceeded;
(i)a description of the testing methods to be used during manufacture;
(j)the results of—
(i)pharmaceutical (physico-chemical, biological or microbiological) tests;
(ii)safety tests and residue tests;
(iii)pre-clinical and clinical trials;
(iv)tests assessing the potential risks to the environment from the product;
(k)a detailed description of the pharmacovigilance system and, where appropriate, the risk management system that the applicant will put in place;
(l)a summary of the product characteristics, mock-ups of all proposed packaging and the proposed package leaflet, if any;
(m)a document showing that the manufacturer is authorised to produce veterinary medicinal products;
(n)copies (which must be updated if there are any changes while the application is being considered) of—
(i)any marketing authorisation obtained in another F33... country for the relevant veterinary medicinal product, and a list of any other [F34countries] in which an application for authorisation of the product has been submitted;
(ii)if the product is already authorised outside the United Kingdom, the summary of product characteristics for each authorisation;
(iii)any decision to refuse authorisation, [F35in any other] country and the reasons for that decision;
(o)proof that the applicant has the services of a qualified person responsible for pharmacovigilance (referred to in these Regulations as a qualified person (pharmacovigilance)) and has the necessary means for the notification of any adverse reaction suspected of occurring [F36 in another] country;
[F37(p)if the veterinary medicinal product is intended for food-producing species and contains one or more pharmacologically active substances for the species in question for which a maximum residue limit has not yet been established under Regulation (EC) No 470/2009 of the European Parliament and of the Council, a document certifying that a valid application for the establishment of maximum residue limits has been submitted.]
(4) All documents relating to the results of tests or trials must be accompanied by a detailed and critical expert report that has been drafted and signed by a person with the requisite technical or professional qualifications and that has a brief curriculum vitae of the person signing the report attached to it.
(5) In the case of immunological products, the applicant must submit a description of the methods used to establish that the manufacturing process will consistently produce a veterinary medicinal product that is in accordance with the marketing authorisation.
Extent Information
E12This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F31Sch. 1 para. 2(2A) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(9) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F32Words in Sch. 1 para. 2(3)(h) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(10)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F33Words in Sch. 1 para. 2(3)(n)(i) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(10)(b)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F34Word in Sch. 1 para. 2(3)(n)(i) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(10)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F35Words in Sch. 1 para. 2(3)(n)(iii) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(10)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F36Words in Sch. 1 para. 2(3)(o) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(10)(d) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F37Sch. 1 para. 2(3)(p) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(10)(e) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Information with the applicationN.I.
2.—(1) An application must include all necessary administrative information, and all scientific documentation necessary for demonstrating the safety, quality and efficacy of the product.
(2) In particular, the applicant must provide all the data required in Annex I to Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products(28), generated in accordance with that Annex.
(3) The application must contain the following information—
(a)the name of the person who will hold the marketing authorisation, that person’s address and, if different, the name and address of all the manufacturers involved in each stage of the manufacture, and the sites where the manufacture will take place;
(b)the name of the veterinary medicinal product, which may be either—
(i)an invented name provided that this is not liable to be confused with the common name of the product or the international non-proprietary name (INN) recommended by the World Health Organization; or
(ii)a common or scientific name accompanied by a trademark or the name of the marketing authorisation holder;
(c)the qualitative and quantitative particulars of all the constituents of the veterinary medicinal product, including its INN recommended by the World Health Organization, where an INN exists, or its chemical name;
(d)a description of the method of manufacture;
(e)all therapeutic indications, contra-indications and adverse reactions;
(f)the dosage for each species of animal for which the veterinary medicinal product is intended, its pharmaceutical form, method and route of administration and proposed shelf life;
(g)any proposed precautionary and safety measures to be taken when storing the veterinary medicinal product, administering it to animals or disposing of waste, together with an indication of potential risks that the veterinary medicinal product might pose to the environment, to human or animal health or to plants, together with the reasons;
(h)in the case of medicinal products intended for food‑producing species, the proposed withdrawal period necessary to ensure that the maximum residue limits specified in Regulation (EC) No 470/2009 of the European Parliament and of the Council are not exceeded;
(i)a description of the testing methods to be used during manufacture;
(j)the results of—
(i)pharmaceutical (physico-chemical, biological or microbiological) tests;
(ii)safety tests and residue tests;
(iii)pre-clinical and clinical trials;
(iv)tests assessing the potential risks to the environment from the product;
(k)a detailed description of the pharmacovigilance system and, where appropriate, the risk management system that the applicant will put in place;
(l)a summary of the product characteristics, mock-ups of all proposed packaging and the proposed package leaflet, if any;
(m)a document showing that the manufacturer is authorised to produce veterinary medicinal products;
(n)copies (which must be updated if there are any changes while the application is being considered) of—
(i)any marketing authorisation obtained in [F194a] member State or in a third country for the relevant veterinary medicinal product, and a list of any F195... member States in which an application for authorisation of the product has been submitted;
(ii)if the product is already authorised outside the United Kingdom, the summary of product characteristics for each authorisation;
(iii)any decision to refuse authorisation, whether in the Community or a third country and the reasons for that decision;
(o)proof that the applicant has the services of a qualified person responsible for pharmacovigilance (referred to in these Regulations as a qualified person (pharmacovigilance)) and has the necessary means for the notification of any adverse reaction suspected of occurring either in the Community or in a third country;
(p)if the veterinary medicinal product is intended for food-producing species and contains one or more pharmacologically active substances not yet included for the species in question in Commission Regulation (EU) No 37/2010, a document certifying that a valid application for the establishment of maximum residue limits has been submitted to the Agency in accordance with Regulation (EC) No 470/2009 of the European Parliament and of the Council.
(4) All documents relating to the results of tests or trials must be accompanied by a detailed and critical expert report that has been drafted and signed by a person with the requisite technical or professional qualifications and that has a brief curriculum vitae of the person signing the report attached to it.
(5) In the case of immunological products, the applicant must submit a description of the methods used to establish that the manufacturing process will consistently produce a veterinary medicinal product that is in accordance with the marketing authorisation.
Extent Information
E84This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F194Word in Sch. 1 para. 2(3)(n)(i) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(b)
F195Word in Sch. 1 para. 2(3)(n)(i) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(b)
Summary of product characteristicsU.K.
3. The summary of product characteristics required under the preceding paragraph must include the following information, set out in the same format—
Summary of product characteristics | ||
| 1 | Name of the veterinary medicinal product, followed by its strength and pharmaceutical form. | |
| 2 | The name and proportion of each active substance, and of any excipient if knowledge of the excipient is needed for safety reasons. | |
| 3 | Pharmaceutical form. | |
| 4 | Clinical particulars— | |
| 4.1 | target species; | |
| 4.2 | indications for use, specifying the target species; | |
| 4.3 | contra-indications; | |
| 4.4 | special warnings for each target species; | |
| 4.5 | special precautions for use, including special precautions to be taken by the person administering the medicinal product to the animals; | |
| 4.6 | adverse reactions (frequency and seriousness); | |
| 4.7 | use during pregnancy, lactation or lay; | |
| 4.8 | interaction with other medicinal products and other forms of interaction; | |
| 4.9 | amounts to be administered and administration route; | |
| 4.10 | overdose (symptoms, emergency procedures, antidotes) if necessary; | |
| 4.11 | withdrawal periods for the various foodstuffs, including those for which the withdrawal period is zero. | |
| 5 | Pharmacological properties— | |
| 5.1 | pharmacodynamic properties; | |
| 5.2 | pharmacokinetic particulars; | |
| 6 | Pharmaceutical particulars— | |
| 6.1 | list of excipients; | |
| 6.2 | major incompatibilities; | |
| 6.3 | shelf life, when necessary after reconstitution of the medicinal product or when the immediate packaging is opened for the first time; | |
| 6.4 | special precautions for storage; | |
| 6.5 | nature and contents of immediate packaging; | |
| 6.6 | special precautions for the disposal of unused veterinary medicinal products or waste materials derived from the use of such products, if appropriate; | |
| 7 | Marketing authorisation holder; | |
| 8 | Marketing authorisation number; | |
| 9 | Date of the first authorisation or date of renewal of the authorisation; | |
| 10 | Date of any revision of the text; | |
| 11 | Any other information required by the Secretary of State. | |
Supply of a copy of the summary of product characteristicsU.K.
4. A holder of a marketing authorisation must supply a copy of the summary of product characteristics to any person on demand.
Time limits for applications for products for use in food-producing animalsE+W+S
5. In the case of a veterinary medicinal product for food-producing animals, a marketing authorisation may not be applied for until at least six months after a valid application has been made for the establishment of a maximum residue limit F38....
Extent Information
E13This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F38Words in Sch. 1 para. 5 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(11) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Time limits for applications for products for use in food-producing animalsN.I.
5. In the case of a veterinary medicinal product for food-producing animals, a marketing authorisation may not be applied for until at least six months after a valid application has been made for the establishment of a maximum residue limit in accordance with Regulation (EC) No 470/2009 of the European Parliament and of the Council.
Extent Information
E85This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
PART 2U.K.Derogations from some of the requirements in Part 1
ScopeU.K.
6. This Part provides for applications for marketing authorisations in which not all the information required in Part 1 is required, but for the avoidance of doubt any applicant may apply for a marketing authorisation using Part 1 if the applicant wishes to do so.
Bibliographic applicationE+W+S
7.—(1) An applicant for a marketing authorisation need not provide the results of safety tests, residue tests, pre-clinical trials or clinical trials if the active substance of the veterinary medicinal product has been in an authorised veterinary medicinal product for that species F39... for at least ten years, and the applicant provides appropriate scientific literature to demonstrate this.
(2) The applicant may use any publicly available document.
(3) If an applicant makes use of scientific literature to obtain authorisation for a food-producing species, and submits, in respect of the same medicinal product and with a view to obtaining authorisation for another food-producing species, new residue studies, together with further clinical trials, a third party may not use those studies or trials in an application for a pharmacologically equivalent product for a period of three years from the grant of the authorisation for the additional species.
Extent Information
E14This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F39Words in Sch. 1 para. 7(1) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(12) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Bibliographic applicationN.I.
7.—(1) An applicant for a marketing authorisation need not provide the results of safety tests, residue tests, pre-clinical trials or clinical trials if the active substance of the veterinary medicinal product has been in an authorised veterinary medicinal product for that species in the Community for at least ten years, and the applicant provides appropriate scientific literature to demonstrate this.
(2) The applicant may use any publicly available document.
(3) If an applicant makes use of scientific literature to obtain authorisation for a food-producing species, and submits, in respect of the same medicinal product and with a view to obtaining authorisation for another food-producing species, new residue studies, together with further clinical trials, a third party may not use those studies or trials in an application for a pharmacologically equivalent product for a period of three years from the grant of the authorisation for the additional species.
Extent Information
E86This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for a product using a new combination of active substancesU.K.
8. If an application is for a veterinary medicinal product containing active substances already used in an authorised veterinary medicinal product but not previously used in that combination in a veterinary medicinal product, the applicant need not provide the safety and efficacy data for the individual active substances.
Application using existing dataU.K.
9. If the Secretary of State has granted a marketing authorisation, the Secretary of State may, with the permission of the holder, use the data submitted in support of that marketing authorisation when assessing an application for another marketing authorisation.
Application for a pharmacologically equivalent medicinal productE+W+S
10.—(1) An applicant need not provide the results of safety tests, residue tests, pre-clinical trials or clinical trials if the applicant can demonstrate that the veterinary medicinal product is pharmacologically equivalent to a veterinary medicinal product already authorised in the [F40United Kingdom].
(2) For the purposes of this paragraph a product is pharmacologically equivalent to an existing product if—
(a)it has the same qualitative and quantitative composition in active substances;
(b)it has the same pharmaceutical form; and
(c)bioequivalence has been demonstrated by means of appropriate bioavailability studies.
(3) For the purposes of this paragraph—
(a)the different salts, esters, ethers, isomers, mixtures of isomers, complexes or derivatives of an active substance are considered to be the same active substance, unless they differ significantly in properties with regard to safety or efficacy; and
(b)if they do differ significantly in properties with regard to efficacy or safety, additional information intended to provide proof of the safety or efficacy of the various salts, esters or derivatives of an authorised active substance must be supplied by the applicant.
(4) Different immediate-release oral pharmaceutical forms are regarded as the same pharmaceutical form.
(5) Bioavailability studies are not required if the bioequivalence guidelines produced by the [F41European Medicines] Agency exempt the product.
(6) In the case of a reference product authorised in another member State but not in the United Kingdom, the Secretary of State must be satisfied that the risk-benefit balance of the original product is appropriate for the product to be placed on the market in the United Kingdom, and if the data provided under Article 13, third paragraph of Directive 2001/82/EC by the member State in which the product is authorised are insufficient for the Secretary of State to be satisfied of this, the Secretary of State may notify the applicant and require the applicant to provide further data.
Extent Information
E15This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F40Words in Sch. 1 para. 10(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(13)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F41Words in Sch. 1 para. 10(5) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(13)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Application for a pharmacologically equivalent medicinal productN.I.
10.—(1) An applicant need not provide the results of safety tests, residue tests, pre-clinical trials or clinical trials if the applicant can demonstrate that the veterinary medicinal product is pharmacologically equivalent to a veterinary medicinal product already authorised in the Community.
(2) For the purposes of this paragraph a product is pharmacologically equivalent to an existing product if—
(a)it has the same qualitative and quantitative composition in active substances;
(b)it has the same pharmaceutical form; and
(c)bioequivalence has been demonstrated by means of appropriate bioavailability studies.
(3) For the purposes of this paragraph—
(a)the different salts, esters, ethers, isomers, mixtures of isomers, complexes or derivatives of an active substance are considered to be the same active substance, unless they differ significantly in properties with regard to safety or efficacy; and
(b)if they do differ significantly in properties with regard to efficacy or safety, additional information intended to provide proof of the safety or efficacy of the various salts, esters or derivatives of an authorised active substance must be supplied by the applicant.
(4) Different immediate-release oral pharmaceutical forms are regarded as the same pharmaceutical form.
(5) Bioavailability studies are not required if the bioequivalence guidelines produced by the Agency exempt the product.
(6) In the case of a reference product authorised in [F196a] member State but not in [F197Northern Ireland], the Secretary of State must be satisfied that the risk-benefit balance of the original product is appropriate for the product to be placed on the market in the United Kingdom, and if the data provided under Article 13, third paragraph of Directive 2001/82/EC by the member State in which the product is authorised are insufficient for the Secretary of State to be satisfied of this, the Secretary of State may notify the applicant and require the applicant to provide further data.
Extent Information
E87This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F196Word in Sch. 1 para. 10(6) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(c)(i)
F197Words in Sch. 1 para. 10(6) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(c)(ii)
Time limits for marketing authorisations granted under the procedure for a pharmacologically equivalent productU.K.
11.—(1) This paragraph establishes the time limits relating to granting a marketing authorisation under the procedure for a pharmacologically equivalent product.
(2) An application for a marketing authorisation cannot be made until two years before the product may be placed on the market in accordance with this paragraph.
(3) The product may not be placed on the market until ten years (or, in the case of medicinal products for fish or bees where the application for a marketing authorisation was submitted after 30th October 2005, thirteen years) have elapsed from the initial authorisation of the reference product.
(4) Time limits in this paragraph are calculated from the first grant of the marketing authorisation for the reference product.
Extension of time limitsU.K.
12.—(1) This paragraph applies in relation to veterinary medicinal products that—
(a)are intended for administration to food-producing species; and
(b)contain a new active substance that was not authorised in the Community by 30th April 2004.
(2) If a person submitted an application for a marketing authorisation for a product on or after 30th October 2005, and within 5 years of the original marketing authorisation being granted, the marketing authorisation is extended to include additional food-producing species, the ten-year protection period is extended by one year for each additional food-producing species added to the marketing authorisation.
(3) The total period may not exceed 13 years.
(4) The extension applies only if the marketing authorisation holder originally applied for determination of the maximum residue limits for the active substance.
Parallel importsE+W+S
13.—(1) The Secretary of State may grant a marketing authorisation in relation to a veterinary medicinal product authorised in another [F42country] and imported into the United Kingdom from that [F42country] in accordance with this paragraph without the data required in Part 1 if the applicant can demonstrate compliance with this paragraph.
(2) If the product is for a food-producing species it must be identical to a product authorised in the United Kingdom.
(3) Other products must be therapeutically the same as a product authorised in the United Kingdom unless the importer can justify any differences.
F43(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(5) The applicant must be established within the [F44United Kingdom].
(6) The applicant must hold (or have a contract with the holder of) a wholesale dealer’s authorisation in the United Kingdom appropriate to the type of product to be imported.
(7) If re-labelling is to take place in the United Kingdom the applicant must also be (or have a contract with) the holder of a suitable manufacturing authorisation in the United Kingdom.
Extent Information
E16This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F42Word in Sch. 1 para. 13(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(15)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F43Sch. 1 para. 13(4) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(15)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F44Words in Sch. 1 para. 13(5) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(15)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Parallel importsN.I.
13.—(1) The Secretary of State may grant a marketing authorisation in relation to a veterinary medicinal product authorised in [F198a] member State and imported into [F199Northern Ireland] from that member State in accordance with this paragraph without the data required in Part 1 if the applicant can demonstrate compliance with this paragraph.
(2) If the product is for a food-producing species it must be identical to a product authorised in [F200Northern Ireland].
(3) Other products must be therapeutically the same as a product authorised in [F201Northern Ireland] unless the importer can justify any differences.
(4) The member State from which it is imported must have authorised the product in accordance with Directive 2001/82/EC.
(5) The applicant must be established within the Community.
(6) The applicant must hold (or have a contract with the holder of) a wholesale dealer’s authorisation in [F202Northern Ireland] appropriate to the type of product to be imported.
(7) If re-labelling is to take place in [F203Northern Ireland] the applicant must also be (or have a contract with) the holder of a suitable manufacturing authorisation in [F203Northern Ireland].
Extent Information
E88This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F198Word in Sch. 1 para. 13(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(i)
F199Words in Sch. 1 para. 13(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
F200Words in Sch. 1 para. 13(2) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
F201Words in Sch. 1 para. 13(3) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
F202Words in Sch. 1 para. 13(6) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
F203Words in Sch. 1 para. 13(7) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
Specific batch control schemeE+W+S
14.—(1) Where a veterinary medicinal product (other than a biological veterinary medicinal product) has been granted a marketing authorisation or an animal test certificate, and any starting material (active substance, excipient or packaging) or any batch of the product does not fully meet the requirements of the authorisation or animal test certificate, the holder may apply to the Secretary of State to place one or more batches on the market notwithstanding this.
(2) The Secretary of State may authorise the placing on the market on being satisfied that the safety, quality and efficacy of the product are not compromised, and that in all the circumstances of the case the product should be placed on the market.
F45(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(4) In this paragraph a biological veterinary medicinal product is a veterinary medicinal product, the active substance of which is a biological substance; and a biological substance is a substance that is produced by or extracted from a biological source and for which a combination of physico-chemical-biological testing and the production process and its control is needed for its characterisation and the determination of its quality.
Extent Information
E17This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F45Sch. 1 para. 14(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(16) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Specific batch control schemeN.I.
14.—(1) Where a veterinary medicinal product (other than a biological veterinary medicinal product) has been granted a marketing authorisation or an animal test certificate, and any starting material (active substance, excipient or packaging) or any batch of the product does not fully meet the requirements of the authorisation or animal test certificate, the holder may apply to the Secretary of State to place one or more batches on the market notwithstanding this.
(2) The Secretary of State may authorise the placing on the market on being satisfied that the safety, quality and efficacy of the product are not compromised, and that in all the circumstances of the case the product should be placed on the market.
(3) This paragraph does not apply in relation to a product recognised in more than one member State.
(4) In this paragraph a biological veterinary medicinal product is a veterinary medicinal product, the active substance of which is a biological substance; and a biological substance is a substance that is produced by or extracted from a biological source and for which a combination of physico-chemical-biological testing and the production process and its control is needed for its characterisation and the determination of its quality.
Extent Information
E89This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Similar immunological productsU.K.
15. Where an immunological veterinary medicinal product is pharmacologically equivalent to a reference product other than differences in raw materials or in the manufacturing process, the results of the appropriate pre-clinical tests or clinical trials must be provided, but the applicant need not provide the results of safety tests or residue tests.
Marketing a product authorised in another countryE+W+S
16. Where the health situation so requires, the Secretary of State may authorise the placing on the market of a veterinary medicinal product that has been authorised [F46in another] country.
Extent Information
E18This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F46Words in Sch. 1 para. 16 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(17) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Marketing a product authorised in another countryN.I.
16. Where the health situation so requires, the Secretary of State may authorise the placing on the market of a veterinary medicinal product that has been authorised by [F204a] member State or, if there is no such authorised product, authorised in a third country.
Extent Information
E90This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F204Word in Sch. 1 para. 16 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(e)
PART 3U.K.Grant of a marketing authorisation
Time limitsU.K.
17. The Secretary of State must ensure that the procedure for granting an authorisation for a veterinary medicinal product is completed within a maximum of 210 days after the submission of the application.
Place of establishment of applicantE+W+S
18. Only an applicant established in [F47the United Kingdom] may be granted a marketing authorisation.
Extent Information
E19This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F47Words in Sch. 1 para. 18 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(18) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Place of establishment of applicantN.I.
18. Only an applicant established in [F205the United Kingdom or] a member State may be granted a marketing authorisation.
Extent Information
E91This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F205Words in Sch. 1 para. 18 inserted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(i)
ProcedureU.K.
19. The Secretary of State may require the applicant to provide additional information or to generate additional data, including laboratory testing, or may require the applicant to provide samples of any medicinal product, its starting materials and intermediate products or other constituent materials for testing in a laboratory.
Products authorised in another member StateE+W+S
F4820. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E20This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F48Sch. 1 para. 20 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(19) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Products authorised in another member StateN.I.
20. Where the Secretary of State is informed or discovers that [F206a] member State has authorised a veterinary medicinal product that is the subject of an application for authorisation by the Secretary of State, the Secretary of State must reject the application unless it was submitted in accordance with the mutual recognition procedure or the decentralised procedure in Part 6.
Extent Information
E92This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F206Word in Sch. 1 para. 20 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(ii)
Assessment reportsU.K.
21. The Secretary of State must produce an assessment of the dossier, consisting of an evaluation of the results of the pharmaceutical, safety and residue tests and the pre-clinical and clinical trials of the veterinary medicinal product concerned, and any additional related information.
Grant of a marketing authorisationU.K.
22. When granting a marketing authorisation, the Secretary of State must inform the applicant of the summary of product characteristics that has been approved, and the distribution category of the product.
Marketing authorisations for food-producing speciesE+W+S
23.—(1) The Secretary of State must not grant a marketing authorisation for a veterinary medicinal product for food-producing species unless [F49maximum residue limits have been established under Regulation (EC) No 470/2009 of the European Parliament and of the Council in respect of ] all its pharmacologically active substances F50....
(2) This does not apply in the case of a marketing authorisation for a veterinary medicinal product for administration to a horse that has been declared on its horse passport as not intended for slaughter for human consumption; but in this case the product must not include an active substance that [F51has been classified under Article 14 of Regulation (EC) No 470/2009 of the European Parliament and of the Council as prohibited for use in food producing animals.]
Extent Information
E21This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F49Words in Sch. 1 para. 23(1) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(20)(a)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F50Words in Sch. 1 para. 23(1) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(20)(a)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F51Words in Sch. 1 para. 23(2) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(20)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Marketing authorisations for food-producing speciesN.I.
23.—(1) The Secretary of State must not grant a marketing authorisation for a veterinary medicinal product for food-producing species unless all its pharmacologically active substances appear in Table 1 in the Annex to Commission Regulation (EU) No 37/2010.
(2) This does not apply in the case of a marketing authorisation for a veterinary medicinal product for administration to a horse that has been declared on its horse passport as not intended for slaughter for human consumption; but in this case the product must not include an active substance that appears in Table 2 in the Annex to Commission Regulation (EU) No 37/2010 and must not be intended for the treatment of a condition for which a veterinary medicinal product is already authorised for horses.
Extent Information
E93This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Refusal of a marketing authorisationE+W+S
24.—(1) The Secretary of State must refuse to grant a marketing authorisation if the application does not comply with these Regulations.
(2) In addition, the Secretary of State must refuse to grant it if—
(a)the data submitted with the application are inadequate;
(b)the risk-benefit balance of the veterinary medicinal product is unfavourable;
(c)the product has insufficient therapeutic effect;
(d)the withdrawal period proposed by the applicant is not long enough to ensure [F52food safety], or is insufficiently substantiated;
(e)the veterinary medicinal product is for a prohibited use;
(f)the way that the product will be used will have an unnecessarily undesirable effect on the environment.
(3) The Secretary of State may refuse to grant a marketing authorisation—
(a)if there is F53... legislation pending that is incompatible with the requested authorisation; or
(b)if additional data have been requested and those data are not provided within such time limit as may be stipulated.
(4) If the Secretary of State, on the grounds of safety, quality or efficacy, intends to refuse an application, or proposes to grant a marketing authorisation that is different from the one applied for, the Secretary of State must notify the applicant accordingly, and the applicant may appeal to the Veterinary Products Committee.
Extent Information
E22This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F52Words in Sch. 1 para. 24(2)(d) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(21)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F53Word in Sch. 1 para. 24(3)(a) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(21)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Refusal of a marketing authorisationN.I.
24.—(1) The Secretary of State must refuse to grant a marketing authorisation if the application does not comply with these Regulations.
(2) In addition, the Secretary of State must refuse to grant it if—
(a)the data submitted with the application are inadequate;
(b)the risk-benefit balance of the veterinary medicinal product is unfavourable;
(c)the product has insufficient therapeutic effect;
(d)the withdrawal period proposed by the applicant is not long enough to ensure that Regulation (EC) No 470/2009 of the European Parliament and of the Council is complied with, or is insufficiently substantiated;
(e)the veterinary medicinal product is for a prohibited use;
(f)the way that the product will be used will have an unnecessarily undesirable effect on the environment.
(3) The Secretary of State may refuse to grant a marketing authorisation—
(a)if there is Community legislation pending that is incompatible with the requested authorisation; or
(b)if additional data have been requested and those data are not provided within such time limit as may be stipulated.
(4) If the Secretary of State, on the grounds of safety, quality or efficacy, intends to refuse an application, or proposes to grant a marketing authorisation that is different from the one applied for, the Secretary of State must notify the applicant accordingly, and the applicant may appeal to the Veterinary Products Committee.
Extent Information
E94This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Publication following the grant of a marketing authorisationU.K.
25.—(1) On granting a marketing authorisation the Secretary of State must publish—
(a)the notice granting the marketing authorisation;
(b)the summary of the product characteristics;
(c)the assessment report that has already been prepared but with any commercially confidential or personal information deleted.
(2) The Secretary of State must update the assessment report whenever new information that is of importance and relates to the quality, safety or efficacy of the veterinary medicinal product becomes available.
(3) The Secretary of State must send a copy of the assessment report, and any update, to the holder of the marketing authorisation before publication to enable the holder to make representations concerning any confidential or personal information that may be in it, and may specify a date by which representations must be made.
Marketing authorisations in exceptional circumstancesU.K.
26.—(1) In exceptional circumstances, and if there is no other product with a full marketing authorisation for the indicated condition in the target species, the Secretary of State may grant an exceptional marketing authorisation consisting of—
(a)a provisional marketing authorisation subject to a requirement for the applicant to provide further data; or
(b)a limited marketing authorisation for a product with a limited market.
(2) The Secretary of State must reassess each provisional or limited marketing authorisation annually.
Provisions of samples and expertiseU.K.
27.—(1) The Secretary of State may require a marketing authorisation holder to provide, at any time and at any stage of the manufacturing process, samples of starting materials or the veterinary medicinal product for testing.
(2) At the request of the Secretary of State, the marketing authorisation holder must provide technical expertise to facilitate any analysis of the product.
Supply of informationU.K.
28.—(1) A marketing authorisation holder must immediately inform the Secretary of State on receipt of any new information that might adversely affect the risk-benefit balance of the veterinary medicinal product.
(2) The holder must immediately inform the Secretary of State of any prohibition or restriction imposed by the competent authorities of any country in which the veterinary medicinal product is authorised.
(3) The Secretary of State may at any time require the marketing authorisation holder to provide data relating to the risk-benefit balance.
Duties on the holder of a marketing authorisation relating to an immunological productE+W+S
29.—[F54(1) Before placing an immunological product on the market the holder of the marketing authorisation must notify the Secretary of State asking for written approval to do so.]
(2) If notified under sub-paragraph [F55(1)] the Secretary of State must give or refuse a written approval as soon as is reasonably practicable.
(3) No person may place an immunological product on the market without a written approval issued by the Secretary of State F56....
Extent Information
E23This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F54Sch. 1 para. 29(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(22)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F55Word in Sch. 1 para. 29(2) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(22)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F56Words in Sch. 1 para. 29(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(22)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Duties on the holder of a marketing authorisation relating to an immunological productN.I.
29.—(1) Before placing an immunological product on the market the holder of the marketing authorisation must either—
(a)notify the Secretary of State asking for written approval to do so; or
(b)if the holder has already received written approval from [F207a] member State permitting the release of the product, send a copy of that approval to the Secretary of State.
(2) If notified under sub-paragraph (1)(a) the Secretary of State must give or refuse a written approval as soon as is reasonably practicable.
(3) No person may place an immunological product on the market without a written approval issued by the Secretary of State or (if the approval was issued by [F208a] member State) without sending a copy of that approval to the Secretary of State.
Extent Information
E95This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F207Word in Sch. 1 para. 29(1)(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(iii)
F208Word in Sch. 1 para. 29(3) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(iii)
Control testsU.K.
30. The holder of a marketing authorisation must give to the Secretary of State on demand evidence that the holder has carried out all control tests required under the marketing authorisation, and the results of those tests.
Placing on the marketE+W+S
31.—(1) A holder of a marketing authorisation must notify the Secretary of State when the veterinary medicinal product is first placed on the market in the United Kingdom, and the date on which it was placed on the market.
(2) A holder of a marketing authorisation who removes the veterinary medicinal product from the market in the United Kingdom must notify the Secretary of State at least two months (or a shorter period in exceptional circumstances) before doing so.
(3) Upon request by the Secretary of State, the marketing authorisation holder must provide—
(a)all data relating to the volume of sales of the veterinary medicinal product by the holder; and
(b)any data in the holder’s possession relating to the number of prescriptions written for the product and the total volume supplied under those prescriptions.
Extent Information
E24This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Placing on the marketN.I.
31.—(1) A holder of a marketing authorisation must notify the Secretary of State when the veterinary medicinal product is first placed on the market in [F209Northern Ireland], and the date on which it was placed on the market.
(2) A holder of a marketing authorisation who removes the veterinary medicinal product from the market in [F210Northern Ireland] must notify the Secretary of State at least two months (or a shorter period in exceptional circumstances) before doing so.
(3) Upon request by the Secretary of State, the marketing authorisation holder must provide—
(a)all data relating to the volume of sales of the veterinary medicinal product by the holder; and
(b)any data in the holder’s possession relating to the number of prescriptions written for the product and the total volume supplied under those prescriptions.
Extent Information
E96This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F209Words in Sch. 1 para. 31(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(iv)
F210Words in Sch. 1 para. 31(2) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(iv)
Duration and validity of a marketing authorisationE+W+S
32.—(1) A marketing authorisation is initially valid for five years.
(2) The authorisation may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance.
(3) An application for renewal must be made at least six months, and not more than nine months, before the marketing authorisation ceases to be valid.
(4) An applicant who applies for the renewal of the marketing authorisation must enclose a list of all documents concerning the product that the applicant has submitted to the Secretary of State since the marketing authorisation was granted.
(5) The Secretary of State may require the applicant to provide a copy of any of the listed documents at any time.
(6) Once renewed, the marketing authorisation is valid indefinitely unless, within five years of the renewal, the Secretary of State notifies the holder, on justified grounds relating to pharmacovigilance, that the authorisation will cease to be valid five years from the first renewal unless the holder applies for a further renewal.
(7) The further renewal is not time-limited.
(8) Any marketing authorisation granted under these Regulations that is not followed within three years of its granting by the actual placing on the market of the authorised veterinary medicinal product in the United Kingdom ceases to be valid.
(9) When a veterinary medicinal product authorised under these Regulations and previously placed on the market in the United Kingdom is not present on the market in the United Kingdom for a period of three consecutive years, its marketing authorisation ceases to be valid.
(10) The Secretary of State may, on human or animal health grounds, grant exemptions from sub-paragraphs (8) and (9).
Extent Information
E25This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Duration and validity of a marketing authorisationN.I.
32.—(1) A marketing authorisation is initially valid for five years.
(2) The authorisation may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance.
(3) An application for renewal must be made at least six months, and not more than nine months, before the marketing authorisation ceases to be valid.
(4) An applicant who applies for the renewal of the marketing authorisation must enclose a list of all documents concerning the product that the applicant has submitted to the Secretary of State since the marketing authorisation was granted.
(5) The Secretary of State may require the applicant to provide a copy of any of the listed documents at any time.
(6) Once renewed, the marketing authorisation is valid indefinitely unless, within five years of the renewal, the Secretary of State notifies the holder, on justified grounds relating to pharmacovigilance, that the authorisation will cease to be valid five years from the first renewal unless the holder applies for a further renewal.
(7) The further renewal is not time-limited.
(8) Any marketing authorisation granted under these Regulations that is not followed within three years of its granting by the actual placing on the market of the authorised veterinary medicinal product in [F211Northern Ireland] ceases to be valid.
(9) When a veterinary medicinal product authorised under these Regulations and previously placed on the market in [F212Northern Ireland] is not present on the market in [F212Northern Ireland] for a period of three consecutive years, its marketing authorisation ceases to be valid.
(10) The Secretary of State may, on human or animal health grounds, grant exemptions from sub-paragraphs (8) and (9).
Extent Information
E97This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F211Words in Sch. 1 para. 32(8) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(v)
F212Words in Sch. 1 para. 32(9) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(v)
PART 4U.K.Variations of marketing authorisations on the application of the holder
Variation of a marketing authorisationU.K.
33.—(1) The Secretary of State is the competent authority for the purposes of Commission Regulation (EC) No 1234/2008(29).
(2) The holder of a marketing authorisation may apply to the Secretary of State for a variation of that marketing authorisation.
(3) An application for a variation under paragraph (2) may only relate to a “single variation” unless the application is submitted in accordance with—
(a)Article 7 of Commission Regulation (EC) No 1234/2008 (“grouped variations”), or
(b)Article 20 of Commission Regulation (EC) No 1234/2008 (“workshare variations”).
(4) The Secretary of State, when granting a variation of a veterinary medicinal product, may (unless there are exceptional circumstances necessary to protect human or animal health or the environment) specify transitional measures to enable products produced in accordance with the previous authorisation to continue to be marketed for the transitional period.
Refusal of a variation of a marketing authorisationU.K.
34.—(1) This paragraph applies in relation to the refusal by the Secretary of State of an application for a variation unless the procedure following the refusal of a variation is one of those set out in Article 13 of Regulation 1234/2008.
(2) The grounds on which the Secretary of State may refuse an application for a variation of a marketing authorisation are those set out in paragraph 24 of this Schedule (refusal of a marketing authorisation).
(3) The Secretary of State must give written reasons for refusing to grant a variation; and if—
(a)those reasons are on the grounds of safety, quality or efficacy; and
(b)the variation is Type II or an extension application (whether or not in each case as part of an application for a worksharing or grouped application),
the applicant may appeal to the Veterinary Products Committee.
Administrative variationsU.K.
35.—(1) The holder of a marketing authorisation may apply for a minor change in a marketing authorisation to be made without the Secretary of State considering any scientific data (an “administrative variation”).
(2) If the Secretary of State grants an administrative variation, and subsequently establishes that this should have been a variation requiring consideration of scientific data, the Secretary of State may notify the marketing authorisation holder, require the holder to submit an application for a variation enabling data to be assessed and revoke the administrative variation.
Changes after a marketing authorisation has been issuedU.K.
36. After a marketing authorisation has been issued, the holder must take account of scientific and technical progress in manufacturing and control methods, and apply to the Secretary of State for any variation in the marketing authorisation that may be required to enable that veterinary medicinal product to be manufactured and checked by means of generally accepted scientific methods.
Compulsory variationU.K.
37.—(1) If the Secretary of State decides, for any of the reasons for suspending a marketing authorisation specified in paragraph 38, or because the classification of a veterinary medicinal product should be changed, that a variation to a marketing authorisation is necessary, the Secretary of State must by a notification in writing to the holder of the marketing authorisation require that person to apply for a variation of the marketing authorisation, giving reasons for requiring the application to be made.
(2) The notification may specify a time limit within which the marketing authorisation holder must apply for the variation.
(3) If the variation is on the grounds of safety, quality or efficacy, the applicant may, within 28 days of the notification, appeal to the Veterinary Products Committee.
(4) If the marketing authorisation holder fails to apply for the variation within that time limit the Secretary of State may suspend or revoke the marketing authorisation.
PART 5U.K.Suspension, etc. of a marketing authorisation
Suspension of a marketing authorisation: groundsU.K.
38.—(1) The Secretary of State may suspend a marketing authorisation at any time on being satisfied that —
(a)this is necessary for the protection of animal or public health or the environment;
(b)the terms of the marketing authorisation have not been complied with; or
(c)the veterinary medicinal product has insufficient therapeutic effect.
(2) The Secretary of State may also suspend a marketing authorisation on being satisfied that a marketing authorisation holder has failed to make an application for a variation to take account of scientific and technical progress in manufacturing and control methods to enable the veterinary medicinal product to be manufactured and checked by means of generally accepted scientific methods.
(3) The Secretary of State must suspend a marketing authorisation on being satisfied that—
(a)the risk-benefit balance is unfavourable;
(b)the withdrawal period does not ensure that residues in foodstuffs obtained from the treated animal comply with Regulation (EC) No 470/2009 of the European Parliament and of the Council;
(c)information given in the application documents is incorrect;
(d)any control tests required have not been carried out;
(e)changes have been made to the manufacturing process without the authority of the Secretary of State; or
(f)any information required to be supplied to the Secretary of State has not been so supplied.
Suspension of a marketing authorisation: procedureE+W+S
39.—(1) If a marketing authorisation is suspended the Secretary of State must notify the holder immediately, and, unless the Secretary of State directs otherwise, the suspension has immediate effect, and continues in effect unless the marketing authorisation is reinstated.
(2) If the suspension is on the grounds of safety, quality or efficacy, the holder may, within 28 days of the notification, appeal to the Veterinary Products Committee.
F57(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(4) When a marketing authorisation is suspended, the Secretary of State may in addition prohibit the supply of the veterinary medicinal product, and if necessary require the marketing authorisation holder to recall the product.
Extent Information
E26This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F57Sch. 1 para. 39(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(23) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Suspension of a marketing authorisation: procedureN.I.
39.—(1) If a marketing authorisation is suspended the Secretary of State must notify the holder immediately, and, unless the Secretary of State directs otherwise, the suspension has immediate effect, and continues in effect unless the marketing authorisation is reinstated.
(2) If the suspension is on the grounds of safety, quality or efficacy, the holder may, within 28 days of the notification, appeal to the Veterinary Products Committee.
(3) If the veterinary medicinal product is authorised in more than one member State, the Secretary of State—
(a)must immediately refer the matter to the Agency, and must comply with a decision of the Commission within 30 days of the decision; and
(b)may suspend the marketing and the use of the veterinary medicinal product in [F213Northern Ireland] pending a decision of the Agency, but must inform the Commission and the F214... member States no later than the following working day of the reasons for the action.
(4) When a marketing authorisation is suspended, the Secretary of State may in addition prohibit the supply of the veterinary medicinal product, and if necessary require the marketing authorisation holder to recall the product.
Extent Information
E98This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F213Words in Sch. 1 para. 39(3)(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(g)(i)
F214Word in Sch. 1 para. 39(3)(b) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(g)(i)
RevocationU.K.
40. The Secretary of State may revoke any marketing authorisation that has been suspended for more than 28 days unless there is a current appeal to the Veterinary Products Committee, and may publicise a revocation in such manner as the Secretary of State sees fit.
Prohibiting the supply of veterinary medicinal productsE+W+S
41.—(1) In addition to the powers to suspend a marketing authorisation, the Secretary of State, on being satisfied that a product has not been manufactured in accordance with the marketing authorisation, may prohibit the supply of a veterinary medicinal product, and if necessary require the marketing authorisation holder to recall it.
(2) The prohibition on supply and the requirement for recall may be confined to specific production batches.
F58(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E27This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F58Sch. 1 para. 41(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(24) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Prohibiting the supply of veterinary medicinal productsN.I.
41.—(1) In addition to the powers to suspend a marketing authorisation, the Secretary of State, on being satisfied that a product has not been manufactured in accordance with the marketing authorisation, may prohibit the supply of a veterinary medicinal product, and if necessary require the marketing authorisation holder to recall it.
(2) The prohibition on supply and the requirement for recall may be confined to specific production batches.
(3) In the case of an immunological veterinary medicinal product manufactured outside the United Kingdom, if a batch has had all the tests that were originally carried out by the manufacturer repeated by the competent authority of [F215a] member State, the Secretary of State may not prohibit the release of that batch if all the results have been submitted to the Secretary of State and the results demonstrate that the product is within the terms of the authorisation.
Extent Information
E99This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F215Word in Sch. 1 para. 41(3) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(g)(ii)
[F59PART 6U.K.Mutual recognition and multiple applications
Textual Amendments
F59Sch. 1 Pt. 6 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(25) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Application for a marketing authorisation where one already exists in [F60a] member StateU.K.
42.—(1) If a veterinary medicinal product has already received a marketing authorisation in [F61a] member State at the time of application, and the holder of the marketing authorisation applies for a marketing authorisation in [F62Northern Ireland], the following procedure (“the mutual recognition procedure”) applies.
(2) The applicant must submit to the Secretary of State a dossier identical to the one submitted to the competent authority of the member State in which the veterinary medicinal product has been authorised (“the reference member State”).
(3) If there is a marketing authorisation current in more than one member State the applicant must identify which member State is acting as the reference member State.
(4) An applicant applying in more than one member State must supply the Secretary of State with a list of all the States in which the applicant is applying.
(5) The Secretary of State must obtain an assessment report from the reference member State and, where appropriate, an explanation of any extension of the period of data protection.
(6) Within 90 days after receipt of the assessment report, the Secretary of State must, subject to the following provisions, either—
(a)approve the assessment report, the summary of product characteristics, the labelling and the package leaflet, and inform the reference member State accordingly; or
(b)notify the reference member State that they have not been approved, and provide the reference member State with a detailed statement of the reasons.
(7) The Secretary of State may only refuse an application on the grounds of serious risk to human or animal health or the environment.
(8) If the assessment report, the summary of product characteristics, the labelling and the package leaflet are approved, the Secretary of State must ensure that a decision whether or not to grant a marketing authorisation can be made within 30 days of the approval.
(9) If the Secretary of State is notified by the reference member State that—
(a)not all member States concerned have within 90 days approved the assessment report, summary of product characteristics, labelling or package leaflet; and
(b)the reference member State has sent a detailed statement of the reasons to the other member States involved in the application, the applicant and the coordination group for action in accordance with Article 33(3) of Directive 2001/82/EC,
the Secretary of State must within 30 days comply with the decision of the coordination group or, if the coordination group refers the matter to the Agency, the decision of the Commission.
(10) The Secretary of State may grant the marketing authorisation even though not all member States have agreed to grant it, but must revoke or vary the authorisation if this is necessary to comply with the decision of the Commission when it is received.
Textual Amendments
F60Word in Sch. 1 para. 42 heading substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(i)(aa)
F61Word in Sch. 1 para. 42(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(i)(aa)
F62Words in Sch. 1 para. 42(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(i)(bb)
Application in [F63a] member StateU.K.
43.—(1) When the Secretary of State has granted a marketing authorisation for a veterinary medicinal product and is notified by the marketing authorisation holder that the marketing authorisation holder has applied to have that veterinary medicinal product authorised in [F64a] member State, the Secretary of State must prepare an assessment report for the product within 90 days of the notification and send it to the member State or States concerned.
(2) If the other member State (or, if there is more than one, all of them) agrees with the assessment report, the summary of product characteristics, the labelling and the package leaflet the Secretary of State need take no further action.
(3) If not all the other member States concerned so agree within a further 90 days the Secretary of State must send a detailed statement setting out why they have disagreed to the other member States, the applicant and the coordination group for action in accordance with Article 33(3) of Directive 2001/82/EC.
(4) The Secretary of State must within 30 days comply with the decision of the coordination group or, if the coordination group refers the matter to the Agency, the decision of the Commission.
Textual Amendments
F63Word in Sch. 1 para. 43 heading substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(ii)(aa)
F64Word in Sch. 1 para. 43(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(ii)(bb)
Application for a marketing authorisation in multiple member States where a marketing authorisation does not exist in any member StateU.K.
44.—(1) If an applicant wishes to apply for a marketing authorisation in more than one member State, and a marketing authorisation does not exist in any member State for the product (“the decentralised procedure”), the applicant must—
(a)apply simultaneously in all the relevant member States;
(b)submit a dossier to the Secretary of State that is identical to the dossier being submitted to all the other member States;
(c)include a list of all member States in which applications have been made; and
(d)nominate one of them to act as the reference member State to prepare a draft assessment report and drafts of the summary of product characteristics, labelling and package leaflet for consideration by the other member States (“the concerned member States”).
(2) If the United Kingdom is the reference member State, the Secretary of State must prepare a draft assessment report and drafts of the summary of product characteristics, labelling and package leaflet within 120 days of the receipt of a valid application and must send them to the other concerned member States and to the applicant.
(3) If the United Kingdom is not the reference member State, within 90 days after receipt of the assessment report and drafts of the summary of product characteristics, labelling and package leaflet from the reference member State, the Secretary of State must, subject to the following provisions, either—
(a)approve the assessment report, the summary of product characteristics, the labelling and the package leaflet, and inform the reference member State accordingly; or
(b)notify the reference member State that the Secretary of State will not approve it, and provide the reference member State with a detailed statement of the reasons.
(4) The Secretary of State may only refuse an application on the grounds of serious risk to human or animal health or the environment.
(5) If all the member States involved agree the assessment report, the summary of product characteristics, the labelling and the package leaflet within 90 days, the Secretary of State must ensure that a decision whether or not to grant a marketing authorisation can be made within 30 days.
(6) If, within 90 days, not all the member States have agreed the assessment report, summary of product characteristics, labelling and package leaflet on grounds of a potential serious risk to human or animal health or to the environment, the Secretary of State (if the United Kingdom is the reference member State) must send a detailed statement of the reasons to the other member States involved in the application, the applicant, and the coordination group to act in accordance with Article 33(3) of Directive 2001/82/EC.
(7) If reference has been made to the coordination group by any member State, the Secretary of State must within 30 days comply with the decision of the coordination group or, if the coordination group refers the matter to the Agency, the decision of the Commission.
(8) If the Secretary of State wishes to do so, the Secretary of State may grant the marketing authorisation even though not all member States have agreed to grant it, but must revoke or vary the authorisation if this is necessary to comply with the decision of the Commission when it is received.]
PART 7U.K.Labelling and package leaflets
Approval by the Secretary of StateU.K.
45. The Secretary of State, when issuing a marketing authorisation, must approve all containers, packaging, labels and package leaflets.
Reference to being authorisedE+W+S
46. A label and package leaflet of an authorised veterinary medicinal product may contain in legible characters the words “UK authorised veterinary medicinal product” or, if the marketing authorisation provides, other wording specified in the authorisation indicating that the product is authorised in the United Kingdom.
Extent Information
E28This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Reference to being authorisedN.I.
46. A label and package leaflet of an authorised veterinary medicinal product may contain in legible characters the words “[F216UK(NI)] authorised veterinary medicinal product” or, if the marketing authorisation provides, other wording specified in the authorisation indicating that the product is authorised in [F217Northern Ireland].
Extent Information
E100This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F216Word in Sch. 1 para. 46 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(i)(i)
F217Words in Sch. 1 para. 46 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(i)(ii)
LanguageU.K.
47.—(1) All labels and package leaflets must be in English, but may contain other languages provided that the information given is identical in all the languages.
(2) This requirement does not apply in the case of a product imported by a veterinary surgeon and administered by or under the responsibility of that same veterinary surgeon.
Labelling with all the information on the immediate packagingE+W+S
48.—(1) If it is reasonably practicable to do so, the following must be provided on the immediate packaging, in legible characters—
(a)the name, strength and pharmaceutical form of the veterinary medicinal product;
(b)the name and strength of each active substance, and of any excipient if this is required under paragraph 2 of the summary of product characteristics;
(c)the route of administration (if not immediately apparent);
(d)the batch number;
(e)the expiry date;
(f)the words “For animal treatment only” and, if appropriate, “To be supplied only on veterinary prescription”;
(g)the contents by weight, volume or number of dose units;
(h)the marketing authorisation number;
(i)the name and address of the marketing authorisation holder [F65, the local representative designated under paragraph 18 of this Schedule] or, if there is a distributor authorised in the marketing authorisation, that distributor;
(j)a suitably labelled space to record discard date (if relevant);
(k)the target species;
(l)the distribution category;
(m)the words “Keep out of reach of children”;
(n)storage instructions;
(o)the in-use shelf-life (if appropriate);
(p)for food-producing species, the withdrawal period for each species or animal product concerned;
(q)any warning specified in the marketing authorisation;
(r)disposal advice;
(s)full indications;
(t)dosage instructions;
(u)contra-indications;
(v)further information required in the marketing authorisation;
(w)if the product is one that requires a dose to be specified for the animal being treated, a space for this.
(2) If all this is on the immediate packaging, there is no need for any outer packaging or a package leaflet.
Extent Information
E29This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F65Words in Sch. 1 para. 48(1)(i) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(7)(b)
Labelling with all the information on the immediate packagingN.I.
48.—(1) If it is reasonably practicable to do so, the following must be provided on the immediate packaging, in legible characters—
(a)the name, strength and pharmaceutical form of the veterinary medicinal product;
(b)the name and strength of each active substance, and of any excipient if this is required under paragraph 2 of the summary of product characteristics;
(c)the route of administration (if not immediately apparent);
(d)the batch number;
(e)the expiry date;
(f)the words “For animal treatment only” and, if appropriate, “To be supplied only on veterinary prescription”;
(g)the contents by weight, volume or number of dose units;
(h)the marketing authorisation number;
(i)the name and address of the marketing authorisation holder or, if there is a distributor authorised in the marketing authorisation, that distributor;
(j)a suitably labelled space to record discard date (if relevant);
(k)the target species;
(l)the distribution category;
(m)the words “Keep out of reach of children”;
(n)storage instructions;
(o)the in-use shelf-life (if appropriate);
(p)for food-producing species, the withdrawal period for each species or animal product concerned;
(q)any warning specified in the marketing authorisation;
(r)disposal advice;
(s)full indications;
(t)dosage instructions;
(u)contra-indications;
(v)further information required in the marketing authorisation;
(w)if the product is one that requires a dose to be specified for the animal being treated, a space for this.
(2) If all this is on the immediate packaging, there is no need for any outer packaging or a package leaflet.
Extent Information
E101This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Products with immediate and outer packagingU.K.
49.—(1) If it is not reasonably practicable to have all the required information on the immediate packaging then this paragraph applies.
(2) The immediate packaging must have at least the following information—
(a)the name of the veterinary medicinal product, including its strength and pharmaceutical form;
(b)the name and proportion of each active substance, and of any excipient if knowledge of the excipient is needed for safety reasons;
(c)the route of administration (if not immediately apparent);
(d)the batch number;
(e)the expiry date;
(f)the words “For animal treatment only” and if appropriate, “To be supplied only on veterinary prescription”;
(g)the words “Keep the container in the outer carton”.
(3) In addition, the immediate packaging must have as much of the required information as is reasonably practicable.
(4) The outer packaging must contain all the required information if it is reasonably practicable to do this, and if it is not reasonably practicable to do this a package leaflet must be supplied with the product in accordance with the following paragraph.
Package leafletsU.K.
50.—(1) If it is not reasonably practicable to have all the required information on the immediate packaging or all of this information on the outer packaging, there must be a package leaflet supplied with the product, containing all the required information except for the batch number and the expiry date, and including the name of both the marketing authorisation holder and, if different, the name of the distributor named in the marketing authorisation.
(2) If there is a package leaflet, the immediate packaging and the outer packaging must both refer the user to it.
(3) A package leaflet must relate solely to the veterinary medicinal product with which it is included.
(4) It must be written in plain English.
(5) Only a package leaflet approved in the marketing authorisation may be included with the veterinary medicinal product.
AmpoulesU.K.
51.—(1) In the case of ampoules or other unit dose forms, where the container cannot bear legibly the required information, only the following information must be shown on the immediate packaging—
(a)the name of the veterinary medicinal product;
(b)the name and strength of the active ingredient;
(c)the route of administration (if not immediately apparent);
(d)the batch number;
(e)the expiry date;
(f)the words “For animal treatment only” and if appropriate, “To be supplied only on veterinary prescription”.
(2) The outer packaging must contain all the required information if it is reasonably practicable to do this, and if it is not reasonably practicable to do this a package leaflet must be supplied with the product, except that the ampoule need not refer to the package leaflet.
Small containers other than ampoulesU.K.
52. As regards small immediate packaging containing a single dose, other than ampoules, on which it is impossible to give the required information, all the required information must appear on the outer packaging or outer packaging and package leaflet, but the immediate packaging must be labelled with the batch number and the expiry date and, if there is room, the other information in the preceding paragraph.
Homeopathic remediesU.K.
53.—(1) A homeopathic remedy registered under these Regulations must be labelled in accordance with this paragraph.
(2) There must be no specific therapeutic indication on the labelling or in any information relating to it.
(3) The labelling (or labelling and package leaflet) must contain the following and no other information—
(a)the words “homeopathic remedy without approved therapeutic indications for veterinary use”;
(b)the scientific name of the stock or stocks followed by the degree of dilution, using the symbols of the pharmacopoeia used (if the homeopathic remedy is composed of more than one stock, the labelling may mention an invented name in addition to the scientific names of the stocks);
(c)the name and address of the registration holder and (on the package leaflet) of the manufacturer;
(d)the method and, if necessary, route of administration;
(e)the expiry date;
(f)the pharmaceutical form;
(g)the contents of the pack;
(h)any special storage precautions;
(i)the target species;
(j)any necessary special warnings;
(k)the batch number; and
(l)the registration number.
VariationsU.K.
54. The Secretary of State may permit variations in the above in any individual marketing authorisation if this is necessary for public or animal health purposes or the protection of the environment.
PART 8U.K.Pharmacovigilance
Qualified persons responsible for pharmacovigilanceE+W+S
55. A marketing authorisation holder must have permanently and continuously the services of an appropriately qualified person responsible for pharmacovigilance (“a qualified person (pharmacovigilance)”) F66....
Extent Information
E30This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F66Words in Sch. 1 para. 55 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(26) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Qualified persons responsible for pharmacovigilanceN.I.
55. A marketing authorisation holder must have permanently and continuously the services of an appropriately qualified person responsible for pharmacovigilance (“a qualified person (pharmacovigilance)”) who resides in a member State [F218or the United Kingdom].
Extent Information
E102This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F218Words in Sch. 1 para. 55 inserted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(i)
Duties relating to the qualified personE+W+S
56. The marketing authorisation holder must ensure that the qualified person (pharmacovigilance)—
(a)establishes and maintains a system that ensures that information about all suspected adverse reactions reported to the marketing authorisation holder is collected and collated in order to be accessible F67...;
(b)answers any request from the Secretary of State for the provision of additional information necessary for the evaluation of the benefits and risks afforded by a veterinary medicinal product fully and within any time limit imposed by the Secretary of State when the information was requested, including the volume of sales of the veterinary medicinal product concerned and, if available, details of prescriptions;
(c)provides to the Secretary of State any other information relevant to the evaluation of the benefits and risks afforded by a veterinary medicinal product, including appropriate information on post-marketing surveillance studies; and in this paragraph “post-marketing surveillance studies” means a pharmacoepidemiological study or a clinical trial carried out in accordance with the terms of the marketing authorisation, conducted with the aim of identifying and investigating a safety hazard relating to an authorised veterinary medicinal product.
Extent Information
E31This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F67Words in Sch. 1 para. 56(a) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(27) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Duties relating to the qualified personN.I.
56. The marketing authorisation holder must ensure that the qualified person (pharmacovigilance)—
(a)establishes and maintains a system that ensures that information about all suspected adverse reactions reported to the marketing authorisation holder is collected and collated in order to be accessible at least at one point in a member State;
(b)answers any request from the Secretary of State for the provision of additional information necessary for the evaluation of the benefits and risks afforded by a veterinary medicinal product fully and within any time limit imposed by the Secretary of State when the information was requested, including the volume of sales of the veterinary medicinal product concerned and, if available, details of prescriptions;
(c)provides to the Secretary of State any other information relevant to the evaluation of the benefits and risks afforded by a veterinary medicinal product, including appropriate information on post-marketing surveillance studies; and in this paragraph “post-marketing surveillance studies” means a pharmacoepidemiological study or a clinical trial carried out in accordance with the terms of the marketing authorisation, conducted with the aim of identifying and investigating a safety hazard relating to an authorised veterinary medicinal product.
Extent Information
E103This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Adverse reactions to a veterinary medicinal product administered in the United KingdomE+W+S
57.—(1) A marketing authorisation holder must act in accordance with this paragraph on learning of any suspected—
(a)serious adverse reaction;
(b)human adverse reaction; or
(c)unintended transmission of an infectious agent through a veterinary medicinal product,
following the administration of the product in the United Kingdom.
(2) The holder must make a record of what happened.
(3) The holder must without delay and in any event within 15 days report it (electronically if this is practicable) to the Secretary of State.
(4) In addition, the holder must supply to the Secretary of State all relevant veterinary pharmacovigilance information that the holder possesses relating to the reaction, giving a full description of the incident and a list of all the symptoms using internationally recognised veterinary and medical terminology, either with the report or, if the information becomes available after the report has been sent, as soon after it becomes available as is reasonably practicable.
(5) In this and the following paragraph—
“human adverse reaction” means a reaction that is noxious and unintended and that occurs in a human being following exposure to a veterinary medicine;
“serious adverse reaction” means an adverse reaction that results in death, is life-threatening, results in significant disability or incapacity, is a congenital anomaly or birth defect, or that results in permanent or prolonged signs in the animals treated.
Extent Information
E32This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Adverse reactions to a veterinary medicinal product administered in [F219Northern Ireland] N.I.
57.—(1) A marketing authorisation holder must act in accordance with this paragraph on learning of any suspected—
(a)serious adverse reaction;
(b)human adverse reaction; or
(c)unintended transmission of an infectious agent through a veterinary medicinal product,
following the administration of the product in [F220Northern Ireland].
(2) The holder must make a record of what happened.
(3) The holder must without delay and in any event within 15 days report it (electronically if this is practicable) to the Secretary of State.
(4) In addition, the holder must supply to the Secretary of State all relevant veterinary pharmacovigilance information that the holder possesses relating to the reaction, giving a full description of the incident and a list of all the symptoms using internationally recognised veterinary and medical terminology, either with the report or, if the information becomes available after the report has been sent, as soon after it becomes available as is reasonably practicable.
(5) In this and the following paragraph—
“human adverse reaction” means a reaction that is noxious and unintended and that occurs in a human being following exposure to a veterinary medicine;
“serious adverse reaction” means an adverse reaction that results in death, is life-threatening, results in significant disability or incapacity, is a congenital anomaly or birth defect, or that results in permanent or prolonged signs in the animals treated.
Extent Information
E104This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F219Words in Sch. 1 para. 57 heading substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(ii)
F220Words in Sch. 1 para. 57(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(ii)
Adverse reactions to a veterinary medicinal product administered in [F68another ] countryE+W+S
58.—(1) A marketing authorisation holder for a veterinary medicinal product authorised in [F69Great Britain] must act in accordance with this paragraph on learning of any suspected—
(a)serious, unexpected adverse reaction (for these purposes a reaction is unexpected if its nature, severity or outcome is not consistent with the summary of the product characteristics);
(b)human adverse reaction; or
(c)unintended transmission of an infectious agent through a veterinary medicinal product,
following the administration of the product in [F70another] country.
(2) The holder must make a record of what happened.
(3) The holder must without delay and in any event within 15 days report the suspected reaction or transmission (electronically if this is practicable) to the Secretary of StateF71....
(4) In addition to the report, the holder must supply to the Secretary of StateF72... all relevant veterinary pharmacovigilance information in the holder’s possession relating to the reaction as in the preceding paragraph.
Extent Information
E33This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F68Word in Sch. 1 para. 58 heading substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(28)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F69Words in Sch. 1 para. 58(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(7)(c)
F70Word in Sch. 1 para. 58(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(28)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F71Words in Sch. 1 para. 58(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(28)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F72Words in Sch. 1 para. 58(4) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(28)(d) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Adverse reactions to a veterinary medicinal product administered in a third countryN.I.
58.—(1) A marketing authorisation holder for a veterinary medicinal product authorised in [F221Northern Ireland] must act in accordance with this paragraph on learning of any suspected—
(a)serious, unexpected adverse reaction (for these purposes a reaction is unexpected if its nature, severity or outcome is not consistent with the summary of the product characteristics);
(b)human adverse reaction; or
(c)unintended transmission of an infectious agent through a veterinary medicinal product,
following the administration of the product in a third country.
(2) The holder must make a record of what happened.
(3) The holder must without delay and in any event within 15 days report the suspected reaction or transmission (electronically if this is practicable) to the Secretary of State, the competent authorities of all member States in which the product is authorised, and the Agency.
(4) In addition to the report, the holder must supply to the Secretary of State, the competent authorities of all F222... member States where the product is authorised and the Agency, all relevant veterinary pharmacovigilance information in the holder’s possession relating to the reaction as in the preceding paragraph.
Extent Information
E105This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F221Words in Sch. 1 para. 58(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(iii)(aa)
F222Word in Sch. 1 para. 58(4) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(iii)(bb)
Periodic safety update reportsE+W+S
59.—(1) The marketing authorisation holder must submit to the Secretary of State records of all adverse reactions (including nil reports) in the form of a periodic safety update report for each marketing authorisation in accordance with this paragraph, including a summary of each incident and a list of all the symptoms using internationally recognised veterinary and medical terminology.
(2) A marketing authorisation holder who has not yet placed a product on the market in the United Kingdom must submit a periodic safety update report immediately upon request of the Secretary of State and at least every six months after authorisation.
(3) Following the placing on the market in the United Kingdom, the marketing authorisation holder must submit a periodic safety update report to the Secretary of State immediately upon request and—
(a)at least every six months during the first two years following the initial placing on the market;
(b)once a year for the following two years; and
(c)thereafter, at three-yearly intervals.
(4) Following the granting of a marketing authorisation, the marketing authorisation holder may apply to the Secretary of State to change the periods of notification.
(5) The periodic safety update report must include a scientific evaluation of the risk-benefit balance of the veterinary medicinal product.
(6) The periodic safety update report must include—
(a)the volume of the product sold in each year covered by the report, calculated on an annual basis beginning 1st January;
(b)the number of adverse reactions for each year of the report;
(c)the ratio of adverse reactions to volume of product sold for each year of the report, together with an explanation of the basis of the calculation;
(d)differentiation of data based on—
(i)target species (if the product is authorised for use in more than one species);
(ii)reaction type (such as serious, non-serious, human, suspected lack of efficacy, unauthorised use or other);
(iii)the country of origin of the report.
(7) If the product is indicated for more than one species, the information in sub-paragraph (6)(c) must be based so far as is practicable on the estimated use of the product.
(8) Data relating to different formulations (either different dosage forms or different strengths) must be provided in separate reports.
Extent Information
E34This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Periodic safety update reportsN.I.
59.—(1) The marketing authorisation holder must submit to the Secretary of State records of all adverse reactions (including nil reports) in the form of a periodic safety update report for each marketing authorisation in accordance with this paragraph, including a summary of each incident and a list of all the symptoms using internationally recognised veterinary and medical terminology.
(2) A marketing authorisation holder who has not yet placed a product on the market in [F223Northern Ireland] must submit a periodic safety update report immediately upon request of the Secretary of State and at least every six months after authorisation.
(3) Following the placing on the market in [F224Northern Ireland], the marketing authorisation holder must submit a periodic safety update report to the Secretary of State immediately upon request and—
(a)at least every six months during the first two years following the initial placing on the market;
(b)once a year for the following two years; and
(c)thereafter, at three-yearly intervals.
(4) Following the granting of a marketing authorisation, the marketing authorisation holder may apply to the Secretary of State to change the periods of notification.
(5) The periodic safety update report must include a scientific evaluation of the risk-benefit balance of the veterinary medicinal product.
(6) The periodic safety update report must include—
(a)the volume of the product sold in each year covered by the report, calculated on an annual basis beginning 1st January;
(b)the number of adverse reactions for each year of the report;
(c)the ratio of adverse reactions to volume of product sold for each year of the report, together with an explanation of the basis of the calculation;
(d)differentiation of data based on—
(i)target species (if the product is authorised for use in more than one species);
(ii)reaction type (such as serious, non-serious, human, suspected lack of efficacy, unauthorised use or other);
(iii)the country of origin of the report.
(7) If the product is indicated for more than one species, the information in sub-paragraph (6)(c) must be based so far as is practicable on the estimated use of the product.
(8) Data relating to different formulations (either different dosage forms or different strengths) must be provided in separate reports.
Extent Information
E106This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F223Words in Sch. 1 para. 59(2) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(iv)
F224Words in Sch. 1 para. 59(3) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(iv)
Release of information by the marketing authorisation holderU.K.
60.—(1) A marketing authorisation holder must not communicate information relating to pharmacovigilance concerns to the general public in relation to its authorised veterinary medicinal product without giving prior or simultaneous notification to the Secretary of State.
(2) The marketing authorisation holder must ensure that such information is presented objectively and is not misleading.
Action taken on account of pharmacovigilanceE+W+S
61.—(1) Where, as a result of the evaluation of veterinary pharmacovigilance data, the Secretary of State considers that a marketing authorisation should be—
(a)suspended;
(b)revoked; or
(c)varied so as to—
(i)restrict the indications;
(ii)change the distribution category;
(iii)amend the dose;
(iv)add a contraindication; or
(v)add a new precautionary measure,
the Secretary of State must forthwith inform F73... and the marketing authorisation holder.
(2) If urgent action is necessary for protecting human or animal health, the Secretary of State may suspend the marketing authorisation of a veterinary medicinal productF74....
F75(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F76(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E35This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F73Words in Sch. 1 para. 61(1) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(29)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F74Words in Sch. 1 para. 61(2) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(29)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F75Sch. 1 para. 61(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(29)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F76Sch. 1 para. 61(4) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(29)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Action taken on account of pharmacovigilanceN.I.
61.—(1) Where, as a result of the evaluation of veterinary pharmacovigilance data, the Secretary of State considers that a marketing authorisation should be—
(a)suspended;
(b)revoked; or
(c)varied so as to—
(i)restrict the indications;
(ii)change the distribution category;
(iii)amend the dose;
(iv)add a contraindication; or
(v)add a new precautionary measure,
the Secretary of State must forthwith inform the Agency, all F225... member States (irrespective of whether the product is authorised in [F226a] member State) and the marketing authorisation holder.
(2) If urgent action is necessary for protecting human or animal health, the Secretary of State may suspend the marketing authorisation of a veterinary medicinal product, but must inform the Agency, the Commission and the F227... member States within one working day.
(3) If, following the opinion of the Agency, the Commission requests the Secretary of State to suspend, withdraw or vary the marketing authorisation, the Secretary of State must comply with that request immediately on a temporary basis.
(4) The Secretary of State must take final measures in accordance with the Decision of the Commission.
Extent Information
E107This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F225Word in Sch. 1 para. 61(1) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(v)(aa)
F226Word in Sch. 1 para. 61(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(v)(aa)
F227Word in Sch. 1 para. 61(2) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(v)(bb)
PART 9U.K.Homeopathic remedies
Meaning of “homeopathic remedy”E+W+S
62. For the purposes of these Regulations, a homeopathic remedy is a veterinary medicinal product (which may contain a number of principles) prepared from homeopathic stocks in accordance with a homeopathic manufacturing procedure described in the European Pharmacopoeia(30) or, if it is not described there, in a pharmacopoeia published by the British Pharmacopoeial Commission F77....
Extent Information
E36This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F77Words in Sch. 1 para. 62 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(30) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Meaning of “homeopathic remedy”N.I.
62. For the purposes of these Regulations, a homeopathic remedy is a veterinary medicinal product (which may contain a number of principles) prepared from homeopathic stocks in accordance with a homeopathic manufacturing procedure described in the European Pharmacopoeia(30) or, if it is not described there, in a pharmacopoeia published by the British Pharmacopoeial Commission or by the competent authority of any member State.
Extent Information
E108This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Placing a homeopathic remedy on the market in accordance with a registrationE+W+S
63.—(1) By way of derogation from the provisions of these Regulations requiring a marketing authorisation for a veterinary medicinal product, a homeopathic remedy may be placed on the market in accordance with a registration by the Secretary of State instead of in accordance with a marketing authorisation if it complies with this paragraph.
(2) It must not be an immunological product.
(3) The route of administration must be as described in the European PharmacopoeiaF78....
(4) There must be a sufficient degree of dilution to guarantee the safety of the product, and in any event it must not contain more than one part in 10,000 of the mother tincture.
(5) All other provisions relating to marketing authorisations apply in the same way to registrations of a homeopathic remedy.
Extent Information
E37This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F78Words in Sch. 1 para. 63(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(31) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Placing a homeopathic remedy on the market in accordance with a registrationN.I.
63.—(1) By way of derogation from the provisions of these Regulations requiring a marketing authorisation for a veterinary medicinal product, a homeopathic remedy may be placed on the market in accordance with a registration by the Secretary of State instead of in accordance with a marketing authorisation if it complies with this paragraph.
(2) It must not be an immunological product.
(3) The route of administration must be as described in the European Pharmacopoeia or, if it is not described there, by a pharmacopoeia currently used officially in any member State.
(4) There must be a sufficient degree of dilution to guarantee the safety of the product, and in any event it must not contain more than one part in 10,000 of the mother tincture.
(5) All other provisions relating to marketing authorisations apply in the same way to registrations of a homeopathic remedy.
Extent Information
E109This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for registrationE+W+S
64.—(1) An applicant for registration must submit the following to the Secretary of State—
(a)the scientific name or other name of the homeopathic stock given in a pharmacopoeia, together with a statement of the various routes of administration, pharmaceutical forms and degree of dilution;
(b)a dossier describing how the homeopathic stock is obtained and controlled, and justifying its homeopathic nature, on the basis of an adequate bibliography;
(c)in the case of a product containing biological substances, a description of the measures taken to ensure the absence of pathogens;
(d)the manufacturing and control file for each pharmaceutical form and a description of the method of dilution and potentisation;
(e)a copy of the manufacturing authorisation for the product;
(f)copies of any registrations or authorisations obtained for the same homeopathic remedyF79...;
(g)a mock-up of the outer packaging and immediate packaging;
(h)stability data;
(i)the proposed withdrawal period necessary to ensure that the provisions of Regulation (EC) No 470/2009 of the European Parliament and of the Council are complied with together with all necessary justification.
(2) These documents must demonstrate the pharmaceutical quality and the batch-to-batch homogeneity of the products concerned.
(3) In the case of a food-producing animal, if the applicant states in the application that the homeopathic remedy contains an active substance, or has been manufactured using an active substance, that substance must be one [F80for which a maximum residue limit has been established under Regulation (EC) No 470/2009 of the European Parliament and of the Council.]
F81(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E38This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F79Words in Sch. 1 para. 64(1)(f) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(32)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F80Words in Sch. 1 para. 64(3) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(32)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F81Sch. 1 para. 64(4) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(32)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Application for registrationN.I.
64.—(1) An applicant for registration must submit the following to the Secretary of State—
(a)the scientific name or other name of the homeopathic stock given in a pharmacopoeia, together with a statement of the various routes of administration, pharmaceutical forms and degree of dilution;
(b)a dossier describing how the homeopathic stock is obtained and controlled, and justifying its homeopathic nature, on the basis of an adequate bibliography;
(c)in the case of a product containing biological substances, a description of the measures taken to ensure the absence of pathogens;
(d)the manufacturing and control file for each pharmaceutical form and a description of the method of dilution and potentisation;
(e)a copy of the manufacturing authorisation for the product;
(f)copies of any registrations or authorisations obtained for the same homeopathic remedy in other member States;
(g)a mock-up of the outer packaging and immediate packaging;
(h)stability data;
(i)the proposed withdrawal period necessary to ensure that the provisions of Regulation (EC) No 470/2009 of the European Parliament and of the Council are complied with together with all necessary justification.
(2) These documents must demonstrate the pharmaceutical quality and the batch-to-batch homogeneity of the products concerned.
(3) In the case of a food-producing animal, if the applicant states in the application that the homeopathic remedy contains an active substance, or has been manufactured using an active substance, that substance must be one that appears in Table 1 in the Annex to Commission Regulation (EU) No 37/2010 and complies with any requirements in that Table relating to that substance.
(4) If a product is registered in [F228a] member State, the Secretary of State may waive some or all of the requirements of this paragraph on being satisfied that it is reasonable to do so.
Extent Information
E110This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F228Word in Sch. 1 para. 64(4) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(k)
Procedure for registrationU.K.
65.—(1) The procedure for registration is the same as the procedure for granting a marketing authorisation in accordance with Part 3, except—
(a)the applicant is not required to provide proof of efficacy;
(b)the product is not required to have a summary of product characteristics;
(c)the Secretary of State is not required to publish an assessment report.
(2) The procedure for variation, suspension and revocation is the same as for a marketing authorisation.
Products on the market before 1994U.K.
66. A homeopathic remedy that was on the market before 1st January 1994 may be placed on the market without being registered.
ClassificationU.K.
67. The registration must specify the classification of the homeopathic remedy, which must be one of the classifications specified for a veterinary medicinal product in Schedule 3.
OffencesU.K.
68. It is an offence to fail to comply with—
(a)a requirement made under paragraph 27(1);
(b)a request made under paragraph 27(2);
(c)paragraph 28(1) or (2);
(d)a requirement made under paragraph 28(3);
(e)paragraph 29(3);
(f)paragraph 30;
(g)paragraph 31(1) or (2);
(h)a request made under paragraph 31(3);
(i)a prohibition or requirement made under paragraph 39(4);
(j)a prohibition or requirement made under paragraph 41(1);
(k)paragraph 55;
(l)paragraph 56;
(m)paragraph 57;
(n)paragraph 58;
(o)paragraph 59; or
(p)paragraph 60.
Regulation 4(4)
[F82SCHEDULE 1AE+W+SConverted EU marketing authorisations
Textual Amendments
F82Sch. 1A inserted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(3), Sch. 8 Pt. 1; 2020 c. 1, Sch. 5 para. 1(1)
1. In this Schedule—E+W+S
“converted EU marketing authorisation” means an EU marketing authorisation to which paragraph 2 applies;
“EU marketing authorisation” means a marketing authorisation for a veterinary medicinal product granted by the European Commission in accordance with Title 3 of Regulation (EC) No 726/2004 of the European Parliament and of the Council laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency.
2. This paragraph applies to an EU marketing authorisation which—E+W+S
(a)was granted before exit day, and
(b)remains in force immediately before exit day.
3. A converted EU marketing authorisation has effect on and after exit day for the purposes of these regulations as if it were a marketing authorisation granted by the Secretary of State under these Regulations on the date it was originally granted—E+W+S
(a)on the terms which were in force immediately before exit day,
(b)with the benefit of any periods of data marketing exclusivity from which the holder benefited immediately before exit day, and
(c)subject to any suspension or post-authorisation obligations which were in force immediately before exit day.
4. Without prejudice to the generality of paragraph 3—E+W+S
(a)the holder of a converted EU marketing authorisation is subject to the annual fee as set out in paragraph 26 of Schedule 7;
(b)a converted EU marketing authorisation is to be treated as having been granted in accordance with regulation 4(3) and Schedule 1 for the purposes of Regulation (EC) No 469/2009.]
Regulation 4(5)
[F83SCHEDULE 1BE.W.S.Qualifying Northern Ireland good (QNIG) certificates
Textual Amendments
F83Sch. 1B inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(8)
1. In this Schedule—E.W.S.
“QNIG certificate” means a certificate issued under paragraph 3;
“QNIG certificate holder”, in relation to a QNIG certificate, means the person to whom that certificate was issued under paragraph 3;
“qualifying Northern Ireland goods” has the meaning given to it from time to time in regulations made under section 8C(6) of the European Union (Withdrawal) Act 2018;
“Northern Ireland VMRs” means the Veterinary Medicines Regulations 2013 as they have effect in Northern Ireland.
2. This Schedule applies to a veterinary medicinal product which is—E.W.S.
(a)a qualifying Northern Ireland good in respect of which there is a marketing authorisation valid in Northern Ireland under the Northern Ireland VMRs,
(b)not a product in respect of which there is a marketing authorisation which is valid in Great Britain (including any marketing authorisation which has effect under paragraph 3 of Schedule 1A),
(c)not a product in respect of which a QNIG certificate issued under this Schedule already applies, and
(d)not a product to which Article 41(1) of the EU withdrawal agreement applies.
3. If the condition in paragraph 4 is met in respect of the veterinary medicinal product, the Secretary of State must issue a QNIG certificate in respect of that product to the person who holds a marketing authorisation in respect of the product which is valid in Northern Ireland under the Northern Ireland VMRs.E.W.S.
4. The condition is that the person who holds a marketing authorisation in respect of the product which is valid in Northern Ireland under the Northern Ireland VMRs, who must be a person established in Northern Ireland, has provided the Secretary of State with the following information—E.W.S.
(a)the Northern Ireland address of that person;
(b)all necessary administrative information, and all scientific documentation necessary for demonstrating the safety, quality and efficacy of the veterinary medicinal product, equivalent to that which would need to be provided under Schedule 1 if an application for a marketing authorisation was to be made in respect of that product under paragraph 1 of that Schedule (allowing for any relevant derogations provided for in Part 2 of that Schedule);
(c)the name and address of a person who resides in the United Kingdom or in a member State who is to provide in respect of the veterinary medicinal product, permanently and continuously, the services of a qualified person (pharmacovigilance) for the purposes of Part 8 of Schedule 1.
5. A QNIG certificate has effect as if it were a marketing authorisation granted by the Secretary of State under these Regulations subject to the modification that the qualified person (pharmacovigilance) for the purposes of Part 8 of Schedule 1 is the person identified under paragraph 4(c).E.W.S.
6. The QNIG certificate holder must provide to the Secretary of State from time to time such further information as is appropriate to ensure that the information provided under paragraph 4 remains accurate and complete.E.W.S.
7. Without prejudice to any other power to suspend a marketing authorisation under Schedule 1, if the Secretary of State considers that a QNIG certificate holder is in breach of these Regulations as modified by paragraph 5, or that the information provided in respect of the matters specified in paragraph 4 is no longer accurate or complete, the Secretary of State may by notice suspend the QNIG certificate.E.W.S.
8. The Secretary of State must publish any notice given under paragraph 7 in such manner as the Secretary of State considers appropriate from time to time.E.W.S.
9. Paragraphs 39 and 40 of Schedule 1 apply to the suspension of a QNIG certificate under paragraph 7 as they would to the suspension of such a certificate under paragraph 38 of that Schedule as read with paragraph 5.]E.W.S.
Regulation 5(2)
SCHEDULE 2U.K.The manufacture of veterinary medicinal products
PART 1 Manufacturing authorisations
1.Application
2.Time limits
3.Granting the authorisation
4.The authorisation
5.Suspension, variation or revocation of the authorisation
6.Inspection of premises
7.Report following inspection
8.Duties on the holder of a manufacturing authorisation
9.Qualified persons for manufacture
10.Refusal or revocation of appointment
11.Duties on a qualified person
12.Register
13.Test sites
PART 2 Authorisation of manufacturers of autogenous vaccines
14.Authorisation to manufacture autogenous vaccines
15.Types of authorisation
16.Labelling
17.Records
18.Adverse reactions
19.Inspection of premises
PART 3 Authorisation of blood banks
20.Authorisation of blood banks
21.Supply and administration of blood from a blood bank
22.Labelling
23.Records
24.Inspection of premises
PART 4 Authorisation of manufacturers of products for administration under the cascade
25.Authorisation to manufacture products for administration under the cascade
26.Labelling
27.Records
28.Adverse reactions
29.Inspection of premises
PART 5 Authorisation of equine stem cell centres
30.Authorisation of stem cell centres
31.Supply and administration of stem cells
32.Labelling
33.Records
34.Inspection of premises
35.Offences
PART 1U.K.Manufacturing authorisations
ApplicationU.K.
1. An application for a manufacturing authorisation must be made to the Secretary of State.
Time limitsU.K.
2.—(1) The Secretary of State must process an application for a manufacturing authorisation within 90 days of receiving it.
(2) The Secretary of State must process an application for a variation of a manufacturing authorisation within 30 days unless the Secretary of State notifies the applicant in writing that the time has been extended to 90 days.
Granting the authorisationU.K.
3. The Secretary of State must grant a manufacturing authorisation on being satisfied that the applicant has suitable and sufficient premises, staff, technical equipment and facilities for the manufacture, control and storage of the products, and will comply with these Regulations.
The authorisationU.K.
4.—(1) The manufacturing authorisation must specify—
(a)the types of veterinary medicinal products and pharmaceutical forms that may be manufactured or imported;
(b)the place where they are to be manufactured or controlled;
(c)the name and address of the person holding the authorisation;
(d)the address of the premises to which it relates;
(e)the names of all qualified persons nominated to act under this Schedule.
(2) It may specify that different activities must be carried out in different premises or parts of premises, and may require the holder of the manufacturing authorisation to restrict access to premises or parts of premises to persons carrying out activities there.
(3) The holder of a manufacturing authorisation must notify the Secretary of State, and if necessary apply for a variation of the authorisation, before making a material alteration to the premises or facilities used under the authorisation, or to the operations for which they are used.
Suspension, variation or revocation of the authorisationU.K.
5.—(1) The Secretary of State may suspend, vary or revoke a manufacturing authorisation if the holder—
(a)has not complied with these Regulations;
(b)has manufactured a veterinary medicinal product not authorised by the manufacturing authorisation;
(c)has produced a veterinary medicinal product outside the terms of a marketing authorisation; or
(d)no longer has suitable premises or equipment.
(2) The Secretary of State may also suspend, vary or revoke it on being satisfied that the qualified person (manufacture) is not fulfilling their duties under these Regulations.
Inspection of premisesE+W+S
6.—(1) The Secretary of State must, from time to time, inspect premises registered under paragraph 3, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
(2) Within 90 days after an inspection, the Secretary of State must issue a certificate of good manufacturing practice to the manufacturer if the inspection established compliance with the principles and guidelines on good manufacturing practice in accordance with Commission Directive 91/412/EEC laying down the principles and guidelines of good manufacturing practice for veterinary medicinal products(31).
F84(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F84(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F84(5) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E39This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F84Sch. 2 para. 6(3)-(5) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Inspection of premisesN.I.
6.—(1) The Secretary of State must, from time to time, inspect premises registered under paragraph 3, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
(2) Within 90 days after an inspection, the Secretary of State must issue a certificate of good manufacturing practice to the manufacturer if the inspection established compliance with the principles and guidelines on good manufacturing practice in accordance with Commission Directive 91/412/EEC laying down the principles and guidelines of good manufacturing practice for veterinary medicinal products(31).
(3) If an inspection is carried out at the request of the European Pharmacopoeia to establish compliance with a monograph, the Secretary of State must issue a certificate of compliance with the monograph, if appropriate.
(4) The Secretary of State must provide details of each certificate of good manufacturing practice issued to the Agency for entry into a database.
(5) If the outcome of the inspection is that the manufacturer does not comply with the principles and guidelines of good manufacturing practice, the Secretary of State must provide details to the Agency for entry into the database.
Extent Information
E111This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Report following inspectionU.K.
7.—(1) After each inspection of manufacturing premises, the inspector must make a written report to the Secretary of State on whether the principles and guidelines on good manufacturing practice and the conditions of these Regulations are being complied with.
(2) The Secretary of State must inform the inspected manufacturer of the content of such reports.
Duties on the holder of a manufacturing authorisationU.K.
8.—(1) A holder of a manufacturing authorisation must ensure that the veterinary medicinal product is manufactured in accordance with the marketing authorisation.
(2) The holder must have permanently at their disposal the services of at least one qualified person (manufacture) who is on the register of qualified persons (manufacture) maintained by the Secretary of State and must place all necessary facilities at the qualified person’s disposal.
(3) The holder must—
(a)have a current Certificate of Good Manufacturing Practice;
(b)have in place a system of Quality Assurance and Quality Control; and
(c)give to the Secretary of State on request proof of all control tests carried out on the veterinary medicinal product or the constituents and intermediate products of the manufacturing process in accordance with the data submitted in support of the application for the marketing authorisation.
(4) A holder who makes up a bulk package of veterinary medicinal products must ensure that the package is labelled, in a way that the label is clearly visible and legible, with—
(a)the name of the veterinary medicinal product, its strength as shown in the summary of product characteristics and its pharmaceutical form;
(b)the batch number;
(c)the expiry date;
(d)any storage requirements; and
(e)any other warning necessary for the safe handling of the package.
(5) A holder must keep an adequate number of representative samples of each batch of a veterinary medicinal product in stock at least until the expiry date of the batch, and must submit any such sample to the Secretary of State if required in writing to do so.
Qualified persons for manufactureU.K.
9.—(1) The Secretary of State may appoint as a qualified person (manufacture) any person who is—
(a)a member of the Royal Pharmaceutical Society or registered with the Pharmaceutical Society of Northern Ireland;
(b)a Chartered Chemist or a Fellow, Member or Associate Member of the Royal Society of Chemistry; or
(c)a Chartered Biologist or a Fellow, Member or Associate Member of the Society of Biology;
who qualified on the basis of a formal course of study lasting not less than three years full-time or equivalent and who has sufficient practical experience to carry out the duties under this Schedule.
(2) The Secretary of State may exceptionally appoint a person who is not a member of one of those institutions to act as a qualified person (manufacture) on being satisfied that that person has the educational qualifications or practical experience to carry out the duties under this Schedule.
Refusal or revocation of appointmentU.K.
10. The Secretary of State may refuse or revoke an appointment if the Secretary of State is not satisfied that a person has fulfilled or will fulfil duties under these Regulations.
Duties on a qualified personE+W+S
11.—(1) The qualified person (manufacture) must ensure that each batch of veterinary medicinal product manufactured under that person’s responsibility is manufactured and checked in compliance with these Regulations and in accordance with the data submitted in support of the application for the marketing authorisation.
(2) If a manufacturer imports a veterinary medicinal product from [F85another] country,F86..., the qualified person (manufacture) must ensure that, following importation, each production batch imported is fully tested F87..., including a full qualitative analysis, a quantitative analysis of at least all the active substances and all the other tests or controls necessary to ensure the quality of a veterinary medicinal product is in accordance with the requirements of the marketing authorisation.
(3) Sub-paragraph (2) does not apply [F88where the exporting country has demonstrated equivalent standards to those of the United Kingdom or ] where appropriate arrangements have been made F89...with the exporting country to ensure that the manufacturer of the veterinary medicinal product applies standards of good manufacturing practice at least equivalent to those laid down in Commission Directive 91/412/EEC and to ensure that the controls in sub‑paragraph (2) have been carried out in the exporting country.
(4) At each stage of manufacture, including release for sale, the qualified person (manufacture) must certify in writing that all control tests required under the marketing authorisation have been carried out, and that the production batch complies with the marketing authorisation.
Extent Information
E40This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F85Word in Sch. 2 para. 11(2) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(b)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F86Words in Sch. 2 para. 11(2) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F87Words in Sch. 2 para. 11(2) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(b)(iii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F88Words in Sch. 2 para. 11(3) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(c)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F89Words in Sch. 2 para. 11(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(c)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Duties on a qualified personN.I.
11.—(1) The qualified person (manufacture) must ensure that each batch of veterinary medicinal product manufactured under that person’s responsibility is manufactured and checked in compliance with these Regulations and in accordance with the data submitted in support of the application for the marketing authorisation.
(2) If a manufacturer imports a veterinary medicinal product from a third country, including a product manufactured in a member State, the qualified person (manufacture) must ensure that, following importation, each production batch imported is fully tested in a member State, including a full qualitative analysis, a quantitative analysis of at least all the active substances and all the other tests or controls necessary to ensure the quality of a veterinary medicinal product is in accordance with the requirements of the marketing authorisation.
(3) Sub-paragraph (2) does not apply where appropriate arrangements have been made by the European Union with the exporting country to ensure that the manufacturer of the veterinary medicinal product applies standards of good manufacturing practice at least equivalent to those laid down in Commission Directive 91/412/EEC and to ensure that the controls in sub‑paragraph (2) have been carried out in the exporting country.
(4) At each stage of manufacture, including release for sale, the qualified person (manufacture) must certify in writing that all control tests required under the marketing authorisation have been carried out, and that the production batch complies with the marketing authorisation.
Extent Information
E112This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
RegisterU.K.
12. The Secretary of State must maintain and publish a register of—
(a)holders of manufacturing authorisations; and
(b)qualified persons (manufacture) appointed under paragraph 9(2).
Test sitesU.K.
13.—(1) The Secretary of State may authorise premises to act as a test site to carry out contract testing for a holder of a manufacturing authorisation.
(2) The premises must have a current certificate of good manufacturing practice.
(3) Authorisation and inspection of the premises are the same as for a manufacturing authorisation.
PART 2U.K.Authorisation of manufacturers of autogenous vaccines
Authorisation to manufacture autogenous vaccinesU.K.
14.—(1) The Secretary of State may authorise a person to manufacture autogenous vaccines and may authorise premises for the purpose of such manufacture by that person.
(2) In order to be authorised the premises must be under the supervision of—
(a)a veterinary surgeon; or
(b)a person who the Secretary of State is satisfied has sufficient qualifications and experience to manufacture the product safely.
(3) Before authorising the premises, the Secretary of State must be satisfied that the production process will produce a consistent, safe product.
(4) No person may manufacture an autogenous vaccine other than in accordance with an authorisation under sub-paragraph (1).
Types of authorisationU.K.
15.—(1) The authorisation must specify the products that may be manufactured.
(2) It may either be for the production of a single batch of product or for ongoing production of the products specified in the authorisation.
(3) If it is for a single batch it must be time limited.
(4) Only the products specified in the authorisation may be manufactured, and in the case of an authorisation for a single batch the product may only be manufactured before the expiry of the authorisation.
LabellingU.K.
16. The operator of the premises must ensure that every container containing autogenous vaccine is labelled with—
(a)the name of the veterinary surgeon who ordered the vaccine;
(b)a precise description of the vaccine;
(c)the date the vaccine was produced;
(d)the name of the authorisation holder and address of the authorised premises;
(e)the expiry date;
(f)any necessary warnings; and
(g)instructions for use.
RecordsU.K.
17. The operator of the premises must, as soon as is reasonably practicable, record—
(a)the name and address of the veterinary surgeon who ordered the vaccine;
(b)the identification of the source animal;
(c)the expiry date;
(d)the date of supply to the veterinary surgeon,
and must keep the records for at least five years.
Adverse reactionsU.K.
18. The authorised person must notify the Secretary of State of any adverse reactions to an autogenous vaccine within 15 days of learning of the reaction.
Inspection of premisesU.K.
19. The Secretary of State must inspect the authorised premises, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
PART 3U.K.Authorisation of blood banks
Authorisation of blood banksU.K.
20.—(1) The Secretary of State may authorise blood banks for—
(a)the collection, storage and supply of blood, or
(b)the storage and supply of blood constituents obtained by the physical separation of donor blood into different fractions within a closed-bag system,
for the treatment of non-food-producing animals.
(2) The authorisation may be for either or both of these activities.
(3) In order to be authorised a blood bank must be under the supervision of—
(a)a veterinary surgeon named in the authorisation; or
(b)a person named in the authorisation who the Secretary of State is satisfied is suitably qualified to operate the blood bank.
(4) Before authorising a blood bank, the Secretary of State must be satisfied—
(a)that the welfare of animals used in the collection of blood will be respected; and
(b)that the production process will produce a consistent, safe product.
(5) The Secretary of State may suspend, vary or revoke an authorisation of a blood bank if—
(a)the holder no longer uses fit and proper processes;
(b)the premises in which the blood bank is being or is to be operated are not suitable;
(c)the equipment is not suitable; or
(d)the holder has not complied with these Regulations.
(6) Blood may only be collected under the responsibility of a veterinary surgeon.
(7) No person may operate a blood bank for treatment of animals other than in accordance with such an authorisation.
Supply and administration of blood from a blood bankU.K.
21.—(1) The operator of a blood bank may only supply blood to a veterinary surgeon.
(2) No person other than a veterinary surgeon or someone acting under a veterinary surgeon’s responsibility may administer blood.
(3) No person may administer blood to a food-producing animal.
LabellingU.K.
22.—(1) The operator of a blood bank must ensure that every container used for the blood is labelled with—
(a)the identification of the donor animal;
(b)the date of collection;
(c)the authorisation number of the blood bank;
(d)any necessary warnings;
(e)the expiry date.
(2) There must be no specific therapeutic indication on the label or on any information relating to the product.
RecordsU.K.
23. The operator of a blood bank must, as soon as is reasonably practicable, record—
(a)the date of collection;
(b)the identification of the donor animal;
(c)the veterinary surgeon who collected it;
(d)the expiry date;
(e)the date the blood was used or, if it was supplied to another veterinary surgeon, the name of that veterinary surgeon and the date it was supplied;
and must keep the records for at least five years.
Inspection of premisesU.K.
24. The Secretary of State must inspect the authorised premises, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
PART 4U.K.Authorisation of manufacturers of products for administration under the cascade
Authorisation to manufacture products for administration under the cascadeU.K.
25.—(1) The Secretary of State may authorise a person and premises to manufacture an unauthorised veterinary medicinal product for administration under the cascade.
(2) In order to be authorised the premises must be under the supervision of a person who the Secretary of State is satisfied has sufficient qualifications and experience to manufacture the product safely.
(3) Before authorising the premises, the Secretary of State must be satisfied that the production process will produce a safe product.
(4) The authorisation must specify what types of product it covers.
(5) No person may manufacture an unauthorised veterinary medicinal product other than in accordance with an authorisation under sub-paragraph (1).
LabellingU.K.
26. The authorised person must ensure that, before a veterinary medicinal product is supplied, every container is labelled with—
(a)the name of the veterinary surgeon who ordered the veterinary medicinal product;
(b)a precise description of the veterinary medicinal product;
(c)the date of production;
(d)the name of the authorisation holder and the address of the authorised premises;
(e)the expiry date;
(f)any necessary warnings; and
(g)instructions for use.
RecordsU.K.
27. The authorised person must, as soon as is reasonably practicable, record—
(a)the name and address of the veterinary surgeon who ordered the veterinary medicinal product;
(b)a precise description of the veterinary medicinal product;
(c)the date of production;
(d)the expiry date; and
(e)the date of supply to the veterinary surgeon,
and must keep the record for at least five years.
Adverse reactionsU.K.
28. The authorised person must notify the Secretary of State of any adverse reactions to a product manufactured by that person within 15 days of learning of the reaction.
Inspection of premisesU.K.
29. The Secretary of State must inspect the authorised premises, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
PART 5U.K.Authorisation of equine stem cell centres
Authorisation of stem cell centresU.K.
30.—(1) The Secretary of State may authorise equine stem cell centres for the collection, storage, processing, production and administration of equine stem cells for use as an autologous treatment for horses.
(2) In order to be authorised a centre must be under the supervision of—
(a)a veterinary surgeon named in the authorisation; or
(b)a person named in the authorisation who the Secretary of State is satisfied is suitably qualified to operate the centre.
(3) Before authorising a centre, the Secretary of State must be satisfied—
(a)that the welfare of animals used in the collection of equine stem cells will be respected; and
(b)that the production process will produce a consistent, safe product.
(4) Equine stem cells may only be collected under the responsibility of a veterinary surgeon.
(5) The Secretary of State may suspend, vary or revoke an authorisation of an equine stem cell centre if—
(a)the holder no longer uses fit and proper processes;
(b)the premises in which the centre is being or is to be operated are not suitable;
(c)the equipment of the centre is not suitable; or
(d)the holder has not complied with these Regulations.
(6) No person may operate an equine stem cell centre other than in accordance with such an authorisation.
Supply and administration of stem cellsU.K.
31.—(1) The operator of an equine stem cell centre may only collect equine stem cells.
(2) The operator of an equine stem cell centre may not collect stem cells from embryonic tissues.
(3) No person other than a veterinary surgeon or someone acting under a veterinary surgeon’s responsibility may administer any product grown from collected equine stem cells.
(4) No person may administer any product grown from collected equine stem cells to a food-producing horse.
LabellingU.K.
32.—(1) The operator of an equine stem cell centre must ensure that every container used for the stem cell product is labelled with—
(a)the identification of the donor animal;
(b)the date of collection;
(c)the authorisation number of the equine stem cell centre;
(d)any necessary warnings;
(e)the expiry date.
(2) The operator of an equine stem cell centre must ensure that no specific therapeutic indication is included on the label or on any information relating to the product.
RecordsU.K.
33. The operator of an equine stem cell centre must, as soon as is reasonably practicable, record for each stem cell product—
(a)the identification of the donor animal;
(b)the date of collection;
(c)the veterinary surgeon under whose responsibility the stem cells were collected;
(d)the expiry date;
(e)the date the product was used or, if it was supplied to another veterinary surgeon, the name of that veterinary surgeon and the date it was supplied,
and must keep the records for at least five years.
Inspection of premisesU.K.
34. The Secretary of State must inspect the authorised premises, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
OffencesU.K.
35. It is an offence to fail to comply with—
(a)paragraph 4(3);
(b)paragraph 11;
(c)paragraph 14(4);
(d)paragraph 16;
(e)paragraph 17;
(f)paragraph 18;
(g)paragraph 20(6) or (7);
(h)paragraph 21;
(i)paragraph 22;
(j)paragraph 23;
(k)paragraph 25(5);
(l)paragraph 26;
(m)paragraph 27;
(n)paragraph 28;
(o)paragraph 30(4) or (6);
(p)paragraph 31;
(q)paragraph 32; or
(r)paragraph 33.
Regulation 7
SCHEDULE 3U.K.Classification and supply, wholesale dealers and sheep dip
PART 1 Classification and supply of authorised veterinary medicinal products
1.Classification of veterinary medicinal products
2.Wholesale supply of veterinary medicinal products
3.Retail supply of veterinary medicinal products
4.Prescriptions by a veterinary surgeon
5.Prescriptions
6.Written prescriptions
7.Duties when a product is prescribed or supplied
8.Supply by a veterinary surgeon from registered premises
9.Supply by a veterinary surgeon
10.Supply by a pharmacist
11.Supply of a veterinary medicinal product for incorporation into feedingstuffs
12.Labelling at the time of retail supply
13.Supply of veterinary medicinal products for use under the cascade
14.Supply by a suitably qualified person
15.Annual audit
PART 2 Requirements for a wholesale dealer’s authorisation
16.Application
17.Time limits
18.Granting the authorisation
19.The authorisation
20.Suspension, variation or revocation of the authorisation
21.Duties on the holder of a wholesale dealer’s authorisation
PART 3 Sheep dip
22.Supply of sheep dip
23.Use of sheep dip
24.Offences
PART 1U.K.Classification and supply of authorised veterinary medicinal products
Classification of veterinary medicinal productsU.K.
1.—(1) There shall be the following categories of authorised veterinary medicinal products—
(a)Prescription Only Medicine–Veterinarian (abbreviated to POM-V);
(b)Prescription Only Medicine–Veterinarian, Pharmacist, Suitably Qualified Person (abbreviated to POM-VPS);
(c)Non-Food Animal–Veterinarian, Pharmacist, Suitably Qualified Person (abbreviated to NFA-VPS);
(d)Authorised Veterinary Medicine–General Sales List (abbreviated to AVM-GSL).
(2) The Secretary of State must specify the classification of the veterinary medicinal product when granting the initial marketing authorisation.
(3) The Secretary of State may change the classification after the marketing authorisation has been granted, either at the request of the marketing authorisation holder or in accordance with paragraph 37 of Schedule 1 (compulsory variation).
(4) When granting the marketing authorisation the Secretary of State must classify the following as POM-V—
(a)products containing narcotic or psychotropic substances;
(b)products intended for administration following a diagnosis or clinical assessment by a veterinary surgeon.
(5) When granting the marketing authorisation the Secretary of State must classify the following as POM-V or POM-VPS—
(a)products for food-producing animals;
(b)products in respect of which special precautions must be taken in order to avoid any unnecessary risk to—
(i)the target species;
(ii)the person administering the products to the animal; and
(iii)the environment;
(c)products that may cause effects that impede or interfere with subsequent diagnostic or therapeutic measures; and
(d)new veterinary medicinal products containing an active substance that has not been included in an authorised veterinary medicinal product for five years.
(6) The requirement in sub-paragraph (5)(a) relating to veterinary medicinal products for food-producing animals does not apply if all the following criteria are met—
(a)the administration of the veterinary medicinal product is restricted to formulations requiring no particular knowledge or skill in using the product;
(b)the veterinary medicinal product does not present a direct or indirect risk, even if administered incorrectly, to the animal or animals treated, to the person administering the product or to the environment;
(c)the summary of product characteristics of the veterinary medicinal product does not contain any warnings of potential serious side effects deriving from its correct use;
(d)neither the veterinary medicinal product nor any other product containing the same active substance has previously been the subject of frequent serious adverse reaction reporting;
(e)the summary of product characteristics does not refer to contra-indications related to other veterinary medicinal products commonly used without prescription;
(f)the veterinary medicinal product is not subject to special storage conditions;
(g)there is no risk for consumer safety as regards residues in food obtained from treated animals even where the veterinary medicinal products are used incorrectly; and
(h)there is no risk to human or animal health as regards the development of resistance to antimicrobials or anthelmintic substances even where the veterinary medicinal products containing those substances are used incorrectly.
Wholesale supply of veterinary medicinal productsU.K.
2.—(1) Only a holder of a marketing authorisation, the holder of a manufacturing authorisation or the holder of a wholesale dealer’s authorisation granted by the Secretary of State may supply a veterinary medicinal product wholesale, or be in possession of it for that purpose.
(2) A person mentioned in sub-paragraph (1) may only supply a veterinary medicinal product if—
(a)the authorisation in question relates to that product, and
(b)the supply is to another person who is entitled to supply that product under these Regulations, either wholesale or retail.
(3) If the supply is to a suitably qualified person, it must be to the premises approved in accordance with paragraph 14.
(4) It is immaterial whether or not the supply is for profit.
(5) This paragraph does not apply in relation to a retailer of veterinary medicinal products who supplies another retailer with such products for the purpose of alleviating a temporary supply shortage that could be detrimental to animal welfare.
(6) A wholesale dealer may break open any package (other than the immediate packaging) of a veterinary medicinal product.
Retail supply of veterinary medicinal productsU.K.
3.—(1) This paragraph applies in relation to retail supply of veterinary medicinal products.
(2) A veterinary medicinal product classified as POM-V may only be supplied by a veterinary surgeon or a pharmacist and must be supplied in accordance with a prescription from a veterinary surgeon.
(3) A veterinary medicinal product classified as POM-VPS may only be supplied by—
(a)a veterinary surgeon;
(b)a pharmacist; or
(c)a suitably qualified person in accordance with paragraph 14,
and must be in accordance with a prescription from one of those persons.
(4) A veterinary medicinal product classified as NFA-VPS may be supplied without prescription, but may only be supplied by—
(a)a veterinary surgeon;
(b)a pharmacist; or
(c)a suitably qualified person in accordance with paragraph 14.
(5) There are no restrictions on the supply of AVM-GSL products.
(6) In this paragraph—
(a)“retail supply” means any supply other than to or from the holder of a wholesale dealer’s authorisation, and whether or not for payment; and
(b)a person may supply a product irrespective of who owns it.
Prescriptions by a veterinary surgeonU.K.
4.—(1) A veterinary surgeon who prescribes a veterinary medicinal product classified as POM-V must first carry out a clinical assessment of the animal, and the animal must be under that veterinary surgeon’s care.
(2) This does not apply in relation to the administration of such a product to a wild animal where the administration is authorised by the Secretary of State.
PrescriptionsU.K.
5.—(1) A prescription may be oral or written, but a veterinary medicinal product classified as POM-V or POM-VPS may only be supplied—
(a)by the person who prescribed it;
(b)under a written prescription that complies with paragraph 6; or
(c)(in the case of POM-VPS) by a suitably qualified person in accordance with paragraph 14(5).
(2) A person supplying such a product under a written prescription—
(a)may only supply the product specified in that prescription;
(b)must take all reasonable steps to be satisfied that the prescription has been written and signed by a person entitled to prescribe the product; and
(c)must take all reasonable steps to ensure that it is supplied to the person named in the prescription.
(3) No person may alter a written prescription unless authorised to do so by the person who signed it.
Written prescriptionsU.K.
6.—(1) A written prescription must include—
(a)the name, address and telephone number of the person prescribing the product;
(b)the qualifications enabling the person to prescribe the product;
(c)the name and address of the owner or keeper;
(d)the identification (including the species) of the animal or group of animals to be treated;
(e)the premises at which the animals are kept if this is different from the address of the owner or keeper;
(f)the date of the prescription;
(g)the signature or other authentication of the person prescribing the product;
(h)the name and amount of the product prescribed;
(i)the dosage and administration instructions;
(j)any necessary warnings;
(k)the withdrawal period if relevant; and
(l)if it is prescribed under the cascade, a statement to that effect.
(2) A written prescription for a controlled drug as specified in Schedules 2 to 4 of the Misuse of Drugs Regulations 2001(32) is valid for 28 days.
(3) A written prescription for any other drug is valid for six months or such shorter period as may be specified in the prescription.
(4) If the prescription is repeatable it must specify the number of times the veterinary medicinal product may be supplied.
Duties when a product is prescribed or suppliedU.K.
7. A person who prescribes a product classified as POM-V or POM-VPS, or supplies a product classified as NFA-VPS—
(a)before doing so, must be satisfied that the person who will use the product is competent to do so safely, and intends to use it for a purpose for which it is authorised;
(b)when doing so, must advise on its safe administration and on any warnings or contra-indications on the label or package leaflet; and
(c)must not prescribe (or, in the case of a NFA-VPS product, supply) more than the minimum amount required for the treatment; but it is a defence to a charge of failing to comply with this paragraph to show that—
(i)the product prescribed or supplied was in a container specified in the marketing authorisation;
(ii)the manufacturer does not supply that veterinary medicinal product in a smaller container; and
(iii)the person prescribing or supplying is not a person authorised to break open the package before supply.
Supply by a veterinary surgeon from registered premisesU.K.
8.—(1) A veterinary surgeon may only supply a veterinary medicinal product from practice premises registered with the Royal College of Veterinary Surgeons as veterinary practice premises at which veterinary medicinal products are stored or supplied.
(2) This paragraph does not apply in relation to a veterinary medicinal product classified as AVM-GSL.
(3) The Royal College of Veterinary Surgeons must, on request, supply the Secretary of State with a copy of the register of veterinary practice premises.
(4) The Secretary of State must, from time to time, inspect premises registered under sub-paragraph (1), basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
(5) Where an inspection under sub-paragraph (4) reveals significant breaches of these Regulations the Secretary of State may require the Royal College of Veterinary Surgeons to remove the premises from the register maintained under sub-paragraph (1).
(6) Where the Secretary of State requires the removal of premises from the register the veterinary surgeon concerned may appeal using the procedure in regulation 30.
(7) Where premises have been removed from the register under sub-paragraph (5) they may not be re-registered without the approval of the Secretary of State.
(8) The Secretary of State may only grant approval under sub-paragraph (7) after a further inspection of the premises.
Supply by a veterinary surgeonU.K.
9.—(1) A veterinary surgeon supplying a veterinary medicinal product (other than one classified as AVM-GSL) must be present when it is handed over unless the veterinary surgeon—
(a)authorises each transaction individually before the product is supplied; and
(b)is satisfied that the person handing it over is competent to do so.
(2) A veterinary surgeon or a person acting under a veterinary surgeon’s responsibility may open any package containing a veterinary medicinal product.
Supply by a pharmacistU.K.
10.—(1) A pharmacist may only supply a veterinary medicinal product classified as POM-V, POM-VPS or NFA-VPS from—
(a)premises registered as a pharmacy with the General Pharmaceutical Council or with the Pharmaceutical Society of Northern Ireland;
(b)premises registered with the Royal College of Veterinary Surgeons as being premises from which a veterinary surgeon supplies veterinary medicinal products; or
(c)(in the case of a veterinary medicinal product classified as POM-VPS or NFA-VPS) from premises approved under paragraph 14.
(2) A pharmacist supplying a veterinary medicinal product (other than one classified as AVM-GSL) must be present when it is handed over unless the pharmacist—
(a)authorises each transaction individually before the product is supplied; and
(b)is satisfied that the person handing it over is competent to do so.
(3) A pharmacist may supply any veterinary medicinal product prepared in a pharmacy in accordance with the prescriptions of a pharmacopoeia and intended to be supplied directly to the end-user.
(4) A pharmacist may supply a homeopathic remedy prepared extemporaneously by a pharmacist in a registered pharmacy (as well as any other homeopathic remedy permitted to be supplied by a pharmacist under these Regulations) provided that it is prepared in accordance with paragraph 63 of Schedule 1 and intended to be supplied directly to the end user.
(5) A pharmacist may break open any package containing a veterinary medicinal product for the purposes of supply other than the immediate packaging of an injectable product.
Supply of a veterinary medicinal product for incorporation into feedingstuffsU.K.
11.—(1) This paragraph applies in relation to the supply of a veterinary medicinal product intended for incorporation into feedingstuffs.
(2) The marketing authorisation holder, an authorised manufacturer of the product or an authorised wholesale dealer may only supply such a veterinary medicinal product to—
(a)a veterinary surgeon, pharmacist or, in the case of a product classified as POM-VPS, a suitably qualified person;
(b)an approved premixture manufacturer; or
(c)an approved feedingstuffs manufacturer if the approval permits the rate of incorporation specified on the label of that veterinary medicinal product (if the manufacturer is the end-user the supply must be in accordance with a prescription).
(3) A veterinary surgeon, pharmacist or, in the case of a product classified as POM-VPS, a suitably qualified person may only supply such a veterinary medicinal product to—
(a)an approved premixture manufacturer; or
(b)an approved feedingstuffs manufacturer if the approval permits the rate of incorporation specified on the label of that veterinary medicinal product (if the manufacturer is the end user the supply must be in accordance with a prescription).
(4) An approved premixture manufacturer or an approved feedingstuffs manufacturer may only supply such a veterinary medicinal product to another approved premixture manufacturer or approved feedingstuff manufacturer if the amount supplied does not exceed five per cent in terms of value of veterinary medicinal product incorporated annually by the person supplying the veterinary medicinal product.
Labelling at the time of retail supplyU.K.
12.—(1) If a veterinary medicinal product is supplied in a container specified in the marketing authorisation, it must not be supplied if any information on the outer packaging (or, if there is no outer packaging, the immediate packaging) is not clearly visible at the time of supply or has been changed in any way.
(2) Sub-paragraph (1) does not apply to a veterinary surgeon who amends a label, or a pharmacist who amends it in accordance with a prescription from a veterinary surgeon, provided that the unamended information remains clearly visible.
(3) If a veterinary medicinal product is supplied in a container other than that specified in the marketing authorisation, the person supplying the veterinary medicinal product must ensure that the container is suitably labelled and must supply sufficient written information (which may include a copy of the summary of product characteristics or the package leaflet) to enable the product to be used safely.
Supply of veterinary medicinal products for use under the cascadeU.K.
13.—(1) A veterinary medicinal product supplied for administration under the cascade may only be supplied in accordance with a prescription from a veterinary surgeon.
(2) Unless the veterinary surgeon who prescribed the veterinary medicinal product both supplies the product and administers it to the animal in person, the person supplying it must label it (or ensure that it is labelled) with at least the following information—
(a)the name and address of the pharmacy, veterinary surgery or approved premises supplying the veterinary medicinal product;
(b)the name of the veterinary surgeon who has prescribed the product;
(c)the name and address of the animal owner;
(d)the identification (including the species) of the animal or group of animals;
(e)the date of supply;
(f)the expiry date of the product, if applicable;
(g)the name or description of the product, which should include at least the name and quantity of active ingredients;
(h)dosage and administration instructions;
(i)any special storage precautions;
(j)any necessary warnings for the user, target species, administration or disposal of the product;
(k)the withdrawal period, if relevant; and
(l)the words “Keep out of reach of children” and “For animal treatment only”.
Supply by a suitably qualified personU.K.
14.—(1) The Secretary of State may recognise bodies that are suitable to maintain a register for suitably qualified persons to prescribe and supply veterinary medicinal products classified as POM-VPS and NFA-VPS.
(2) In order to recognise such a body, the Secretary of State must be satisfied that the body—
(a)has in place a system for ensuring that persons applying for registration have adequate training to act as a suitably qualified person under these Regulations;
(b)has adequate standards in deciding whether or not to register someone as a suitably qualified person;
(c)maintains a programme of continuing professional development for persons registered with it;
(d)operates an adequate appeal system if it intends to refuse to register anyone with appropriate qualifications or to remove anyone from the register.
(3) For the purposes of these Regulations, a suitably qualified person is a person who has passed examinations specified by such a body, and is registered with such a body as a suitably qualified person.
(4) A suitably qualified person may only supply a veterinary medicinal product classified as POM-VPS, NFA-VPS or AVM-GSL, and may only supply it from—
(a)premises approved by the Secretary of State as being suitable for the storage and supply of veterinary medicinal products by a suitably qualified person;
(b)premises registered as a pharmacy with the General Pharmaceutical Council or with the Pharmaceutical Society of Northern Ireland; or
(c)practice premises registered under these Regulations as being premises from which a veterinary surgeon supplies veterinary medicinal products.
(5) A suitably qualified person who supplies a product classified as POM-VPS or NFA-VPS must either—
(a)hand over or despatch the product personally;
(b)ensure that, when the product is handed over or despatched, the suitably qualified person is in a position to intervene if necessary; or
(c)check the product after it has been allocated for supply to a customer, and be satisfied that the person handing over or dispatching it is competent to do so.
(6) A suitably qualified person supplying products from premises approved under this regulation by the Secretary of State who considers that the premises no longer comply with the approval must notify the Secretary of State without unreasonable delay.
(7) The Secretary of State may issue a Code of Practice for suitably qualified persons, and a body recognised under this paragraph must take appropriate action in accordance with any disciplinary code that applies to that body if a suitably qualified person registered with it does not comply with the Code of Practice.
(8) The Secretary of State must publish a list of—
(a)suitably qualified persons; and
(b)the trading names and the addresses of premises approved under this paragraph(33).
(9) A suitably qualified person may break open any package (other than the immediate packaging) of a veterinary medicinal product.
(10) The Secretary of State may suspend or revoke the approval of approved premises on being satisfied that they are no longer suitable for the storage and supply of veterinary medicinal products.
Annual auditU.K.
15. At least once a year every person entitled to supply a veterinary medicinal product on prescription must carry out a detailed audit, and incoming and outgoing veterinary medicinal products must be reconciled with products currently held in stock, any discrepancies being recorded.
PART 2U.K.Requirements for a wholesale dealer’s authorisation
ApplicationU.K.
16. An application for a wholesale dealer’s authorisation must be made to the Secretary of State.
Time limitsU.K.
17. The Secretary of State must process an application for a wholesale dealer’s authorisation within 90 days of receiving it.
Granting the authorisationU.K.
18.—(1) The Secretary of State must grant a wholesale dealer’s authorisation on being satisfied that this paragraph is complied with.
(2) The authorised site must be—
(a)weatherproof;
(b)secure and lockable;
(c)clean; and
(d)free from contaminants.
(3) If the veterinary medicinal products covered by the authorisation are subject to specific storage conditions, the site must be capable of fulfilling those requirements.
(4) The authorisation holder must—
(a)have the services of technically competent staff; and
(b)have an effective emergency recall plan.
The authorisationU.K.
19.—(1) The wholesale dealer’s authorisation must specify—
(a)the types of veterinary medicinal products and pharmaceutical forms that may be dealt in;
(b)the place where they are to be stored;
(c)the name and address of the person holding the authorisation;
(d)the address of the premises to which it relates;
(e)the name of the qualified person nominated to act under the Guidelines on Good Distribution Practice for Human Use(34).
(2) It may cover more than one site.
(3) It lapses if the holder does not deal in veterinary medicinal products for five years.
(4) The holder of a wholesale dealer’s authorisation must notify the Secretary of State, and if necessary apply for a variation of the authorisation, before making a material alteration to the premises or facilities used under the authorisation, or in the operations for which they are used.
Suspension, variation or revocation of the authorisationU.K.
20. The Secretary of State may suspend, vary or revoke a wholesale dealer’s authorisation if the holder—
(a)has not complied with these Regulations; or
(b)no longer has suitable premises or equipment.
Duties on the holder of a wholesale dealer’s authorisationU.K.
21. The holder of a wholesale dealer’s authorisation must—
(a)store veterinary medicinal products in accordance with the terms of the marketing authorisation for each product;
(b)comply with the Guidelines on Good Distribution Practice of Medicinal Products for Human Use as if the veterinary medicinal products were authorised human medicinal products;
(c)carry out a detailed stock audit at least once a year; and
(d)supply information and samples to the Secretary of State on demand.
PART 3U.K.Sheep dip
Supply of sheep dipU.K.
22.—(1) A person who supplies by retail sheep dip which contains a veterinary medicinal product must supply it in accordance with this paragraph.
(2) The supply must be to a person (or a person acting on that person’s behalf) who is qualified to use it in accordance with paragraph 23.
(3) The supplier must make a record of that person’s certificate or award number as soon as is reasonably practicable, and keep it for at least three years.
(4) If the active ingredient of the veterinary medicinal product is an organophosphorus compound, the supplier must give to the buyer—
(a)a double-sided laminated notice meeting the specifications in the following sub-paragraph (unless the notice has been provided to the buyer within the previous twelve months and the supplier knows or has reasonable cause to believe that the buyer still has it available for use); and
(b)two pairs of gloves either as described in the notice or providing demonstrably superior protection to the proposed user against exposure to the dip than would be provided by gloves as so described.
(5) The notice must be at least A4 size with a laminated transparent cover and must tell the user of the sheep dip—
(a)to read and act in accordance with the label, including instructions on measuring and diluting concentrate;
(b)that sheep dip is absorbed through the skin;
(c)always to wear the recommended protective clothing, including gloves, and have spare protective clothing available;
(d)always to wash protective clothing before taking it off; and
(e)to direct any questions to the supplier or manufacturer.
(6) The notice must contain a diagram showing recommended protective clothing.
Use of sheep dipU.K.
23.—(1) No person may use sheep dip which contains a veterinary medicinal product unless the person is acting under the supervision and in the presence of, a person who holds either—
(a)a Certificate of Competence in the Safe Use of Sheep Dips showing that Parts 1 and 2 or units 1 and 2 of the assessment referred to in the Certificate have been satisfactorily completed; or
(b)NPTC Level 2 Award in the Safe Use of Sheep Dip (QCF).
(2) The certificate must be issued—
(a)in England, Wales and Northern Ireland; by—
(i)the National Proficiency Tests Council;
(ii)NPTC Part of the City & Guilds Group; or
(iii)City and Guilds NPTC;
(b)in Scotland, by one of those organisations or the Scottish Skills Testing Service.
OffencesU.K.
24. It is an offence to fail to comply with—
(a)paragraph 2;
(b)paragraph 3;
(c)paragraph 4(1);
(d)paragraph 5;
(e)paragraph 7;
(f)paragraph 8(1);
(g)paragraph 9(1);
(h)paragraph 10;
(i)paragraph 11;
(j)paragraph 12(1) or (3);
(k)paragraph 13;
(l)paragraph 14(4), (5) or (6);
(m)paragraph 15;
(n)paragraph 19(4);
(o)paragraph 21;
(p)paragraph 22; or
(q)paragraph 23(1).
Regulation 8
SCHEDULE 4U.K.Administration of a veterinary medicinal product outside the terms of a marketing authorisation
1.Administration under the cascade
2.Withdrawal periods
3.Administration to food-producing horses
4.Immunological products for serious epizootic disease
5.Immunological products for an imported or exported animal
6.Administration by veterinary surgeons from other member States
7.Treatment in exceptional circumstances
8.Administration of a homeopathic remedy
9.Administration under an animal test certificate
10.Offences
Administration under the cascadeE+W+S
1.—(1) A veterinary surgeon acting under this paragraph who prescribes a veterinary medicinal product may either administer it personally or may direct another person to do so under the responsibility of the veterinary surgeon.
(2) If there is no authorised veterinary medicinal product in the United Kingdom for a condition the veterinary surgeon responsible for the animal may, in particular to avoid unacceptable suffering, treat the animal concerned with the following (“the cascade”), cascaded in the following order—
(a)a veterinary medicinal product authorised in the United Kingdom for use with another animal species, or for another condition in the same species; or
(b)if there is no such product that is suitable, either—
(i)a human medicinal product authorised in the United Kingdom; or
(ii)a veterinary medicinal product not authorised in the United Kingdom but authorised in another [F90country] for use with any animal species (in the case of a food-producing animal, it must be a food-producing species); or
(c)if there is no such product that is suitable, a veterinary medicinal product prepared extemporaneously by a pharmacist, a veterinary surgeon or a person holding a manufacturing authorisation authorising the manufacture of that type of product.
(3) In the case of a veterinary medicinal product imported from another [F91country], if the veterinary surgeon has not obtained a certificate from the Secretary of State under regulation 25(5) permitting importation, the veterinary surgeon must obtain a certificate from the Secretary of State before administration.
(4) Any pharmacologically active substances included in a medicinal product administered to a food-producing animal under the cascade must [F92be substances for which a maximum residue limit has been established under Regulation (EC) No 470/2009 of the European Parliament and of the Council].
Extent Information
E41This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F90Word in Sch. 4 para. 1(2)(b)(ii) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(a)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F91Word in Sch. 4 para. 1(3) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(a)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F92Words in Sch. 4 para. 1(4) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(a)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Administration under the cascadeN.I.
1.—(1) A veterinary surgeon acting under this paragraph who prescribes a veterinary medicinal product may either administer it personally or may direct another person to do so under the responsibility of the veterinary surgeon.
(2) If there is no authorised veterinary medicinal product in [F229Northern Ireland] for a condition the veterinary surgeon responsible for the animal may, in particular to avoid unacceptable suffering, treat the animal concerned with the following (“the cascade”), cascaded in the following order—
(a)a veterinary medicinal product authorised in [F229Northern Ireland] for use with another animal species, or for another condition in the same species; or
(b)if there is no such product that is suitable, either—
(i)a human medicinal product authorised in the United Kingdom; or
(ii)a veterinary medicinal product not authorised in [F229Northern Ireland] but authorised in another member State for use with any animal species (in the case of a food-producing animal, it must be a food-producing species); or
(c)if there is no such product that is suitable, a veterinary medicinal product prepared extemporaneously by a pharmacist, a veterinary surgeon or a person holding a manufacturing authorisation authorising the manufacture of that type of product.
(3) In the case of a veterinary medicinal product imported from another member State, if the veterinary surgeon has not obtained a certificate from the Secretary of State under regulation 25(5) permitting importation, the veterinary surgeon must obtain a certificate from the Secretary of State before administration.
(4) Any pharmacologically active substances included in a medicinal product administered to a food-producing animal under the cascade must be listed in Table 1 in the Annex to Commission Regulation (EU) No 37/2010.
Extent Information
E113This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F229Words in Sch. 4 para. 1 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(a)
Withdrawal periodsE+W+S
2.—(1) A veterinary surgeon prescribing or administering a veterinary medicinal product to a food-producing animal under the cascade must specify an appropriate withdrawal period.
(2) The withdrawal period must ensure that, if there is a maximum residue limit [F93established for the active substance under Regulation (EC) No 470/2009 of the European Parliament and of the Council] , the level of residue of the active substance does not exceed that limit.
(3) In any event, unless the Secretary of State has specified in writing a different withdrawal period for a particular veterinary medicinal product, the withdrawal period (irrespective of whether or not a maximum residue limit [F94has been established]) must not be less than—
(a)7 days for eggs;
(b)7 days for milk;
(c)28 days for meat from poultry and mammals including fat and offal;
(d)500 degree days(35) for fish meat.
Extent Information
E42This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F93Words in Sch. 4 para. 2(2) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(b)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F94Words in Sch. 4 para. 2(3) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Withdrawal periodsN.I.
2.—(1) A veterinary surgeon prescribing or administering a veterinary medicinal product to a food-producing animal under the cascade must specify an appropriate withdrawal period.
(2) The withdrawal period must ensure that, if there is a maximum residue limit specified for the active substance in Table 1 in the Annex to Commission Regulation (EU) No 37/2010, the level of residue of the active substance does not exceed that limit.
(3) In any event, unless the Secretary of State has specified in writing a different withdrawal period for a particular veterinary medicinal product, the withdrawal period (irrespective of whether or not a maximum residue limit is specified in Table 1 in the Annex to Commission Regulation (EU) No 37/2010) must not be less than—
(a)7 days for eggs;
(b)7 days for milk;
(c)28 days for meat from poultry and mammals including fat and offal;
(d)500 degree days(35) for fish meat.
Extent Information
E114This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Administration to food-producing horsesU.K.
3.—(1) If there is no authorised veterinary medicinal product for a food-producing horse (as shown on its horse passport) and treatment under the cascade is unsuitable, substances may be administered in accordance with Commission Regulation (EC) No 122/2013 (establishing, in accordance with Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products, a list of substances essential for the treatment of equidae (36)).
(2) The person administering the substance must comply with Article 3(2) of Commission Regulation (EC) No 122/2013 (recording the details of the treatment in the animal’s passport).
Immunological products for serious epizootic diseaseE+W+S
4. In the event of serious epizootic diseases, the Secretary of State may permit in writing the administration of immunological veterinary medicinal products without a marketing authorisation, in the absence of a suitable medicinal product F95... and may publicise any permit as the Secretary of State sees fit.
Extent Information
E43This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F95Words in Sch. 4 para. 4 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Immunological products for serious epizootic diseaseN.I.
4. In the event of serious epizootic diseases, the Secretary of State may permit in writing the administration of immunological veterinary medicinal products without a marketing authorisation, in the absence of a suitable medicinal product and after informing the Commission of the detailed conditions of use and may publicise any permit as the Secretary of State sees fit.
Extent Information
E115This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Immunological products for an imported or exported animalE+W+S
5. If an animal is imported from, or exported to, [F96another] country, the Secretary of State may permit the administration to that animal of an immunological veterinary medicinal product that is not covered by a marketing authorisation in the United Kingdom but is authorised under the legislation of [F97that other] country.
Extent Information
E44This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F96Word in Sch. 4 para. 5 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(d)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F97Words in Sch. 4 para. 5 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(d)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Immunological products for an imported or exported animalN.I.
5. If an animal is imported from, or exported to, a third country, the Secretary of State may permit the administration to that animal of an immunological veterinary medicinal product that is not covered by a marketing authorisation in [F230Northern Ireland] but is authorised under the legislation of the third country.
Extent Information
E116This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F230Words in Sch. 4 para. 5 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(b)
Administration by veterinary surgeons from other [F98countries] E+W+S
6.—(1) Veterinary surgeons practising in another [F99country with equivalent medicines regulation standards to those of the United Kingdom] may bring into the United Kingdom and administer to animals small quantities of veterinary medicinal products that are not authorised for use in the United Kingdom if—
(a)the quantity does not exceed the requirements for the treatment of specific animals;
(b)the product is authorised in the [F100country] in which the veterinary surgeon is established;
(c)the product is transported by the veterinary surgeon in the original manufacturer’s packaging;
(d)in the case of administration to food-producing animals, there is a veterinary medicinal product authorised in the United Kingdom that has the same qualitative and quantitative composition in terms of active substances;
(e)the veterinary surgeon is acquainted with the Code of Professional Conduct for veterinary surgeons issued by the Royal College of Veterinary Surgeons(37).
(2) The veterinary surgeon must only supply to the owner or keeper enough veterinary medicinal product to complete the treatment of animals concerned.
(3) The veterinary surgeon must—
(a)ensure that the withdrawal period specified on the label of the product is complied with, or the United Kingdom withdrawal period for the equivalent product authorised in the United Kingdom if this is longer than the one on the label; and
(b)keep detailed records of the animals treated, the diagnosis or clinical assessment, the products administered, the dosage administered, the duration of treatment and the withdrawal period applied, and must keep them in the United Kingdom for at least three years.
(4) The overall range and quantity of veterinary medicinal products carried by the veterinary surgeon must not exceed that generally required for the daily needs of good veterinary practice.
(5) This paragraph does not apply in relation to immunological veterinary medicinal products.
Extent Information
E45This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F98Word in Sch. 4 para. 6 heading substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(e)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F99Words in Sch. 4 para. 6(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(e)(ii)(aa) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F100Word in Sch. 4 para. 6(1)(b) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(e)(ii)(bb) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Administration by veterinary surgeons from F231... member StatesN.I.
6.—(1) Veterinary surgeons practising in [F232a] member State may bring into [F233Northern Ireland] and administer to animals small quantities of veterinary medicinal products that are not authorised for use in [F233Northern Ireland] if—
(a)the quantity does not exceed the requirements for the treatment of specific animals;
(b)the product is authorised in the member State in which the veterinary surgeon is established;
(c)the product is transported by the veterinary surgeon in the original manufacturer’s packaging;
(d)in the case of administration to food-producing animals, there is a veterinary medicinal product authorised in [F234Northern Ireland] that has the same qualitative and quantitative composition in terms of active substances;
(e)the veterinary surgeon is acquainted with the Code of Professional Conduct for veterinary surgeons issued by the Royal College of Veterinary Surgeons(37).
(2) The veterinary surgeon must only supply to the owner or keeper enough veterinary medicinal product to complete the treatment of animals concerned.
(3) The veterinary surgeon must—
[F235(a)ensure that the withdrawal period specified on the label of the product is complied with, or the Northern Ireland withdrawal period for the equivalent product authorised in Northern Ireland if this is longer than the one on the label; and]
(b)keep detailed records of the animals treated, the diagnosis or clinical assessment, the products administered, the dosage administered, the duration of treatment and the withdrawal period applied, and must keep them in [F236Northern Ireland] for at least three years.
(4) The overall range and quantity of veterinary medicinal products carried by the veterinary surgeon must not exceed that generally required for the daily needs of good veterinary practice.
(5) This paragraph does not apply in relation to immunological veterinary medicinal products.
Extent Information
E117This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F231Word in Sch. 4 para. 6 heading omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(i)
F232Word in Sch. 4 para. 6(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(ii)
F233Words in Sch. 4 para. 6(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(iii)
F234Words in Sch. 4 para. 6(1)(d) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(iii)
F235Sch. 4 para. 6(3)(a) substituted (N.I) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(iv)
F236Words in Sch. 4 para. 6(3)(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(iii)
Treatment in exceptional circumstancesE+W+S
7.—(1) Where the health situation so requires, and where there is no suitable veterinary medicinal product available either as an authorised product or under the cascade, a veterinary surgeon may treat an animal with a medicinal product authorised in [F101another] country; but a veterinary surgeon who has not obtained a certificate from the Secretary of State under regulation 25(5) permitting importation must obtain a certificate from the Secretary of State before treating the animal.
(2) The certificate may be granted subject to any condition the Secretary of State thinks fit.
Extent Information
E46This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F101Word in Sch. 4 para. 7(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(f) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Treatment in exceptional circumstancesN.I.
7.—(1) Where the health situation so requires, and where there is no suitable veterinary medicinal product available either as an authorised product or under the cascade, a veterinary surgeon may treat an animal with a medicinal product authorised in a third country; but a veterinary surgeon who has not obtained a certificate from the Secretary of State under regulation 25(5) permitting importation must obtain a certificate from the Secretary of State before treating the animal.
(2) The certificate may be granted subject to any condition the Secretary of State thinks fit.
Extent Information
E118This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Administration of a homeopathic remedyU.K.
8.—(1) A registered homeopathic remedy or a homeopathic remedy prepared and supplied by a pharmacist under paragraph 10 of Schedule 3 may be administered to an animal by anyone, subject to any restrictions specified in its registration.
(2) A homeopathic remedy that was on the market before 1st January 1994 may be administered by anyone.
(3) A veterinary surgeon may administer, either personally or under the veterinary surgeon’s responsibility—
(a)a homeopathic remedy authorised for human use, or
(b)a homeopathic remedy prepared extemporaneously by a veterinary surgeon, a pharmacist or a person holding a manufacturing authorisation authorising the manufacture of that type of product.
Administration under an animal test certificateU.K.
9.—(1) A medicinal product may be administered in accordance with an animal test certificate granted for research purposes by the Secretary of State.
(2) An application for an animal test certificate may be refused if this is necessary for the protection of animal or public health or the environment, and the animal test certificate may be varied, suspended or revoked in the same way as a marketing authorisation.
(3) The holder of an animal test certificate may not supply a product for administration that is not within the terms of the animal test certificate.
(4) The holder of an animal test certificate test who becomes aware of any serious adverse reaction following the administration of a product under an animal test certificate must report the reaction to the Secretary of State within 15 days of learning of it.
OffencesU.K.
10. It is an offence to fail to comply with—
(a)paragraph 3(2);
(b)paragraph 6; or
(c)paragraph 9(3) or (4).
Regulation 14
SCHEDULE 5U.K.Medicated feedingstuffs and specified feed additives
Scope and interpretationU.K.
1.—(1) This Schedule applies in relation to the following (referred to in this Schedule as “specified feed additives”) when used as feed additives—
(a)coccidiostats;
(b)histomonostats; and
(c)all other zootechnical additives except—
(i)digestibility enhancers;
(ii)gut flora stabilisers; and
(iii)substances incorporated with the intention of favourably affecting the environment.
(2) It also applies in relation to the manufacture and placing on the market of feedingstuffs containing a veterinary medicinal product.
(3) In this Schedule—
“premixture” means a mixture of a veterinary medicinal product or a specified feed additive with feedingstuffs materials, intended for further mixing with feedingstuffs before being fed to animals;
“zootechnical additive” means any additive used to maintain animals in good health or favourably affect their performance.
Enforcement of Regulation (EC) No 178/2002U.K.
2.—(1) For the purposes of [F102Regulation (EC) No 178/2002] the competent authority is the Secretary of State.
(2) No person may fail to comply with any of the following provisions of that Regulation—
(a)Article 11 (requirements relating to imports);
(b)Article 12 (requirements relating to exports);
(c)Article 15(1) (prohibition on the placing on the market or feeding unsafe feedingstuffs);
(d)Article 16 so far as it prohibits misleading labelling, advertising or presentation of feedingstuffs;
(e)Article 18(2) and (3) (requirements of traceability) in so far as it relates to feed business operators; and
(f)Article 20 (responsibilities of feed business operators).
Textual Amendments
F102Words in Sch. 5 para. 2(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(a)
Enforcement of Regulation (EC) No 1831/2003E+W+S
3.—(1) For the purposes of [F103Regulation (EC) No 1831/2003] the competent authority is the Secretary of State.
(2) An authorisation under Article 3(2) of that Regulation must be in writing.
(3) No person may possess a specified feed additive, or a premixture or feedingstuffs containing a specified feed additive, unless the specified feed additive has been authorised under Regulation (EC) No 1831/2003 or is for export to [F104another] country.
(4) No person may fail to comply with any of the following provisions of that Regulation—
(a)Article 3(1) or Article 3(3) (the authorisation, conditions of use and labelling of specified feed additives);
(b)Article 12(1) or (2) (conditions relating to specified feed additives);
(c)Article 16(1) (labelling);
(d)Article 16(3) (additional labelling requirement);
(e)Article 16(4) (premixtures containing specified feed additives);
(f)Article 16(5) (packaging).
Extent Information
E47This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F103Words in Sch. 5 para. 3(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(b)
F104Word in Sch. 5 para. 3(3) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Enforcement of Regulation (EC) No 1831/2003N.I.
3.—(1) For the purposes of [F237Regulation (EC) No 1831/2003] the competent authority is the Secretary of State.
(2) An authorisation under Article 3(2) of that Regulation must be in writing.
(3) No person may possess a specified feed additive, or a premixture or feedingstuffs containing a specified feed additive, unless the specified feed additive has been authorised under Regulation (EC) No 1831/2003 or is for export to a third country.
(4) No person may fail to comply with any of the following provisions of that Regulation—
(a)Article 3(1) or Article 3(3) (the authorisation, conditions of use and labelling of specified feed additives);
(b)Article 12(1) or (2) (conditions relating to specified feed additives);
(c)Article 16(1) (labelling);
(d)Article 16(3) (additional labelling requirement);
(e)Article 16(4) (premixtures containing specified feed additives);
(f)Article 16(5) (packaging).
Extent Information
E119This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F237Words in Sch. 5 para. 3(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(b)
Enforcement of [F105Regulation (EU) 2017/625] E+W+S
4. For the purposes of [F106 Regulation (EU) 2017/625] the competent authority is the Secretary of State.
Extent Information
E48This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F105Words in Sch. 5 para. 4 heading substituted (E.) (14.12.2019) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) Regulations 2019 (S.I. 2019/1488), regs. 1(1), 27(d)(ii); and said words substituted (W.) (31.1.2020) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) (Wales) Regulations 2020 (S.I. 2020/44), regs. 1(2), 24(1)(d)(ii); and said words substituted (S.N.I.) (31.12.2020) by The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(4)(b)
F106Words in Sch. 5 para. 4 substituted (E.) (14.12.2019) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) Regulations 2019 (S.I. 2019/1488), regs. 1(1), 27(d)(ii); and said words substituted (W.) (31.1.2020) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) (Wales) Regulations 2020 (S.I. 2020/44), regs. 1(2), 24(1)(d)(i); and said words substituted (S.N.I.) (31.12.2020) by The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(4)(b)
[F238Enforcement of Regulation (EU) 2017/625N.I.
4. For the purposes of Regulation (EU) 2017/625 the competent authority is the Secretary of State.]
Extent Information
E120This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
Enforcement of Regulation (EC) No 183/2005U.K.
5.—(1) For the purposes of [F107Regulation (EC) No 183/2005] the competent authority is the Secretary of State.
(2) No person may fail to comply with any of the following provisions of that Regulation—
(a)Article 5(2), (5) or (6) (specific obligations);
(b)Article 6(1) as read with (2) and (3) (HACCP system);
(c)Article 7(1) (documents concerning the HACCP system);
(d)Article 9(2) (official controls, notification and registration);
(e)Article 10(1) (approval of feed business establishments);
(f)Article 11 (prohibition on operating without approval or registration);
(g)Article 17(2) (exemption from on-site visits);
(h)Article 18(3) (declaration of compliance);
(i)Article 23(1) (conditions relating to imports from third countries);
(j)Article 25 (feedingstuffs produced for export to third countries).
(3) A manufacturer must ensure that, so far as is reasonably practicable, the active ingredient is evenly incorporated throughout the feedingstuffs.
(4) In the case of the refusal, suspension or revocation of an approval under the Regulation the appeals procedure relating to a manufacturing authorisation in regulation 30 applies.
Textual Amendments
F107Words in Sch. 5 para. 5(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(d)
Enforcement of Regulation (EC) No 767/2009U.K.
6. No person may contravene Article 8 of Regulation (EC) No 767/2009 of the European Parliament and of the Council in relation to feedingstuffs containing specified feed additives.
Approval of manufacturers and distributors of feedingstuffs containing veterinary medicinal productsU.K.
7.—(1) For the purposes of Directive 90/167/EEC laying down the conditions governing the preparation, placing on the market and use of medicated feedingstuffs in the Community(38) the competent authority is the Secretary of State.
(2) No person may incorporate a veterinary medicinal product into a premixture or feedingstuff, or act as a distributor of premixtures or feedingstuffs containing a veterinary medicinal product, without being approved to do so by the Secretary of State.
(3) The conditions which govern approval of feed business establishments under Regulation (EC) No 183/2005 laying down requirements for feed hygiene(39) also govern approval of manufacturers and distributors under sub-paragraph (2).
(4) The Secretary of State shall conduct inspections of manufacturers and distributors approved under sub-paragraph (2) basing the frequency of inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
(5) A manufacturer must ensure that, so far as is reasonably practical, the veterinary medicinal product is evenly incorporated throughout the feedingstuffs.
(6) The provisions of this paragraph do not apply in relation to any person breeding or selling ornamental fish not intended for human consumption provided that the person does not use more than a total of 1kg of veterinary medicinal product annually for that purpose.
(7) In the case of the refusal, suspension or revocation of an approval under this paragraph the appeals procedure relating to a manufacturing authorisation in regulation 30 applies.
Incorporation of a veterinary medicinal product into a premixtureU.K.
8. Any person who incorporates a veterinary medicinal product into a premixture—
(a)must do so in accordance with the summary of product characteristics, and must take account of any interactions listed there; and
(b)must ensure that the veterinary medicinal product does not contain the same active substance as any other additive.
Top dressingU.K.
9. No person may promote or label any veterinary medicinal product, or anything containing a veterinary medicinal product, as being suitable for top dressing (that is, sprinkling it on to feedingstuffs without thoroughly incorporating it) unless the summary of product characteristics specifically permits this use.
Incorporation of a veterinary medicinal product into feedingstuffsU.K.
10. Any person who incorporates a veterinary medicinal product (or a premixture containing a veterinary medicinal product) into feedingstuffs—
(a)must do so in accordance with the summary of product characteristics, and must take account of any interactions listed there;
(b)must ensure that the veterinary medicinal product does not contain the same active substance as any other additive;
(c)must ensure that the veterinary medicinal product is incorporated in accordance with its marketing authorisation (unless it has been prescribed under the cascade) and the prescription;
(d)must ensure that the daily dose of the veterinary medicinal product is contained in a quantity of medicated feedingstuffs corresponding to at least half the daily feedingstuffs ration of the animals treated or, in the case of ruminants, corresponding to at least half the daily requirements of non-mineral complementary feedingstuffs.
Additional record keeping requirements relating to veterinary medicinal productsU.K.
11.—(1) Any person who—
(a)incorporates a veterinary medicinal product into a premixture;
(b)incorporates a premixture containing a veterinary medicinal product into feedingstuffs; or
(c)incorporates a veterinary medicinal product into feedingstuffs,
must make a daily record of—
(d)the types and quantities of all veterinary medicinal products (and specified feed additives, if any) and premixture used in the manufacturing process; and
(e)the quantity of feedingstuffs and premixture containing veterinary medicinal product manufactured that day.
(2) An approved distributor must make a daily record of—
(a)the types and quantities of all premixtures and feedingstuffs containing veterinary medicinal products bought and sold that day; and
(b)the quantity held.
(3) A manufacturer and distributor must also record, as soon as reasonably practicable, for each consignment supplied—
(a)the date of delivery;
(b)the name and address of each consignee (or, in the case of a manufacturer supplying to a distributor, the name and address of the distributor);
(c)the type of feedingstuffs or premixture supplied;
(d)the quantity;
(e)the type of veterinary medicinal product incorporated into the feedingstuffs; and
(f)the expiry date.
(4) Records must be kept for five years.
Labelling a premixture containing a veterinary medicinal productU.K.
12.—(1) A premixture containing a veterinary medicinal product must be clearly and legibly labelled with the following—
(a)the words “MEDICATED PREMIXTURE” (or, if it is to be labelled as “complementary feedingstuffs” under legislation implementing Council Directive 79/373/EEC on the marketing of compound feedingstuffs(40), “MEDICATED COMPLEMENTARY FEEDINGSTUFFS” ) in upper case letters;
(b)the proprietary name of the veterinary medicinal product and the authorisation number;
(c)the name and amount of the active substance (mg/kg) in the premixture;
(d)the range of acceptable inclusion rates of the premixture into the final feedingstuffs, the range of acceptable levels of the active ingredients in the final feedingstuffs and the words “refer to the prescription for the exact inclusion rate” or equivalent wording;
(e)warnings and contra-indications;
(f)the withdrawal period, and a statement that, if the prescription requires a longer withdrawal period, that is the one that applies;
(g)the expiry date;
(h)any special storage instructions;
(i)where a prescription is required, a statement to this effect.
(2) If there is more than one veterinary medicinal product used, the longest withdrawal period must be shown on the label.
(3) If the premixture also contains a specified feed additive to which this Schedule applies it must also contain the information required under Article 16 of Regulation (EC) No 1831/2003(41).
(4) No person may supply such a premixture unless it is labelled in accordance with this paragraph.
Labelling of feedingstuffs containing a specified feed additiveU.K.
13. No person may contravene the labelling requirements of Article 15 and Article 17 of Regulation (EC) No 767/2009 of the European Parliament and of the Council.
Labelling of feedingstuffs containing a veterinary medicinal productU.K.
14.—(1) Feedingstuffs containing a veterinary medicinal product must be clearly and legibly labelled with the following—
(a)the words “MEDICATED COMPLETE FEED” in upper case letters, or where feedingstuffs are to be labelled as a complementary feedingstuff and intended to be fed to animals without further mixing with feed materials, the words “MEDICATED COMPLEMENTARY FEEDINGSTUFF”;
(b)the proprietary name, authorisation number and inclusion rate (kg/tonne or mg/kg) of the veterinary medicinal product incorporated into the feedingstuffs;
(c)the name and amount of the active substance (mg/kg) in the feedingstuffs;
(d)the species of animal for which the feedingstuffs are intended;
(e)warnings and contra-indications;
(f)the withdrawal period, and a statement that, if the prescription requires a longer withdrawal period, that is the one that applies;
(g)the expiry date;
(h)any special storage instructions required by the marketing authorisation;
(i)a statement to the effect that the feedingstuffs must only be fed in accordance with its prescription;
(j)the name and approval number of the manufacturer or the distributor.
(2) If there is more than one veterinary medicinal product used, the longest withdrawal period must be shown on the label.
(3) If the feedingstuff also contains a specified feed additive to which this Schedule applies it must also contain the information required by Articles 15 and 17 of Regulation (EC) No 767/2009 of the European Parliament and of the Council.
(4) No person may supply feedingstuffs unless they are labelled in accordance with this paragraph.
Supply of specified feed additivesU.K.
15.—(1) No person other than the person who manufactured a specified feed additive or an approved distributor may supply a specified feed additive.
(2) The person who manufactured the specified feed additive may only supply it to—
(a)an approved distributor;
(b)an approved premixture manufacturer or an approved complementary feedingstuffs manufacturer; or
(c)a feedingstuff manufacturer approved to mix a specified feed additive directly into feedingstuff.
(3) An approved distributor may only supply it to—
(a)another approved distributor;
(b)an approved premixture manufacturer or an approved complementary feedingstuffs manufacturer; or
(c)a feedingstuff manufacturer approved to mix a specified feed additive directly into feedingstuff.
Supply of premixtureU.K.
16.—(1) No person other than the person who manufactured a premixture or an approved distributor may supply a premixture.
(2) The person who manufactured the premixture may only supply it to—
(a)an approved distributor; or
(b)a feedingstuff manufacturer approved to incorporate that premixture.
(3) An approved distributor may only supply it to—
(a)another approved distributor; or
(b)a feedingstuff manufacturer approved to incorporate that premixture.
Supply of a complementary feedingstuffU.K.
17.—(1) No person other than—
(a)the person who manufactured a complementary feedingstuff containing a specified feed additive; or
(b)an approved distributor
may supply a complementary feedingstuff containing a specified feed additive.
(2) The person who manufactured such complementary feedingstuff may only supply it to—
(a)an approved distributor; or
(b)a feedingstuff manufacturer registered to incorporate that complementary feedingstuff or approved to incorporate a premixture.
(3) An approved distributor may only supply it to—
(a)another approved distributor, or
(b)a feedingstuff manufacturer registered to incorporate that complementary feedingstuff or approved to incorporate a premixture.
(4) In this paragraph “complementary feedingstuff” has the meaning given in Article 3 of Regulation EC No 767/2009.
Supply of feedingstuffs containing a veterinary medicinal productU.K.
18.—(1) No person other than the person who manufactured the feedingstuffs or an approved distributor may supply feedingstuffs containing a veterinary medicinal product.
(2) The person who manufactured the feedingstuff may only supply it to—
(a)an approved distributor; or
(b)a person who keeps animals for feeding to those animals.
(3) A distributor may only supply it to—
(a)another approved distributor; or
(b)a person who keeps animals for feeding to those animals.
(4) Supply to a person who keeps animals must be in accordance with a written prescription as specified in the following paragraph.
(5) If a prescription is for a period of longer than one month, the supplier may not provide more than one month’s supply at any one time.
(6) No manufacturer or distributor may supply a feedingstuff to anyone not specified in this paragraph, or otherwise than in accordance with this paragraph.
(7) The person supplying the feedingstuff must keep the prescription for five years.
Prescriptions for feedingstuffs containing a veterinary medicinal productU.K.
19.—(1) A prescription for feedingstuffs containing a veterinary medicinal product must contain the following—
(a)the name and address of the person prescribing the product;
(b)the qualifications enabling the person to prescribe the product;
(c)the name and address of the keeper of the animals to be treated;
(d)the species of animal, identification and number of the animals;
(e)the premises at which the animals are kept if this is different from the address of the keeper;
(f)the date of the prescription;
(g)the signature or other authentication of the person prescribing the product;
(h)the name and amount of the product prescribed;
(i)the dosage and administration instructions;
(j)any necessary warnings;
(k)the withdrawal period;
(l)the manufacturer or the distributor of the feedingstuffs (who must be approved for the purpose);
(m)if the validity exceeds one month, a statement that not more than 31 days’ supply may be provided at any time;
(n)the name, type and quantity of feedingstuffs to be used;
(o)the inclusion rate of the veterinary medicinal product and the resulting inclusion rate of the active substance;
(p)any special instructions;
(q)the percentage of the prescribed feedingstuffs to be added to the daily ration; and
(r)if it is prescribed under the cascade, a statement to that effect.
(2) It is valid for three months or such shorter period as may be specified in the prescription.
(3) It must be sufficient for only one course of treatment.
Writing the prescriptionU.K.
20.—(1) The person who writes the prescription must—
(a)give a copy to the person incorporating the veterinary medicinal product into the feedingstuffs or to the distributor of the feedingstuffs;
(b)give one copy to the keeper of the animals to be treated;
(c)keep a copy.
(2) The person must be satisfied that—
(a)there is no undesirable interaction between the veterinary medicinal product and any feed additive used in the feedingstuffs; and
(b)the active substance of the veterinary medicinal product is not the same as an active substance in any feed additive used in the feedingstuffs.
[F108(3) The person must prescribe a veterinary medicinal product authorised for incorporation in feedingstuffs but may, if there is no veterinary medicinal product authorised for a condition in a particular species—
(a)prescribe a veterinary medicinal product authorised for another species or for another condition in the same species, and
(b)prescribe more than one veterinary medicinal product,
provided all veterinary medicinal products prescribed are authorised for incorporation in feedingstuffs.]
Textual Amendments
F108Sch. 5 para. 20(3) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(2)
PossessionU.K.
21.—(1) No person other than a person holding the appropriate approval under this Schedule may be in possession of any—
(a)specified feed additive or veterinary medicinal product to which this Schedule applies;
(b)premixtures containing such an additive or a veterinary medicinal product; or
(c)feedingstuffs or complementary feedingstuffs containing such an additive or a veterinary medicinal product unless supplied under these Regulations.
(2) No person other than a manufacturer or distributor may be in possession of feedingstuffs incorporating a veterinary medicinal product unless it has been supplied under a prescription.
Sampling and analysisU.K.
22.—(1) If any enforcement action is taken under this Schedule based on a sample, that sample must have been taken and analysed in accordance with [F109Regulation (EC) No 152/2009 laying down methods of sampling and analysis for the official control of feedingstuffs.]
(2) Unless otherwise specified in the marketing authorisation, it is a defence if the active ingredient in the medicated feedingstuff sample is within the following tolerances—
Tolerance table for medicated feedingstuff
| Level of active ingredient specified on the label | Tolerance |
|---|---|
| ≤50 mg/kg | ± 50% |
| >50 mg/kg ≤ 500 mg/kg | ± 40% |
| >500 mg/kg ≤ 5g/kg | ± 30% |
| >5g/kg ≤50g/kg | ± 20% |
| >50g/kg | ± 10% |
(3) Unless otherwise specified in the Commission Regulation authorising the specified feed additive in question, it is a defence if the active ingredient of a specified feed additive in a feedingstuff sample is within the tolerances set out in Articles 11(5) and paragraph 2(e) of Annex IV to Regulation (EC) No 767/2009 of the European Parliament and of the Council.
Textual Amendments
F109Words in Sch. 5 para. 22(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(e)
StorageU.K.
23. No person may store a veterinary medicinal product intended for incorporation into feedingstuffs, or a premixture or feedingstuffs containing a veterinary medicinal product, except in—
(a)a suitable storage area that is locked when not in use; or
(b)a hermetic container designed to store those products.
Packages and other containersU.K.
24. No person may place on the market feedingstuffs containing a veterinary medicinal product except in packages or containers that are sealed in such a way that, when the package or container is opened, the seal is damaged.
TransportU.K.
25.—(1) No person may transport feedingstuffs by road tankers or in bulk unless the labelling requirements are set out in a document accompanying the feedingstuffs, and the transporter must hand over details when delivering the feedingstuffs unless these have already been provided to the purchaser.
(2) Any person transporting feedingstuffs containing veterinary medicinal products or specified feed additives in road tankers or similar containers must ensure that the vehicle or container is cleaned before any re-use if this is necessary to prevent undesirable interaction or contamination.
(3) In the case of feedingstuffs containing a veterinary medicinal product the transporter must ensure that the vehicle is accompanied by documentation stating this.
(4) Any person operating an undertaking transporting feedingstuffs containing veterinary medicinal products or specified feed additives must give written instructions to drivers on how to load and unload vehicles so as to avoid cross-contamination, and take reasonable steps to ensure that the driver complies with those instructions.
Possession, placing on the market and use of feedingstuffsU.K.
26.—(1) No person may possess, place on the market or feed to animals any feedingstuffs incorporating veterinary medicinal products or specified feed additives unless they have been incorporated in accordance with this Schedule.
(2) No person may feed to any animal, or buy, possess or supply for the purpose of feeding to any animal, any feedingstuff containing a veterinary medicinal product or specified feed additive unless—
(a)that veterinary medicinal product or specified feed additive is authorised for that species of animal and for the purpose for which it is used; or
(b)in the case of a veterinary medicinal product, it was prescribed for that animal.
(3) This paragraph does not apply in relation to feedingstuffs if the veterinary medicinal product has been incorporated in accordance with an animal test certificate or the feedingstuff has been imported in accordance with this Schedule.
Imports from third countriesE+W+S
F11027. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E49This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F110Sch. 5 para. 27 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Imports from third countriesN.I.
27. No person may import a feedingstuff containing a veterinary medicinal product from a third country.
Extent Information
E121This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Trade between [F111countries]E+W+S
28. No person may bring in from another [F112country] a feedingstuff containing a veterinary medicinal product unless—
F113(a). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(b)it only contains a veterinary medicinal product that has the same quantitative and qualitative composition as a veterinary medicinal product authorised in [F114Great Britain].
Extent Information
E50This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F111Word in Sch. 5 para. 28 heading substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(c)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F112Word in Sch. 5 para. 28 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(c)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F113Sch. 5 para. 28(a) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(c)(iii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F114Words in Sch. 5 para. 28(b) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(9)
Trade [F239with] member StatesN.I.
28. No person may bring in from [F240a] member State a feedingstuff containing a veterinary medicinal product unless—
(a)the feedingstuff has been manufactured in accordance with the provisions of Council Directive 90/167/EEC (laying down the conditions governing the preparation, placing on the market and use of medicated feedingstuffs in the Community(42)) and Regulation (EC) No 183/2005 of the European Parliament and of the Council laying down requirements for food hygiene; and
(b)it only contains a veterinary medicinal product that has the same quantitative and qualitative composition as a veterinary medicinal product authorised in [F241Northern Ireland].
Extent Information
E122This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F239Word in Sch. 5 para. 28 heading substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(c)(i)
F240Word in Sch. 5 para. 28 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(c)(ii)
F241Words in Sch. 5 para. 28(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(c)(iii)
Import for incorporation into premixture or feedingstuffs for exportE+W+S
29.—[F115(1) A manufacturer of premixture or feedingstuffs who imports a veterinary medicinal product authorised in another F116... country for the purposes of incorporating it into premixture or feedingstuffs for export does not commit an offence under regulation 43(q) (importation of an unauthorised veterinary medicinal product) or regulation 43(r) (possession of an unauthorised veterinary medicinal product)]
(2) No person may place that premixture or feedingstuff on the market in the United Kingdom once the veterinary medicinal product has been incorporated into it.
Extent Information
E51This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F115Sch. 5 para. 29(1) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(3)
F116Words in Sch. 5 para. 29(1) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(d) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Import for incorporation into premixture or feedingstuffs for exportN.I.
29.—[F242(1) A manufacturer of premixture or feedingstuffs who imports a veterinary medicinal product authorised in [F243a] Member State or third country for the purposes of incorporating it into premixture or feedingstuffs for export does not commit an offence under regulation 43(q) (importation of an unauthorised veterinary medicinal product) or regulation 43(r) (possession of an unauthorised veterinary medicinal product)]
(2) No person may place that premixture or feedingstuff on the market in [F244Northern Ireland] once the veterinary medicinal product has been incorporated into it.
Extent Information
E123This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F242Sch. 5 para. 29(1) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(3)
F243Word in Sch. 5 para. 29(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(d)(i)
F244Words in Sch. 5 para. 29(2) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(d)(ii)
Animals on domestic premisesU.K.
30.—(1) The requirements of paragraph 7 (approval of manufacturers and distributors of feedingstuffs containing veterinary medicinal product) do not apply in relation to a person who incorporates a veterinary medicinal product into feedingstuffs in domestic premises for feeding, on those premises—
(a)non-food-producing animals; or
(b)food-producing animals provided that the animals or products from those animals are not sold or supplied commercially.
(2) Notwithstanding paragraphs 16 and 18 of this Schedule, a veterinary surgeon, a pharmacist or a suitably qualified person who is registered in accordance with paragraph 14 of Schedule 3 may be supplied with and may supply a premixture containing a veterinary medicinal product, or feedingstuffs containing a veterinary medicinal product, to such a producer.
(3) The requirement for a written prescription does not apply in relation to such supply, but the provisions of Schedule 3 relating to supply of a veterinary medicinal product apply in relation to the supply of premixture and feedingstuffs in the same way as they apply to a veterinary medicinal product.
OffencesE+W+S
31. It is an offence to fail to comply with—
(a)paragraph 2(2);
(b)paragraph 3(3) or (4);
(c)paragraph 5(2) or (3);
(d)paragraph 6;
(e)paragraph 7(2) or (5);
(f)paragraph 8;
(g)paragraph 9;
(h)paragraph 10;
(i)paragraph 11;
(j)paragraph 12(4);
(k)paragraph 13;
(l)paragraph 14(4);
(m)paragraph 15;
(n)paragraph 16;
(o)paragraph 17;
(p)paragraph 18;
(q)paragraph [F11720] ;
(r)paragraph 21;
(s)paragraph 23;
(t)paragraph 24;
(u)paragraph 25;
(v)paragraph 26(1) or (2);
F118(w). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(x)paragraph 28; or
(y)paragraph 29(2).
Extent Information
E52This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F117Word in Sch. 5 para. 31(q) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(4)
F118Sch. 5 para. 31(w) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(e) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
OffencesN.I.
31. It is an offence to fail to comply with—
(a)paragraph 2(2);
(b)paragraph 3(3) or (4);
(c)paragraph 5(2) or (3);
(d)paragraph 6;
(e)paragraph 7(2) or (5);
(f)paragraph 8;
(g)paragraph 9;
(h)paragraph 10;
(i)paragraph 11;
(j)paragraph 12(4);
(k)paragraph 13;
(l)paragraph 14(4);
(m)paragraph 15;
(n)paragraph 16;
(o)paragraph 17;
(p)paragraph 18;
(q)paragraph [F24520] ;
(r)paragraph 21;
(s)paragraph 23;
(t)paragraph 24;
(u)paragraph 25;
(v)paragraph 26(1) or (2);
(w)paragraph 27;
(x)paragraph 28; or
(y)paragraph 29(2).
Extent Information
E124This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F245Word in Sch. 5 para. 31(q) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(4)
Regulation 15(4)
SCHEDULE 6U.K.Exemptions for small pet animals
Animals to which this Schedule appliesU.K.
1. This Schedule applies in relation to veterinary medicinal products intended solely for the following animals kept exclusively as a pet—
(a)aquarium animals;
(b)cage birds;
(c)ferrets;
(d)homing pigeons;
(e)rabbits;
(f)small rodents; and
(g)terrarium animals.
Placing on the market, importing and administering the productU.K.
2. A veterinary medicinal product intended solely for an animal to which this Schedule applies may be placed on the market, imported or administered without a marketing authorisation if it complies with this Schedule.
ManufactureE+W+S
3.—[F119(1)] The product must have been manufactured by—
(a)the holder of a manufacturing authorisation if manufactured in [F120Great Britain];
F121(b). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F122(c). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(d)in the case of any other country, a manufacturer whose premises have been inspected and approved by an officer of the Secretary of State.
[F123(2) Sub-paragraph (1) does not apply where the exporting country has demonstrated equivalent standards to the United Kingdom or where appropriate arrangements have been made with the exporting country to ensure that the manufacturer of the veterinary medicinal product applies standards of good manufacturing practice at least equivalent to those laid down in Commission Directive 91/412/EEC.]
Extent Information
E53This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F119Sch. 6 para. 3 renumbered as Sch. 6 para. 3(1) (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F120Words in Sch. 6 para. 3(1)(a) substittued (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(b)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(e)(i)); 2020 c. 1, Sch. 5 para. 1(1)
F121Sch. 6 para. 3(1)(b) omitted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(e)(i)); 2020 c. 1, Sch. 5 para. 1(1)
F122Sch. 6 para. 3(1)(c) omitted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(e)(i)); 2020 c. 1, Sch. 5 para. 1(1)
F123Sch. 6 para. 3(2) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(c) (as amended S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(e)(ii))); 2020 c. 1, Sch. 5 para. 1(1)
ManufactureN.I.
3. The product must have been manufactured by—
(a)the holder of a manufacturing authorisation if manufactured in the United Kingdom;
(b)the holder of a manufacturing authorisation issued under Directive 2001/82/EC if manufactured in [F246a] member State;
(c)in the case of Australia, Canada, New Zealand, or Switzerland, the holder of an authorisation from the competent authority permitting the manufacture of medicinal products;
(d)in the case of any other country, a manufacturer whose premises have been inspected and approved by an officer of the Secretary of State.
Extent Information
E125This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F246Word in Sch. 6 para. 3(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(16)
Approval of the active substanceU.K.
4.—(1) The Secretary of State may approve an active substance for use in a veterinary medicinal product manufactured under this Schedule.
(2) The Secretary of State may not grant an approval if the active substance requires veterinary control.
(3) The approval must specify the species of animals for which it is approved, and may specify how the active substance or a product containing it is to be administered.
(4) The Secretary of State may suspend or revoke the approval (or limit it to a smaller number of species) if—
(a)it is demonstrated that the substance requires veterinary control;
(b)serious adverse reactions are reported making suspension or revocation necessary; or
(c)it is demonstrated that the substance—
(i)is carcinogenic;
(ii)is genotoxic; or
(iii)shows developmental toxicity (including teratogenicity).
(5) The procedure for the refusal, suspension or revocation of an approval under this paragraph is the same as the procedure for a marketing authorisation.
The productU.K.
5.—(1) The active substance in the veterinary medicinal product must be approved under paragraph 4.
(2) The veterinary medicinal product must not be an antibiotic.
(3) It must not contain any narcotic or psychotropic substance.
(4) It must not be intended for treatments or pathological processes that require a precise prior diagnosis or the use of which may cause effects that impede or interfere with subsequent diagnostic or therapeutic measures.
LabellingU.K.
6.—(1) The product must be clearly labelled as being exempt from the requirements of these Regulations in relation to a marketing authorisation.
(2) The labelling must show the following—
(a)the name of the veterinary product, including, if it is part of the name, its strength and pharmaceutical form;
(b)the authorisation number of the manufacturer;
(c)the name and strength of each active substance;
(d)the route of administration;
(e)the batch number;
(f)the expiry date;
(g)the words “For animal treatment only”;
(h)the contents by weight, volume or number of dose units;
(i)the name and address of the manufacturer or distributor;
(j)the target species;
(k)the words “Keep out of reach of children”;
(l)storage instructions;
(m)the shelf-life after the immediate packaging has been opened for the first time;
(n)disposal advice;
(o)full indications, including—
(i)therapeutic indications;
(ii)contra-indications;
(iii)interaction with other medicines and other forms of interaction; and
(p)dosage instructions.
(3) If there is insufficient room on the label, the information may instead be in a package leaflet, but the leaflet must contain all the information in the preceding sub-paragraph other than the batch number and the expiry date, but the label on the product must contain at least the following—
(a)the name of the veterinary medicinal product;
(b)its active substance and its strength;
(c)the route of administration;
(d)the batch number;
(e)the expiry date; and
(f)the words “For animal treatment only”.
AdministrationU.K.
7. The method of administration must be oral or topical or (in the case of a product for fish) by addition to the water.
Pack sizeU.K.
8. The pack size must only be sufficient for a single course of treatment or, in the case of a veterinary medicinal product for aquarium fish, sufficient for a single course of treatment of no more than 7 administrations to an aquarium of 25,000 litres.
Adverse reactionsU.K.
9.—(1) The manufacturer, importer or retailer of a veterinary medicinal product must—
(a)notify the Secretary of State within 15 days of learning of any serious adverse reactions (as defined in paragraph 57 of Schedule 1); and
(b)make a record of each adverse reaction and serious adverse reaction on becoming aware of it and keep it for three years.
(2) It is an offence to fail to comply with this paragraph.
Regulation 16
SCHEDULE 7U.K.Fees
PART 1 Introduction
1.Interpretation
2.Payment of fees
3.Time of payment
4.Multiple inspections
5.Expenses for inspections outside the United Kingdom
6.Translation
PART 2 Fees relating to marketing authorisations
7.Specified pharmaceutical applications
8.Decentralised pharmaceutical application where the United Kingdom is the reference member State
9.Application for a marketing authorisation for an immunological or biosimilar product
10.Decentralised immunological application where the United Kingdom is the reference member State
11.Applications for a marketing authorisation using data already assessed
12.Application for an exceptional marketing authorisation (pharmaceutical)
13.Fees for an application for an exceptional marketing authorisation (immunological)
14.Fee for the conversion from an exceptional to a full marketing authorisation
15.Application for a marketing authorisation relating to a parallel import
16.Application to change the distribution category of a product authorised through the centralised procedure
17.Application for a variation to a marketing authorisation dealt with under national or mutual recognition variation procedures.
18.Application for a variation to a marketing authorisation dealt with under worksharing procedures
19.Application for an extension dealt with under the decentralised procedure where the United Kingdom is the reference member State
20.Provision of information relating to the recognition of a United Kingdom marketing authorisation or an extension
21.Exception for a variation relating to animal testing
22.Application for the renewal of a national marketing authorisation
23.Application for the renewal of a marketing authorisation obtained through mutual recognition or the decentralised procedure
24.Registration of a homeopathic remedy
25.Renewal of a homeopathic remedy
26.Annual fees for marketing authorisations
27.Auditor’s certificate
PART 3 Fees payable by manufacturers
28.Application for a manufacturing authorisation
29.Application for a variation of a manufacturing authorisation
30.Application for an authorisation to manufacture an autogenous vaccine or a product for administration under the cascade
31.Annual fees
32.Site inspections – type of site
33.Inspection of a site where immunological veterinary medicinal products are manufactured
34.Inspection of a site where sterile veterinary medicinal products are manufactured
35.Inspection of a site where no immunological or sterile veterinary medicinal products are manufactured
36.Inspection of a site where veterinary medicinal products are assembled
37.Test sites
38.Animal blood bank or equine stem cell centre authorisations
PART 4 Fees relating to a wholesale dealer’s authorisation
39.Application for a wholesale dealer’s authorisation
40.Variation of a wholesale dealer’s authorisation
41.Annual fee for a wholesale dealer’s authorisation
42.Inspection of a wholesale dealer’s premises
PART 5 Fees relating to feedingstuffs
43.Fees for approvals and annual fees relating to feedingstuffs in Great Britain
44.Inspection fees relating to feedingstuffs in Great Britain
45.Fees payable in relation to feedingstuffs in Northern Ireland
46.Fees relating to premises for supply by suitably qualified persons
PART 6 General
47.Testing samples
48.Animal test certificates
49.Importation of a veterinary medicinal product for treatment under the cascade
50.Wholesale dealer’s import certificate
51.Specific batch control
52.Submission of control tests of an immunological product
53.Export certificates
54.Provision of advice
55.Appeals to the Veterinary Products Committee
56.Fee relating to an appointed person
57.Fees relating to a veterinary surgeon’s practice premises
58.Refund of fees relating to the Veterinary Products Committee or appointed persons
59.Fees relating to an improvement notice
60.Non-payment of fees
61.Waiver or reduction of fees
62.Reduction of fees when an application is withdrawn
PART 1U.K.Introduction
InterpretationE+W+S
1. In this Schedule—
F124...
“pharmaceutical product” means any veterinary medicinal product other than an immunological product;
“simultaneous application” is an application in which, at the time an authorisation for a product is applied for, one or more additional applications are submitted for products that are identical to the first product except that—
in the case of an immunological product, they have a lesser number of antigens than the first product, but only contain antigens contained in the first product; and
in the case of a pharmaceutical product, they have different strengths of the active substance,
F125...
Extent Information
E54This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F124Words in Sch. 7 para. 1 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(a)(i); 2020 c. 1, Sch. 5 para. 1(1)
F125Words in Sch. 7 para. 1 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(a)(ii); 2020 c. 1, Sch. 5 para. 1(1)
InterpretationN.I.
1. In this Schedule—
“national application” means an application for a marketing authorisation that does not involve another member State;
“pharmaceutical product” means any veterinary medicinal product other than an immunological product;
“simultaneous application” is an application in which, at the time an authorisation for a product is applied for, one or more additional applications are submitted for products that are identical to the first product except that—
in the case of an immunological product, they have a lesser number of antigens than the first product, but only contain antigens contained in the first product; and
in the case of a pharmaceutical product, they have different strengths of the active substance,
and, in the case of an application involving more than one member State, the additional applications do not include a member State that was not included in the first application.
Extent Information
E126This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Payment of feesU.K.
2. All fees under this Schedule are payable to the Secretary of State.
Time of paymentU.K.
3. All fees are payable on invoice unless otherwise specified.
Multiple inspectionsU.K.
4. If a site, premises or establishment is inspected for more than one type of authorisation, approval or registration at the same time, the fee is the sum of —
(a)the highest fee payable; and
(b)50% of each of the other fees.
Expenses for inspections outside the United KingdomU.K.
5. Whenever premises outside the United Kingdom are inspected, the travel and subsistence costs of the inspectors and interpreters’ fees are payable in addition to the inspection fee specified.
TranslationU.K.
6. All translation costs are charged additionally.
PART 2U.K.Fees relating to marketing authorisations
[F126Application for a marketing authorisation for a pharmaceutical veterinary medicinal product]E+W+S
7. The following table sets out the fees relating to a pharmaceutical veterinary medicinal product for—
(a)[F127an application] for a marketing authorisation that is—
(i)a full application under Part 1 of Schedule 1;
(ii)a bibliographic application; or
(iii)an application based on pharmacological equivalence;
F128(b). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(c)F129...
| Application | Full F130...application under Part 1 of Schedule 1 (£) | Bibliographic F131...application (£) | F132... | |||
|---|---|---|---|---|---|---|
| [F133Pharmacologically equivalent application] | F132... | |||||
| Base Fee: | 13,530 | 12,115 | 7,195 | F132 | F132 | |
| Additional fee if any of the target species is a food-producing animal: | 3,905 | 3,585 | 2,155 | F132 | F132 | |
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom— | ||||||
| food-producing animal: | 7,465 | 6,595 | 5,885 | F132 | F132 | |
| non-food-producing animal: | 6,525 | 5,855 | 5,590 | F132 | F132 | |
| Additional fee for each additional pack type: | 740 | 740 | 605 | F132 | F132 | |
| Additional fee for each additional active ingredient (food-producing animal): | 6,465 | 6,125 | 4,040 | F132 | F132 | |
| Additional fee for each additional active ingredient (non-food-producing animal): | 4,310 | 4,105 | 3,235 | F132 | F132 | |
| Additional fee if there is more than one target species, for each additional species (food-producing animal): | 3,970 | 3,565 | 2,425 | F132 | F132 | |
| Additional fee if there is more than one target species, for each additional species (non- food-producing animal): | 2,495 | 2,090 | 1,550 | F132 | F132 | |
| Additional fee for each additional recommended route of administration (food-producing animal): | 2,695 | 2,490 | 1,620 | F132 | F132 | |
| Additional fee for each additional recommended route of administration (non- food-producing animal): | 1,215 | 1,010 | 740 | F132 | F132 | |
| Simultaneous applications: fee for each additional product in the application: | 2,895 | 2,895 | 2,895 | F132 | F132 | |
Extent Information
E55This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F126Sch. 7 para. 7 heading substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(i); 2020 c. 1, Sch. 5 para. 1(1)
F127Words in Sch. 7 para. 7(a) substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(ii); 2020 c. 1, Sch. 5 para. 1(1)
F128Sch. 7 para. 7(b) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(iii); 2020 c. 1, Sch. 5 para. 1(1)
F129Sch. 7 para. 7(c) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(iii); 2020 c. 1, Sch. 5 para. 1(1)
F130Word in Sch. 7 para. 7 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(iv)(aa); 2020 c. 1, Sch. 5 para. 1(1)
F131Word in Sch. 7 para. 7 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(iv)(bb); 2020 c. 1, Sch. 5 para. 1(1)
F132Words in Sch. 7 para. 7 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(iv)(dd); 2020 c. 1, Sch. 5 para. 1(1)
F133Words in Sch. 7 para. 7 Table substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(iv)(cc); 2020 c. 1, Sch. 5 para. 1(1)
Specified pharmaceutical applicationsN.I.
7. The following table sets out the fees relating to a pharmaceutical veterinary medicinal product for—
(a)a national application for a marketing authorisation that is—
(i)a full application under Part 1 of Schedule 1;
(ii)a bibliographic application; or
(iii)an application based on pharmacological equivalence;
(b)an application for a marketing authorisation using the decentralised procedure where the United Kingdom is a concerned member State;
(c)an application for the mutual recognition of a product authorised in another member State.
| Application | Full national application under Part 1 of Schedule 1 (£) | Bibliographic national application (£) | Pharmacologically equivalent national application | Decentralised application where the UK is a concerned member State or recognition of a product authorised in another member State (£) | ||
|---|---|---|---|---|---|---|
| Reference product authorised in UK (£) | Reference product not authorised in UK (£) | |||||
| Base Fee: | 13,530 | 12,115 | 7,195 | 9,220 | 6,515 | |
| Additional fee if any of the target species is a food-producing animal: | 3,905 | 3,585 | 2,155 | 2,760 | 1,415 | |
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom— | ||||||
| food-producing animal: | 7,465 | 6,595 | 5,885 | 7,495 | 2,630 | |
| non-food-producing animal: | 6,525 | 5,855 | 5,590 | 7,155 | 2,295 | |
| Additional fee for each additional pack type: | 740 | 740 | 605 | 775 | 330 | |
| Additional fee for each additional active ingredient (food-producing animal): | 6,465 | 6,125 | 4,040 | 5,165 | 2,085 | |
| Additional fee for each additional active ingredient (non-food-producing animal): | 4,310 | 4,105 | 3,235 | 4,135 | 1,475 | |
| Additional fee if there is more than one target species, for each additional species (food-producing animal): | 3,970 | 3,565 | 2,425 | 3,100 | 1,280 Applies for a maximum of 2 additional species | |
| Additional fee if there is more than one target species, for each additional species (non- food-producing animal): | 2,495 | 2,090 | 1,550 | 1,980 | 805 Applies for a maximum of 2 additional species | |
| Additional fee for each additional recommended route of administration (food-producing animal): | 2,695 | 2,490 | 1,620 | 2,070 | 940 | |
| Additional fee for each additional recommended route of administration (non- food-producing animal): | 1,215 | 1,010 | 740 | 945 | 405 | |
| Simultaneous applications: fee for each additional product in the application: | 2,895 | 2,895 | 2,895 | 3,705 | 1,685 | |
Extent Information
E127This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Decentralised pharmaceutical application where the United Kingdom is the reference member StateE+W+S
F1348. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E56This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F134Sch. 7 para. 8 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(c); 2020 c. 1, Sch. 5 para. 1(1)
Decentralised pharmaceutical application where the United Kingdom is the reference member StateN.I.
8. The fee for a decentralised application for a pharmaceutical product where the United Kingdom is the reference member State is the same as for a national application as set out in the table in paragraph 7, with the addition of the fees in the following table.
Fees for decentralised pharmaceutical application where the United Kingdom is the reference member State
| Application | Additional fee for a pharmacologically equivalent product (£) | Additional fee otherwise (£) | |
|---|---|---|---|
| Food-producing animal: one member State: | 5,230 | 3,705 | |
| Non-food-producing animal: one member State: | 3,985 | 3,220 | |
| Each additional member State: | 530 | 530 | |
| Simultaneous application: fee for each additional product in the application: | |||
| one member State: | 6,670 | 6,670 | |
| each additional member State: | 120 | 120 | |
Extent Information
E128This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for a marketing authorisation for an immunological or biosimilar productE+W+S
9.—(1) The fee for [F135an application] for a marketing authorisation relating to an immunological or biosimilar product,F136... is in accordance with the following table.
(2) In this paragraph a biosimilar application means an application made in accordance with Article 13(4) of Directive 2001/82/EC and a biosimilar product means a product which is the subject of such an application.
Fees for specified immunological and biosimilar applications
| Application | F137... application for a marketing authorisation(£) | F138... | |
|---|---|---|---|
| 1. Immunological or biosimilar product other than in paragraph 2 below: Base fee: | 11,775 | F138... | |
| The following fees are in addition to the base fee— | F138... | ||
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom, and for each new combination of active ingredients: | 7,405 | F138... | |
| Additional fee for each adjuvant or preservative not previously included in a veterinary medicinal product authorised in the United Kingdom and for each new combination of adjuvants or preservatives: | 1,345 | F138... | |
| More than one antigenic component – fee for each additional component: | 1,350 | F138... | |
| More than one species – fee for each additional species: | 5,380 | F138... | |
| More than one route of administration – fee for each additional route of administration: | 5,380 | F138... | |
| Simultaneous application - fee for each additional product in the application: | 2,895 | F138... | |
| 2. Immunological or product that is identical to a product already authorised in the United Kingdom but with a lesser number of antigens and that only contains antigens contained in that product: | 10,430 | F138... | |
Extent Information
E57This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F135Words in Sch. 7 para. 9(1) substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(d)(i)(aa); 2020 c. 1, Sch. 5 para. 1(1)
F136Words in Sch. 7 para. 9(1) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(d)(i)(bb); 2020 c. 1, Sch. 5 para. 1(1)
F137Word in Sch. 7 para. 9 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(d)(ii)(aa); 2020 c. 1, Sch. 5 para. 1(1)
F138Words in Sch. 7 para. 9 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(d)(ii)(bb); 2020 c. 1, Sch. 5 para. 1(1)
Application for a marketing authorisation for an immunological or biosimilar productN.I.
9.—(1) The fee for a national application for a marketing authorisation relating to an immunological or biosimilar product, a decentralised application where the United Kingdom is the concerned member State or the mutual recognition of a product authorised in another member State is in accordance with the following table.
(2) In this paragraph a biosimilar application means an application made in accordance with Article 13(4) of Directive 2001/82/EC and a biosimilar product means a product which is the subject of such an application.
Fees for specified immunological and biosimilar applications
| Application | National application for a marketing authorisation(£) | Decentralised application where the UK is a concerned member State or recognition of a product authorised in another member State (£) | |
|---|---|---|---|
| 1. Immunological or biosimilar product other than in paragraph 2 below: Base fee: | 11,775 | 5,785 | |
| The following fees are in addition to the base fee— | |||
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom, and for each new combination of active ingredients: | 7,405 | 2,490 | |
| Additional fee for each adjuvant or preservative not previously included in a veterinary medicinal product authorised in the United Kingdom and for each new combination of adjuvants or preservatives: | 1,345 | 675 | |
| More than one antigenic component – fee for each additional component: | 1,350 | 405 | |
| More than one species – fee for each additional species: | 5,380 | 1,615 Applies for a maximum of 2 additional species | |
| More than one route of administration – fee for each additional route of administration: | 5,380 | 1,615 | |
| Simultaneous application - fee for each additional product in the application: | 2,895 | 1,685 | |
| 2. Immunological or product that is identical to a product already authorised in the United Kingdom but with a lesser number of antigens and that only contains antigens contained in that product: | 10,430 | 5,380 | |
Extent Information
E129This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Decentralised immunological application where the United Kingdom is the reference member StateE+W+S
F13910. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E58This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F139Sch. 7 para. 10 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(e); 2020 c. 1, Sch. 5 para. 1(1)
Decentralised immunological application where the United Kingdom is the reference member StateN.I.
10. The fee for a decentralised application for a marketing authorisation for an immunological product where the United Kingdom is the reference member State is the same as for a national application set out in the previous table, with the addition of the fees in the following table—
Fees for decentralised immunological application where the United Kingdom is the reference member State
| Application | Additional fee (£) | |
|---|---|---|
| One member State: | 3,470 | |
| Each additional member State: | 530 | |
| Simultaneous applications: fee for each additional product in the application: | ||
| one member State: | 6,670 | |
| each additional member State: | 120 | |
Extent Information
E130This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F140Application for a marketing authorisation based on informed consent]E+W+S
11. The [F141fee ] for applications for marketing authorisations using identical data submitted simultaneously or on the basis of information provided under Article 13(c) of Directive 2001/82/EC [F142is £945 per application.]
Extent Information
E59This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F140Sch. 7 para. 11 heading substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(f)(i); 2020 c. 1, Sch. 5 para. 1(1)
F141Word in Sch. 7 para. 11 substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(f)(ii); 2020 c. 1, Sch. 5 para. 1(1)
F142Words in Sch. 7 para. 11 substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(f)(iii); 2020 c. 1, Sch. 5 para. 1(1)
Applications for a marketing authorisation using data already assessedN.I.
11. The fees for applications for marketing authorisations using identical data submitted simultaneously or on the basis of information provided under Article 13(c) of Directive 2001/82/EC are in accordance with the following table.
Fees for a marketing authorisation using data already assessed
| Application | Fee (£)per authorisation | |
|---|---|---|
| Decentralised application where the United Kingdom is the reference member State— | ||
| one member State: | 4,165 | |
| each additional member State: | 530 | |
| Any other application: | 945 | |
Extent Information
E131This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for an exceptional marketing authorisation (pharmaceutical)U.K.
12. The fee for an application for an exceptional marketing authorisation for a pharmaceutical product is in accordance with the following table.
Fees for an exceptional marketing authorisation for a pharmaceutical product
| Application | Provisional (£) | Limited (£) | |
|---|---|---|---|
| Base Fee: | 12,015 | 6,765 | |
| The following fees are in addition to the base fee— | |||
| Additional fee if any of the target species is a food-producing animal: | 3,905 | 1,952 | |
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom— | |||
| food-producing animal: | 5,850 | 3,732 | |
| non-food-producing animal: | 4,910 | 3,262 | |
| Additional fee for each additional pack type: | 710 | 370 | |
| Additional fee for each additional active ingredient (food-producing animal): | 5,955 | 3,232 | |
| Additional fee for each additional active ingredient (non-food-producing animal): | 3,800 | 2,155 | |
| Additional fee if there is more than one target species, for each additional species (food-producing animal): | 2,965 | 1,985 | |
| Additional fee if there is more than one target species, for each additional species (non-food-producing animal): | 1,485 | 1,247 | |
| Additional fee for each additional recommended route of administration (food-producing animal): | 2,185 | 1,347 | |
| Additional fee for each additional recommended route of administration (non-food-producing animal): | 710 | 608 | |
| Simultaneous applications— fee for each additional product in the application: | 2,895 | 1,447 | |
Fees for an application for an exceptional marketing authorisation (immunological)U.K.
13. The fee for an application for an exceptional marketing authorisation for an immunological product is in accordance with the following table.
Fees for an exceptional marketing authorisation for an immunological product
| Application | Provisional (£) | Limited (£) |
|---|---|---|
| Base fee: | 10,810 | 5,887 |
| The following fees are in addition to the base fee— | ||
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom, and for each new combination of active ingredients: | 5,650 | 3,702 |
| Additional fee for each adjuvant or preservative not previously included in a veterinary medicinal product authorised in the United Kingdom and for each new combination of adjuvants or preservatives: | 1,350 | 672 |
| More than one antigenic component – fee for each additional component: | 1,190 | 675 |
| More than one species – fee for each additional species: | 4,060 | 2,690 |
| More than one route of administration – fee for each additional route of administration: | 4,060 | 2,690 |
| Simultaneous application - fee for each additional product in the application: | 2,895 | 1,447 |
Fee for the conversion from an exceptional to a full marketing authorisationU.K.
14. The fee for the conversion of an exceptional marketing authorisation to a full marketing authorisation is £3,000.
Application for a marketing authorisation relating to a parallel importE+W+S
15. The fee for a marketing authorisation for a parallel import is in accordance with the following table.
[F143Parallel imports
| Application | Fee (£) |
|---|---|
| Application where the imported product is identical to a product which is authorised for sale in the United Kingdom | 2,130 |
| Application where the imported product is therapeutically similar to a product which is authorised for sale in the United Kingdom (can only be applied to imported products for non-food producing species) | 4,710] |
Extent Information
E60This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F143Sch. 7 para. 15 Table substituted (E.W.S) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), Sch. 8 Pt. 2 ( as amended by S.I. 2020/1461, regs. 1(2)(a), 2(4)); 2020 c. 1, Sch. 5 para. 1(1)
Application for a marketing authorisation relating to a parallel importN.I.
15. The fee for a marketing authorisation for a parallel import is in accordance with the following table.
Parallel imports
| Application | Fee (£) | |
|---|---|---|
| Application where the imported product has been authorised in accordance with the mutual recognition procedure or decentralised procedure, and the United Kingdom is included in these procedures— | ||
| import from one or more member States: | 1,755 | |
| Application to add an additional member State after the marketing authorisation has been granted – fee for each member State: | 455 | |
| Application where the imported product has not been authorised in accordance with the mutual recognition procedure or the decentralised procedure but where the imported product originates from the same manufacturing site as the product authorised in the United Kingdom to which the imported product is considered to be essentially similar: | 2,130 | |
| Any other application – fee for each member State from which the product is imported: | 4,710 | |
Extent Information
E132This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application to change the distribution category of a product authorised through the centralised procedureE+W+S
F14416. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E61This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F144Sch. 7 para. 16 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(h); 2020 c. 1, Sch. 5 para. 1(1)
Application to change the distribution category of a product authorised through the centralised procedureN.I.
16. The fee to change the distribution category of a product authorised through the centralised procedure is £3,135.
Extent Information
E133This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for a variation to a marketing authorisation F145....E+W+S
17.—(1) This paragraph applies in relation to an application for a variation to one or more marketing authorisations except where paragraph 18, 19 or 21 applies.
(2) The fees for the variations to which this paragraph applies are set out in the following table.
(3) Where applications are made at the same time seeking an identical change to the terms of more than one marketing authorisation, and those applications are based on identical data, fees are payable as for a grouped variation.
(4) References in this paragraph to a grouped variation being “led” by a particular type of variation indicate that the principal variation in that group is a variation of that type.
| Type of variation | F146... | F147... | F147... | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Single variations; one change for each product | |||||||||
| Extension: | |||||||||
| Change of strength or potency or the addition of a new strength or potency: | 6,670 | F147... | F147... | ||||||
| Change of pharmaceutical form or the addition of a new pharmaceutical form: | 8,415 | F147... | F147... | ||||||
| Change of route of administration, or the addition of a new one, of— | |||||||||
| (i) an immunological product, or a pharmaceutical product for a [F148non-food-producing] animal: | 5,390 | F147... | F147... | ||||||
| (ii) a pharmaceutical product for a food-producing animal: | 7,135 | F147... | F147... | ||||||
| Change or addition of a food producing target species: | 9,620 | F147... | F147... | ||||||
| Change of active substance, including: | 8,415 | F147... | F147... | ||||||
| use of a different salt, ester, complex or derivative of the same therapeutic moiety: | |||||||||
| use of a different biologically active substance with a slightly different molecular structure: | |||||||||
| modification of the vector used to produce the antigen or the source material, including a new master cell bank from a different source: | |||||||||
| use of a new ligand or coupling mechanism for a radiopharmaceutical: | |||||||||
| change of the extraction solvent or change of the ratio of herbal drug to herbal drug preparation: | |||||||||
| Change of bioavailability: | 8,415 | F147... | F147... | ||||||
| Change of pharmacokinetics: | 8,415 | ...F147... | F147... | ||||||
| Simultaneous application: fee for each additional product in the application: | 2,895 | F147... | F147... | ||||||
| Type II: | 2,895 | F147... | F147... | ||||||
| Type IB: | 885 | F147... | F147... | ||||||
| Type IA: | 455 | F147... | F147... | ||||||
| Grouped variations | |||||||||
| Extension-led: | |||||||||
| The fee for an application for an extension-led grouped variation is the fee for that extension as specified above plus — | |||||||||
| (a) if there is one variation in addition to the extension, the fee for that variation as specified above; or | |||||||||
| (b) if there is more than one variation in addition to the extension, the fee that would be payable for a grouped variation of that type as specified below. | |||||||||
| Type II led: | |||||||||
| For the first nine changes: | 6,280 | F147... | F147... | ||||||
| For each subsequent group of up to ten changes: | 4,500 | F147... | F147... | ||||||
| Type IB led: | |||||||||
| For the first nine changes: | 1,770 | F147... | F147... | ||||||
| For each subsequent group of up to ten changes: | 4,500 | F147... | F147... | ||||||
| Type IA led: | |||||||||
| For the first nine changes: | 885 | F147... | F147... | ||||||
| For each subsequent group of up to ten changes: | 4,500 | F147... | F147... | ||||||
Extent Information
E62This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F145Words in Sch. 7 para. 17 heading omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(i); 2020 c. 1, Sch. 5 para. 1(1)
F146Words in Sch. 7 para. 17 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(i)(ii)(aa); 2020 c. 1, Sch. 5 para. 1(1)
F147Words in Sch. 7 para. 17 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(i)(ii)(bb); 2020 c. 1, Sch. 5 para. 1(1)
F148Words in Sch. 7 para. 17 substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 4(2)
Application for a variation to a marketing authorisation dealt with under national or mutual recognition variation procedures.N.I.
17.—(1) This paragraph applies in relation to an application for a variation to one or more marketing authorisations except where paragraph 18, 19 or 21 applies.
(2) The fees for the variations to which this paragraph applies are set out in the following table.
(3) Where applications are made at the same time seeking an identical change to the terms of more than one marketing authorisation, and those applications are based on identical data, fees are payable as for a grouped variation.
(4) References in this paragraph to a grouped variation being “led” by a particular type of variation indicate that the principal variation in that group is a variation of that type.
| Type of variation | National | UK is the reference member State | UK is a concerned member State | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Single variations; one change for each product | |||||||||
| Extension: | |||||||||
| Change of strength or potency or the addition of a new strength or potency: | 6,670 | - | 1,998 | ||||||
| Change of pharmaceutical form or the addition of a new pharmaceutical form: | 8,415 | - | 2,301 | ||||||
| Change of route of administration, or the addition of a new one, of— | - | ||||||||
| (i) an immunological product, or a pharmaceutical product for a [F247non-food-producing] animal: | 5,390 | - | 1,737 | ||||||
| (ii) a pharmaceutical product for a food-producing animal: | 7,135 | - | 2,058 | ||||||
| Change or addition of a food producing target species: | 9,620 | - | 2,547 | ||||||
| Change of active substance, including: | 8,415 | - | 2,301 | ||||||
| use of a different salt, ester, complex or derivative of the same therapeutic moiety: | |||||||||
| use of a different biologically active substance with a slightly different molecular structure: | |||||||||
| modification of the vector used to produce the antigen or the source material, including a new master cell bank from a different source: | |||||||||
| use of a new ligand or coupling mechanism for a radiopharmaceutical: | |||||||||
| change of the extraction solvent or change of the ratio of herbal drug to herbal drug preparation: | |||||||||
| Change of bioavailability: | 8,415 | - | 2,301 | ||||||
| Change of pharmacokinetics: | 8,415 | - | 2,301 | ||||||
| Simultaneous application: fee for each additional product in the application: | 2,895 | - | 1,011 | ||||||
| Type II: | 2,895 | 6,030 | 1,872 | ||||||
| Type IB: | 885 | 1,325 | 531 | ||||||
| Type IA: | 455 | 685 | 273 | ||||||
| Grouped variations | |||||||||
| Extension-led: | |||||||||
| The fee for an application for an extension-led grouped variation is the fee for that extension as specified above plus — | |||||||||
| (a) if there is one variation in addition to the extension, the fee for that variation as specified above; or | |||||||||
| (b) if there is more than one variation in addition to the extension, the fee that would be payable for a grouped variation of that type as specified below. | |||||||||
| Type II led: | |||||||||
| For the first nine changes: | 6,280 | 12,060 | 3,768 | ||||||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 2,700 | ||||||
| Type IB led: | |||||||||
| For the first nine changes: | 1,770 | 2,650 | 1,062 | ||||||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 2,700 | ||||||
| Type IA led: | |||||||||
| For the first nine changes: | 885 | 1,325 | 531 | ||||||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 2,700 | ||||||
Extent Information
E134This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F247Words in Sch. 7 para. 17 substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 4(2)
Application for a variation to a marketing authorisation dealt with under worksharing proceduresE+W+S
18.—(1) This paragraph applies in relation to an application for a variation to a marketing authorisation dealt with in accordance with worksharing procedures as set out in Article 20 of Commission Regulation (EC) No 1234/.
[F149(2) The fees for a worksharing application are specified in the following table.]
F150(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F150(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F150(5) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
| Type of application | F151... | F152... | F152... | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F152... | F152... | F152... | F152... | F152... | F152... | ||||
Worksharing applications The following fees apply for each change to each product: | |||||||||
| Type II | |||||||||
| For the first nine changes: | 6,240 | F152... | F152... | F152... | F152... | F152... | F152... | ||
| For each subsequent group of up to ten changes: | 4,500 | F152... | F152... | F152... | F152... | F152... | F152... | ||
| Type IB | ... | ||||||||
| For the first nine changes: | 1,770 | F152... | F152... | F152... | F152... | F152... | F152... | ||
| For each subsequent group of up to ten changes: | 4,500 | F152... | F152... | F152... | F152... | F152... | F152... | ||
Textual Amendments
F149Sch. 7 para. 18(2) substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(j)(i); 2020 c. 1, Sch. 5 para. 1(1)
F150Sch. 7 para. 18(3)-(5) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(j)(ii); 2020 c. 1, Sch. 5 para. 1(1)
F151Words in Sch. 7 para. 18 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(j)(iii)(aa); 2020 c. 1, Sch. 5 para. 1(1)
F152Words in Sch. 7 para. 18 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(j)(iii)(bb); 2020 c. 1, Sch. 5 para. 1(1)
Application for a variation to a marketing authorisation dealt with under worksharing proceduresN.I.
18.—(1) This paragraph applies in relation to an application for a variation to a marketing authorisation dealt with in accordance with worksharing procedures as set out in Article 20 of Commission Regulation (EC) No 1234/.
(2) The fee for a worksharing application, involving marketing authorisations obtained by a national procedure in the United Kingdom only, is the fee specified in the following table in the column headed “UK Only”.
(3) The fee for a worksharing application, involving marketing authorisations obtained through a national procedure in the United Kingdom and any other member State, is specified in the following table by reference to the United Kingdom’s role in the procedure, as “UK Reference Authority”, “UK Co-Reference Authority” or “Other”.
(4) The fee for a worksharing application, involving at least one marketing authorisation obtained through the mutual recognition or decentralised procedure, is specified in the following table by reference to the United Kingdom’s role in the procedure, as “UK Reference Authority”, “UK Co-Reference Authority” or “UK Concerned member State”.
(5) The fee for any kind of variation where the Agency co-ordinates worksharing is £455 for each marketing authorisation.
| Type of application | UK Only | Where the application involves nationally authorised products in more than one member State | Application involves mutually recognised products | ||||||
|---|---|---|---|---|---|---|---|---|---|
UK Only | UK Reference Authority | UK Co-Reference Authority | Other | UK Reference Authority | UK Co-Reference Authority | UK Concerned member State | |||
Worksharing applications The following fees apply for each change to each product: | |||||||||
| Type II | |||||||||
| For the first nine changes: | 6,240 | 12,060 | 7,485 | 12,060 | 13,265 | 6,745 | 3,372 | ||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 4,500 | 4,500 | 4,500 | 4,500 | 2,700 | ||
| Type IB | |||||||||
| For the first nine changes: | 1,770 | 2,650 | 2,120 | 2,650 | 2,915 | 1,905 | 954 | ||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 4,500 | 4,500 | 4,500 | 4,500 | 2,700 | ||
Extent Information
E135This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for an extension dealt with under the decentralised procedure where the United Kingdom is the reference member StateE+W+S
F15319. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E63This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F153Sch. 7 para. 19 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(k); 2020 c. 1, Sch. 5 para. 1(1)
Application for an extension dealt with under the decentralised procedure where the United Kingdom is the reference member StateN.I.
19. The fee for a decentralised application for an extension where the United Kingdom is the reference member State is the same as for a national application as set out in the table in paragraph 17, with the addition of the supplementary fees in the following table (save that, where the application is for the addition of more than one species, only one supplementary fee applies).
Decentralised application for an extension where the United Kingdom is the reference member State
| Application | Supplementary fee (£) | |
|---|---|---|
| Pharmaceutical product for a food-producing animal – one member State: | 3,705 | |
| Pharmaceutical product for a non-food-producing animal – one member State: | 3,220 | |
| Immunological product – one member State: | 3,460 | |
| Each additional member State: | 530 | |
| Simultaneous application: fee for each additional product in the application: | ||
| one member State: | 6,670 | |
| each additional member State: | 120 | |
Extent Information
E136This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Provision of information relating to the recognition of a United Kingdom marketing authorisation or an extensionE+W+S
F15420. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E64This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F154Sch. 7 para. 20 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(k); 2020 c. 1, Sch. 5 para. 1(1)
Provision of information relating to the recognition of a United Kingdom marketing authorisation or an extensionN.I.
20.—(1) Where an application is made for the Secretary of State to provide information to other member States to enable them to recognise a marketing authorisation already granted by the United Kingdom the following fees are payable.
(2) Those fees also apply where a marketing authorisation has been granted in more than one member State, the holder applies for an extension for that marketing authorisation and the United Kingdom acts as reference member State.
(3) Where a valid application to provide information to another member State is received within six months of the original grant of the marketing authorisation, or where the Secretary of State has already provided the information to a member State, and a further valid application is made to provide the information to an additional member State within six months of the date the last information was provided, the fees are—
| Type of application | Fee for a pharmacologically equivalent product(a) | Fee (other products) (£) | ||
|---|---|---|---|---|
(a) This fee is payable if the application for the marketing authorisation was on the basis that the product was pharmacologically equivalent to another veterinary medicinal product. | ||||
| Pharmaceutical product for a food-producing animal – one member State: | 3,940 | 2,440 | ||
| Pharmaceutical product for a non-food-producing animal ‑ one member State: | 2,645 | 1,895 | ||
| Immunological product – one member State: | 2,130 | 2,130 | ||
| Each additional member State: | 535 | 535 | ||
(4) Where the information to be provided relates to a product granted a marketing authorisation using identical data submitted simultaneously or on the basis of information provided under Article 13(c) of Directive 2001/82/EC the fees are—
| Application | Fee (£) | |
|---|---|---|
| Provision of information to— | ||
| one member State: | 4,165 | |
| each additional member State: | 530 | |
(5) In any other case the fees are—
| Type of application | Fee for a pharmacologically equivalent product (£)(a) | Fee (other products) (£) | |
|---|---|---|---|
(a) This fee is payable if the application for the marketing authorisation was on the basis that the product was pharmacologically equivalent to another veterinary medicinal product. | |||
| Pharmaceutical product for a food-producing animal – one member State: | 12,015 | 10,515 | |
| Pharmaceutical product for a non-food-producing animal – one member State: | 8,115 | 7,365 | |
| Immunological product – one member State: | 8,940 | 8,940 | |
| Each additional member State: | 535 | 535 | |
(6) In the case of simultaneous applications, the above fees are payable for each additional product in the application for one member State, with a fee of £115 for each additional product for each additional member State.
Extent Information
E137This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Exception for a variation relating to animal testingE+W+S
21. If the only purpose of a variation is to remove animal testing or to reduce the numbers of animals used in testing, no fee is payable for the variation F155....
Extent Information
E65This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F155Words in Sch. 7 para. 21 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(l); 2020 c. 1, Sch. 5 para. 1(1)
Exception for a variation relating to animal testingN.I.
21. If the only purpose of a variation is to remove animal testing or to reduce the numbers of animals used in testing, no fee is payable for the variation in the case of a national authorisation, and the United Kingdom element of the fee for the variation is not payable for an authorisation obtained through the mutual recognition procedure or the decentralised procedure.
Extent Information
E138This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for the renewal of a F156... marketing authorisationE+W+S
22.—(1) The fee for an application for the renewal of a marketing authorisation is £1,360.
(2) The fee for the first reassessment of an exceptional marketing authorisation is £305, and the fee for each subsequent reassessment is £1,360.
Extent Information
E66This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F156Word in Sch. 7 para. 22 heading omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(m); 2020 c. 1, Sch. 5 para. 1(1)
Application for the renewal of a national marketing authorisationN.I.
22.—(1) The fee for an application for the renewal of a marketing authorisation is £1,360.
(2) The fee for the first reassessment of an exceptional marketing authorisation is £305, and the fee for each subsequent reassessment is £1,360.
Extent Information
E139This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for the renewal of a marketing authorisation obtained through mutual recognition or the decentralised procedureE+W+S
F15723. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E67This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F157Sch. 7 para. 23 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(n); 2020 c. 1, Sch. 5 para. 1(1)
Application for the renewal of a marketing authorisation obtained through mutual recognition or the decentralised procedureN.I.
23. The fee for an application for the renewal of a marketing authorisation obtained through mutual recognition or the decentralised procedure is —
(a)£1,835 if the United Kingdom is the reference member State; and
(b)£1,225 if the United Kingdom is a concerned member State.
Extent Information
E140This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Registration of a homeopathic remedyE+W+S
24. The fee for an application for the registration of a homeopathic remedy is in accordance with the following table.
Fee for the registration of a homeopathic remedy
| Type of application | Fees(£) | ||
|---|---|---|---|
| If all stocks and the formulation have already been assessed by the Secretary of State— | |||
| not more than five stocks: | 160 | ||
| more than five stocks: | 375 | ||
| If either all the stocks have already been assessed by the Secretary of State but there is a new formulation, or if the formulation has already been assessed by the Secretary of State but one or more of the stocks have not been already assessed— | |||
| not more than five stocks: | 455 | ||
| more than five stocks: | 665 | ||
| If the formulation and at least one of the stocks has not already been assessed by the Secretary of State— | |||
| not more than five stocks: | 760 | ||
| more than five stocks: | 985 | ||
| If the product is already authorised for human use in the United Kingdom, or for human or veterinary use in the United Kingdom F158...— | |||
| not more than five stocks: | 160 | ||
| more than five stocks: | 375 | ||
Extent Information
E68This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F158Words in Sch. 7 para. 24 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(o); 2020 c. 1, Sch. 5 para. 1(1)
Registration of a homeopathic remedyN.I.
24. The fee for an application for the registration of a homeopathic remedy is in accordance with the following table.
Fee for the registration of a homeopathic remedy
| Type of application | Fees(£) | ||
|---|---|---|---|
| If all stocks and the formulation have already been assessed by the Secretary of State— | |||
| not more than five stocks: | 160 | ||
| more than five stocks: | 375 | ||
| If either all the stocks have already been assessed by the Secretary of State but there is a new formulation, or if the formulation has already been assessed by the Secretary of State but one or more of the stocks have not been already assessed— | |||
| not more than five stocks: | 455 | ||
| more than five stocks: | 665 | ||
| If the formulation and at least one of the stocks has not already been assessed by the Secretary of State— | |||
| not more than five stocks: | 760 | ||
| more than five stocks: | 985 | ||
| If the product is already authorised for human use in the United Kingdom, or for human or veterinary use in the United Kingdom or in another member State— | |||
| not more than five stocks: | 160 | ||
| more than five stocks: | 375 | ||
Extent Information
E141This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Renewal of a homeopathic remedyU.K.
25. The fee for the renewal of a homeopathic remedy is £320.
Annual fees for marketing authorisationsU.K.
26.—(1) Within 30 days of receiving a written demand from the Secretary of State, a holder of a marketing authorisation must provide the Secretary of State with a statement of turnover for the previous calendar year.
(2) The annual fee, rounded to the next £1, is—
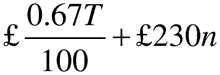
where—
T is the annual turnover in the previous calendar year;
and n is the number of active marketing authorisations held at any time during the previous calendar year.
(3) In the case of an authorisation holder with a turnover relating to all marketing authorisations held of less than £230,000, the annual fee, rounded to the next £1 is—
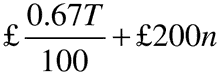
where—
T is the annual turnover in the previous calendar year;
and n is the number of active marketing authorisations held at any time during the previous calendar year.
(4) In this paragraph—
“turnover” means the sales value at manufacturers’ prices of all authorised veterinary medicinal products sold or supplied in the United Kingdom;
“manufacturers’ prices” means the prices charged (excluding value added tax) for authorised products by manufacturers to wholesalers, except to the extent that—
the products are supplied by manufacturers direct to retailers, in which case it means the prices charged for the products by the manufacturers to the retailers reduced by such sum as, in the opinion of the Secretary of State, represents the difference between the prices paid by the retailers and those which could be expected to be charged by the manufacturers to wholesalers according to the practice prevailing during the period in question with regard to such products;
a marketing authorisation holder sells or supplies products that the marketing authorisation holder has neither manufactured nor obtained from the manufacturer, in which case it means the prices paid by the marketing authorisation holder for those products.
Auditor’s certificateU.K.
27.—(1) The Secretary of State may at any time require an audit certificate in support of a statement of turnover.
(2) If the holder of the marketing authorisation does not provide an audit certificate before the date stipulated in the demand, an additional fee is payable for that year of £11,300 plus an additional £2,245 in respect of each marketing authorisation held.
(3) If the Secretary of State is not satisfied that the audit certificate provides sufficient assurance that the figures fairly present the financial records of the company, the Secretary of State may require the marketing authorisation holder to produce a further certificate and specify what further assurances are needed; and if these are not provided by the required date, the additional fee specified in sub-paragraph (2) is payable.
(4) Nothing in this paragraph limits the powers of an inspector to examine financial records.
PART 3U.K.Fees payable by manufacturers
Application for a manufacturing authorisationU.K.
28. The fee for an application for a manufacturing authorisation for a veterinary medicinal product is—
(a)£3,040; or
(b)£530 if the authorisation only covers veterinary medicinal products manufactured under Schedule 6 (exemptions for small pet animals).
Application for a variation of a manufacturing authorisationU.K.
29. The fee for an application for the variation of a manufacturing authorisation is—
(a)£636 if the variation requires scientific or pharmaceutical assessment;
(b)£443 if the variation only involves a change of ownership;
(c)£210 if the authorisation only covers veterinary medicinal products manufactured under Schedule 6 (exemptions for small pet animals); and
(d)otherwise £350.
Application for an authorisation to manufacture an autogenous vaccine or a product for administration under the cascadeU.K.
30.—(1) The fee for an application for a standard authorisation to manufacture an autogenous vaccine or a veterinary medicinal product for administration under the cascade is—
(a)£3,435 for a site in the United Kingdom;
(b)£3,270 for a site outside the United Kingdom.
(2) The fee for each inspection after a standard authorisation has been granted is (in each case) the same as the fee specified in paragraph (1).
(3) In the case of an application for an individual authorisation to manufacture a single batch of autogenous vaccine, or a single batch of veterinary medicinal product for administration under the cascade the fee is £1,635.
(4) The fee to vary an authorisation is £305 if no further inspection is required, and otherwise is the full application fee.
Annual feesU.K.
31.—(1) An annual fee of £550 is payable in respect of each manufacturing authorisation held (other than as specified in this paragraph).
(2) The annual fee for a manufacturing authorisation for an autogenous vaccine or a veterinary medicinal product for administration under the cascade is 0.67% of the turnover in the previous calendar year rounded to the next £1, with a minimum fee of £10.
(3) There is no annual fee for a manufacturing authorisation for a veterinary medicinal product manufactured in accordance with Schedule 6 for small pet animals.
(4) In this paragraph “turnover” means the sales value at manufacturers’ prices net of value added tax of all authorised veterinary medicinal products sold or supplied in the United Kingdom.
Site inspections – type of siteU.K.
32. For the purposes of deciding the fee for a site inspection—
“super site” is a site at which 250 or more relevant persons are employed;
“major site” is a site at which 60 or more, but fewer than 250, relevant persons are employed;
“standard site” is a site at which 10 or more, but fewer than 60 relevant persons are employed;
“minor site” is a site at which fewer than 10 relevant persons are employed;
“relevant person” means a person employed on the premises and systems inspected.
Inspection of a site where immunological veterinary medicinal products are manufacturedU.K.
33. The fees for the inspection of a site where immunological veterinary medicinal products are manufactured are in accordance with the following table.
Sites where immunological veterinary medicinal products are manufactured
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside United Kingdom | |
| Super site | 24,071 | 22,867 |
| Major site | 16,785 | 15,946 |
| Standard site | 6,661 | 6,327 |
| Minor site | 4,757 | 4,519 |
Inspection of a site where sterile veterinary medicinal products are manufacturedU.K.
34. The following fees are payable for the inspection of a site where no immunological veterinary medicinal products are manufactured, but where sterile products are manufactured.
Sites where sterile veterinary medicinal products are manufactured
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 23,324 | 22,157 |
| Major site | 13,010 | 12,359 |
| Standard site | 8,244 | 7,832 |
| Minor site | 5,022 | 4,770 |
Inspection of a site where no immunological or sterile veterinary medicinal products are manufacturedU.K.
35. The following fees are payable for the inspection of a site where only non-immunological and non-sterile veterinary medicinal products are manufactured—
Site where no immunological or sterile veterinary medicinal products are manufactured
| Type of site | Fee (£) | ||
|---|---|---|---|
| United Kingdom site | Site outside the United Kingdom | ||
| Super site | 14,180 | 13,471 | |
| Major site | 8,325 | 7,909 | |
| Standard site | 6,854 | 6,511 | |
| Minor site | 3,789 | 3,600 | |
| If the site is only involved in the manufacture of veterinary medicinal products authorised under Schedule 6 (exemptions for small pet animals— | |||
| Standard site | 5,055 | 4,802 | |
| Minor site | 2,728 | 2,592 | |
Inspection of a site where veterinary medicinal products are assembledU.K.
36. The following fees are payable for the inspection of a site where the only manufacturing process in relation to veterinary medicinal products is their assembly after the product has been put into its immediate container.
Site where medicinal products are assembled
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 11,025 | 10,474 |
| Major site | 5,949 | 5,652 |
| Standard site | 4,917 | 4,671 |
| Minor site | 2,035 | 1,933 |
Test sitesU.K.
37. The fee for the inspection of a test site is £3,344, or £3,177 for a site outside the United Kingdom.
Animal blood bank or equine stem cell centre authorisationsU.K.
38.—(1) The fee for an authorisation to operate a blood bank is—
(a)on a first inspection £3,113; and
(b)on each subsequent inspection—
(i)£3,113 for a site in the United Kingdom; and
(ii)£2,966 for a site outside the United Kingdom.
(2) The fee for an authorisation to operate an equine stem cell centre is £3,427, and £3,092 for each subsequent inspection.
(3) The fee for a variation to an authorisation to operate a blood-bank or equine stem cell centre is £320.
PART 4U.K.Fees relating to a wholesale dealer’s authorisation
Application for a wholesale dealer’s authorisationU.K.
39.—(1) The fee for an application for a wholesale dealer’s authorisation is—
(a)£1,745;
(b)£785 if the application is accompanied by an estimate that the first year’s turnover will be less than £35,000; or
(c)£785 if the authorisation only relates to products classified as AVM-GSL, homeopathic remedies, or products authorised under Schedule 6 (exemptions for small pet animals).
(2) An applicant who has paid a fee of £785 on the grounds of turnover must send a declaration of turnover for the first year of trading on the anniversary of the grant of the authorisation, and if the figure is more than £35,000 must pay the balance of £960 within 30 days.
(3) If the applicant paid £1,745 but the turnover for the first year of trading was lower than £35,000, if the applicant sends a declaration certifying the turnover, the Secretary of State must refund the excess.
(4) Nothing in this paragraph limits the powers of an inspector to examine financial records.
(5) In this paragraph “turnover” means the sales value net of value added tax of all veterinary medicinal products (whether or not authorised for use in the United Kingdom) sold by way of wholesale dealing by the holder in the United Kingdom.
Variation of a wholesale dealer’s authorisationU.K.
40. The fee for an application to vary a wholesale dealer’s authorisation is—
(a)£515 if the variation requires scientific or pharmaceutical assessment;
(b)£430 if the variation only involves a change of ownership; and
(c)otherwise £300.
Annual fee for a wholesale dealer’s authorisationU.K.
41.—(1) The annual fee for a wholesale dealer’s authorisation is—
(a)£483; or
(b)£315, if—
(i)the holder certifies when making the payment that the turnover during the previous year was less than £35,000; or
(ii)the authorisation only relates to products classified as AVM-GSL or homeopathic remedies;
(c)£215 if the authorisation only relates to products authorised under Schedule 6 (exemptions for small pet animals).
(2) In this paragraph “turnover” means the sales value net of value added tax of all veterinary medicinal products (whether or not authorised for use in the United Kingdom) sold by way of wholesale dealing by the holder in the United Kingdom.
Inspection of a wholesale dealer’s premisesU.K.
42. The fee for the inspection of a wholesale dealer’s premises is—
(a)£3,058; or
(b)£1,442 if—
(i)the authorisation only relates to products classified as AVM-GSL or homeopathic remedies; or
(ii)the turnover relating to all veterinary medicinal products in the calendar year preceding the inspection was less than £35,000;
(c)£830 if the authorisation only relates to products authorised under Schedule 6 (exemptions for small pet animals).
PART 5U.K.Fees relating to feedingstuffs
Fees for approvals and annual fees relating to feedingstuffs in Great BritainU.K.
43.—(1) Subject to sub-paragraph (3) the fee for the application for approval of establishments manufacturing feedingstuffs and approval of distributors of feedingstuffs in Great Britain is £70.
(2) An annual fee of £70 is payable in respect of any such approval.
(3) No fee is payable under sub-paragraph (1) in respect of an establishment where specified feed additives are manufactured if a veterinary medicinal product intended to be incorporated into feedingstuffs is manufactured at that establishment in accordance with a manufacturing authorisation.
(4) Fees relating to feedingstuffs are payable with the application or on invoice for the subsequent annual fee.
(5) Where more than one manufacturing activity is carried out at one establishment only one fee (the highest) is payable.
Inspection fees relating to feedingstuffs in Great BritainU.K.
44. Fees for the inspection of establishments manufacturing or distributing feedingstuffs in Great Britain are in accordance with the following table.
Inspection fees
| Type of establishment inspected | Fee payable (£) | |
|---|---|---|
(1) No fee is payable for premises that already have a manufacturing authority relating to veterinary medicinal products for incorporating into feedingstuffs. | ||
| 1 | Establishment manufacturing a specified feed additive:(1) | 1,810 |
| 2 | Establishment manufacturing a premixture: | 1,090 |
| 3 | Establishment manufacturing feedingstuffs using specified feed additives and veterinary medicinal products directly at any concentration, or using premixtures or specified feed additive complementary feedingstuffs: | 1,090 |
| 4 | Establishment manufacturing feedingstuffs for placing on the market using a veterinary medicinal product or premixture where the concentration of veterinary medicinal product in the feedingstuffs is 2 kg per tonne or more: | 961 |
| 5 | Establishment manufacturing feedingstuffs using premixtures or specified feed additive complementary feedingstuffs containing specified feed additives when the feedingstuffs are to be placed on the market: | 405 |
| 6 | Establishment manufacturing feedingstuffs for the manufacturers own use using a veterinary medicinal product or premixture where the concentration of veterinary medicinal product in the feedingstuffs is 2 kg per tonne or more: | 320 |
| 7 | Establishment manufacturing feedingstuffs using premixtures containing specified feed additives when the feedingstuffs are to be used by the person manufacturing the feedingstuffs: | 240 |
| 8 | Establishment distributing specified feed additives, premixtures or feedingstuffs containing specified feed additives, or premixtures or complementary feedingstuffs containing veterinary medicinal products: | 227 |
Fees payable in relation to feedingstuffs in Northern IrelandE+W+S
F15945. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E69This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F159Sch. 7 para. 45 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(10)
Fees payable in relation to feedingstuffs in Northern IrelandN.I.
45.—(1) The annual fees payable for the approval of establishments manufacturing and distributing feedingstuffs in Northern Ireland are in accordance with the following table.
(2) Fees are payable with the application or, for the subsequent annual fee, on invoice.
(3) Where more than one manufacturing activity is carried out at one establishment only the highest fee is payable.
Approval fees
| Type of establishment | Fee payable (£) | |
|---|---|---|
(a) No fee is payable for establishments that already have a manufacturing authority relating to veterinary medicinal products for incorporating into feedingstuffs. | ||
| 1 | Establishment manufacturing a specified feed additive(a): | 545 |
| 2 | Establishment manufacturing a premixture: | 435 |
| 3 | Establishment manufacturing feedingstuffs using specified feed additives and veterinary medicinal products directly at any concentration, or using premixtures or specified feed additive complementary feedingstuffs: | 435 |
| 4 | Establishment manufacturing feedingstuffs for placing on the market using a veterinary medicinal product or premixture where the concentration of veterinary medicinal product in the feedingstuffs is 2 kg per tonne or more: | 320 |
| 5 | Establishment manufacturing feedingstuffs using premixtures or specified feed additive complementary feedingstuffs containing specified feed additives when the feedingstuffs are to be placed on the market: | 170 |
| 6 | Establishment manufacturing feedingstuffs for the manufacturers own use using a veterinary medicinal product or premixture where the concentration of veterinary medicinal product in the feedingstuffs is 2 kg per tonne or more: | 131 |
| 7 | Establishment manufacturing feedingstuffs using premixtures containing specified feed additives when the feedingstuffs are to be used by the person manufacturing the feedingstuffs: | 110 |
| 8 | Establishment distributing specified feed additives, premixtures or feedingstuffs containing specified feed additives, or premixtures or feedingstuffs containing veterinary medicinal products: | 70 |
Extent Information
E142This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Fees relating to premises for supply by suitably qualified personsU.K.
46.—(1) The fee to approve of premises for the retail supply of veterinary medicinal products by suitably qualified persons is—
(a)£265; or
(b)if the premises are only authorised to supply veterinary medicinal products for the treatment of—
(i)horses (or horses and companion animals) £145; or
(ii)companion animals £110.
(2) The subsequent annual fee is—
(a)£185; or
(b)if the premises are only authorised to supply veterinary medicinal products for the treatment of—
(i)horses (or horses and companion animals) £95; or
(ii)companion animals £70.
PART 6U.K.General
Testing samplesU.K.
47. The fee for testing a sample required to be submitted by the Secretary of State is the full economic cost of the test.
Animal test certificatesE+W+S
48.—(1) The fee for an animal test certificate is [F160£815].
(2) The fee for an animal test certificate to administer medicinal products in a small scale trial to test them for clinical safety or efficacy is £30.
F161(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(4) The fee for an application for a variation of the certificate is £265 for each change.
(5) The fee for an application to renew a certificate is £130.
(6) The Secretary of State may waive the fee if satisfied that the application is in relation to developing a veterinary medicinal product for a limited market (for example, for a minor species, a minor use, or for a disease with restricted regional distribution).
Extent Information
E70This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F160Sum in Sch. 7 para. 48(1) substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(p)(i); 2020 c. 1, Sch. 5 para. 1(1)
F161Sch. 7 para. 48(3) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(p)(ii); 2020 c. 1, Sch. 5 para. 1(1)
Animal test certificatesN.I.
48.—(1) The fee for an animal test certificate is £345 in the case of—
(a)an immunological veterinary medicinal product that has been authorised in another member State for the species on which the proposed test will be conducted;
(b)a pharmaceutical veterinary medicinal product that has been authorised in another member State for use with a food-producing species on which the proposed test will be conducted where the same or similar dosage regime and method of administration is to be used in the medicinal test as is authorised; or
(c)a pharmaceutical veterinary medicinal product authorised in another member State for human or animal use where the test is to be conducted on companion animals only.
(2) The fee for an animal test certificate to administer medicinal products in a small scale trial to test them for clinical safety or efficacy is £30.
(3) In any other case the fee is £815.
(4) The fee for an application for a variation of the certificate is £265 for each change.
(5) The fee for an application to renew a certificate is £130.
(6) The Secretary of State may waive the fee if satisfied that the application is in relation to developing a veterinary medicinal product for a limited market (for example, for a minor species, a minor use, or for a disease with restricted regional distribution).
Extent Information
E143This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Importation of a veterinary medicinal product for treatment under the cascadeE+W+S
49.—(1) The fee for a certificate to import (if necessary) and be in possession of and administer a veterinary medicinal product under the cascade is—
F162(a). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(b)£30 if the veterinary medicinal product is authorised in [F163another] country.
(2) The fee is payable in respect of each animal treated, but in the case of administration to and treatment of a discrete group of animals, the Secretary of State may notify the applicant in writing that a fee for only one animal is payable.
(3) There is no fee if the application is made using the website of the Veterinary Medicines Directorate.
Extent Information
E71This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F162Sch. 7 para. 49(1)(a) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(q)(i); 2020 c. 1, Sch. 5 para. 1(1)
F163Word in Sch. 7 para. 49(1)(b) substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(q)(ii); 2020 c. 1, Sch. 5 para. 1(1)
Importation of a veterinary medicinal product for treatment under the cascadeN.I.
49.—(1) The fee for a certificate to import (if necessary) and be in possession of and administer a veterinary medicinal product under the cascade is—
(a)£15 if the veterinary medicinal product is authorised in another member State;
(b)£30 if the veterinary medicinal product is authorised in a third country.
(2) The fee is payable in respect of each animal treated, but in the case of administration to and treatment of a discrete group of animals, the Secretary of State may notify the applicant in writing that a fee for only one animal is payable.
(3) There is no fee if the application is made using the website of the Veterinary Medicines Directorate.
Extent Information
E144This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Wholesale dealer’s import certificateU.K.
50.—(1) The fee payable by the holder of a wholesale dealer’s authorisation for a certificate to import and store a veterinary medicinal product not authorised in the United Kingdom to enable it to be supplied for administration under Schedule 4 is [F164£760] .
(2) The fee is only payable if, in the twelve month period immediately before the application, the applicant has supplied the veterinary medicinal product to which the certificate relates in accordance with at least 100 certificates.
Textual Amendments
F164Word in Sch. 7 para. 50 substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 4(3)
Specific batch controlU.K.
[F16551. The fee for an authorisation to release a veterinary medicinal product under specific batch control is—
(a)£560; and
(b)£100 for each additional batch affected by the same issue where the specific batch control application is made at the same time.]
Textual Amendments
F165Sch. 7 para. 51 substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 4(4)
Submission of control tests of an immunological productU.K.
52. The fee for the submission of the results of tests carried out on a batch of immunological products other than autogenous vaccines prior to release is £80.
Export certificatesU.K.
53. The fee for an application for an export certificate is £30, and £15 for each certified copy.
Provision of adviceU.K.
54. The fee for an application for written advice from the Secretary of State as to whether or not a product requires a marketing authorisation is £885.
Appeals to the Veterinary Products CommitteeU.K.
55. The fee for an appeal to the Veterinary Products Committee is £1,500.
Fee relating to an appointed personU.K.
56. The appellant is liable for the full economic cost of a referral to an appointed person subject to a maximum of £5,000.
Fees relating to a veterinary surgeon’s practice premisesU.K.
57.—(1) The fee for the inspection of a veterinary surgeon’s practice premises is £350.
(2) The initial registration and annual fee for the registration of veterinary practice premises with the Royal College of Veterinary Surgeons to supply veterinary medicinal products is £34.
(3) Notwithstanding paragraph 2 of this Schedule, this is payable to the Royal College of Veterinary Surgeons.
Refund of fees relating to the Veterinary Products Committee or appointed personsU.K.
58. The Secretary of State must refund the fee payable in relation to an appeal to the Veterinary Products Committee or to an appointed person if, as a result of the appeal, the Secretary of State changes the decision that was the subject of the appeal.
Fees relating to an improvement noticeU.K.
59. If an improvement notice is served under these Regulations, the fee for any subsequent inspection necessary as a result of the notice is the full economic cost of the inspection, payable by the person on whom the notice was served.
Non-payment of feesU.K.
60. Where any fee (other than any fee relating to a manufacturing authorisation or wholesale dealer’s authorisation) is not paid, the Secretary of State may, after giving one month’s written warning, suspend the processing of any application from the person who has not paid the fee.
Waiver or reduction of feesU.K.
61.—(1) If the Secretary of State is satisfied that for reasons of human or animal health or the protection of the environment it is desirable that a product should be authorised for veterinary use or that an authorised product should remain on the market the Secretary of State may waive or reduce any fees payable under these Regulations.
(2) An applicant or the holder of a marketing authorisation must provide full written justification for any waiver or reduction.
Reduction of fees when an application is withdrawnU.K.
62.—(1) Where an application for a marketing authorisation, or any variation referred to in paragraph 17 or 18 as a Type II variation, an extension, an extension-led grouped variation or a Type II led grouped variation is withdrawn before determination, the fee is reduced in accordance with this paragraph.
(2) If no assessment (veterinary, scientific or pharmaceutical) has begun, the reduction is 90%.
(3) If assessment has begun but the Secretary of State has not yet requested further data, the reduction is 50%.
(4) If the Secretary of State has requested further information but it has not yet been provided, the reduction is 25%.
(5) If the further information requested has been supplied but has not yet been fully assessed or the application has not been referred to the Veterinary Products Committee, the reduction is 10%
(6) Once the further information has been fully assessed, or the application has been referred to the Veterinary Products Committee, there is no reduction.
EXPLANATORY NOTE
(This note is not part of the Regulations)
These Regulations revoke and remake with amendments the Veterinary Medicines Regulations 2011 (S.I. 2011/2159).
Principal changes to the 2011 RegulationsU.K.
The major change to the Regulations is the adjustment of the fees with a view to achieving full cost recovery while avoiding cross-subsidy of one activity by another.
In Great Britain food businesses will pay a much lower fee on application for approval but will pay a larger fee for any inspection. Premises will be selected for inspection on the basis of risk analysis.
The fees for appeals to the Veterinary Products Committee are simplified.
Criminal offences have also been amended. Instead of creating an individual offence in relation to every obligation there is now a single offence governing all relevant obligations in the body of the Regulations and a single offence in each of Schedules 1 to 5.
Other changesU.K.
Regulation 35 extends inspectors’ power of seizure to cover anything they reasonably believe to be, or which purports to be, a veterinary medicine.
Veterinary practice premises must be registered with the Royal College of Veterinary Surgeons and paragraph 8 of Schedule 3 gives the Secretary of State a power to require the removal of premises from this register where they fail to meet the necessary standard.
The RegulationsU.K.
The Regulations make provision for the authorisation, manufacture, classification, distribution and administration of veterinary medicinal products.
They implement the following EU instruments that are Directives:
(a)Council Directive 90/167/EEC laying down the conditions governing the preparation, placing on the market and use of medicated feedingstuffs in the Community, so far it is not superseded by Regulation (EC) No 183/2005;
(b)Commission Directive 91/412/EEC laying down the principles and guidelines of good manufacturing practice for veterinary medicinal products; and
(c)Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products.
They provide for the enforcement of the following EU instruments that are Regulations besides that mentioned above:
(d)Regulation (EC) No 178/2002 of the European Parliament and of the Council laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety (OJ No L 31, 1.2.2002 p. 1), in so far as it applies to veterinary medicinal products used in feedingstuffs
(e)Regulation (EC) No 1831/2003 of the European Parliament and of the Council on additives for use in animal nutrition (OJ No L 268, 18.10.2003 p. 29), in so far as it applies to veterinary medicinal products used in feedingstuffs;
(f)Regulation (EC) No 882/2004 of the European Parliament and of the Council on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules (OJ NO L 191, 28.5.2004, p.1), in so far as it applies to veterinary medicinal products used in feedingstuffs;
(g)Regulation (EC) No 183/2005 of the European Parliament and of the Council laying down requirements for feed hygiene (OJ No L 35, 8.2.2005, p. 1), in so far as it applies to veterinary medicinal products used in feedingstuffs; and
(h)Regulation (EC) No 470/2009 of the European Parliament and of the Council, laying down Community procedures for the establishment of residue limits of pharmacologically active substances in foodstuffs of animal origin (OJ No L152, 16.6.2009, p. 11).
They provide that a veterinary medicinal product must have a marketing authorisation granted by the Secretary of State before being placed on the market, and they make provision for the grant of a marketing authorisation (regulation 4 and Schedule 1).
They specify that a veterinary medicinal product must be manufactured by a person holding a manufacturing authorisation, and make provision for granting an authorisation (regulation 5 and Schedule 2).
They regulate the supply and possession of veterinary medicinal products, and introduce new classifications of those products (regulation 7 and Schedule 3).
They provide that a veterinary medicinal product may only be administered as specified in its marketing authorisation or, in the case of administration by a veterinary surgeon, administration under the rules of the “cascade” (regulation 8 and Schedule 4).
They control bringing a veterinary medicinal product into the United Kingdom (regulation 9) and advertising (regulation 10 to 12).
They control wholesale dealing (regulation 13 and Schedule 3).
They control medicated feedingstuffs and feedingstuffs containing additives specified in the Regulations (regulation 14 and Schedule 5).
They provide for exemptions (regulation 15 and Schedule 6).
They provide for fees (regulation 16 and Schedule 7).
They require records to be kept (regulations 17 to 24).
They create an offence of importation, possession or supply of unauthorised veterinary medicinal products (regulation 43(q) to (s)).
They make provision for the existence of the Veterinary Products Committee (regulation 28). They make provision for an appeals procedure in the case of a refusal, etc., of a marketing authorisation (regulation 30).
They create administrative arrangements for the enforcement of the Regulations (regulations 32 to 36 and 38 to 42) and create offences of obstructing a person acting in the execution of these Regulations (regulation 43(u)) and of failing to comply with an improvement notice (regulation 43(v)).
Under regulation 44 breach of the Regulations is an offence punishable—
(i)on summary conviction, by a fine not exceeding the statutory maximum or by imprisonment for a term not exceeding three months or both, or
(j)on conviction on indictment, by a fine or to imprisonment for a term not exceeding two years or both.
Regulation 46 requires the Secretary of State to review the operation and effect of these Regulations, other than regulation 16 and Schedule 7 (which relate to fees), and lay a report before Parliament within five years after they come into force and within every five years after that. Following a review it will fall to the Secretary of State to consider whether the Regulations should remain as they are, or be revoked or be amended. A further instrument would be needed to revoke the Regulations or to amend them.
Regulation 47 revokes the Veterinary Medicines Regulations 2011.
A full impact assessment has been prepared and placed in the libraries of both Houses of Parliament. It is available, together with a transposition note and a table showing fee changes, on www.vmd.defra.gov.uk at “Publications, Veterinary Medicines Regulations and Guidance”. It is also published with the Explanatory Memorandum alongside the instrument on www.legislation.gov.uk.
OJ No L31, 1.2.2002, p. 1.
OJ No L136, 30.4.2004, p. 1.
OJ No L334, 12.12.2008, p.7.
OJ No L 15, 20.1.2010, p. 1.
OJ No L 149, 7.6.2008, p. 3.
OJ No L152, 16.6.2009, p. 11.
OJ No L229, 1.9.2009, p. 1. Regulation (EC) No 767 2009 was last amended by Regulation (EC) 939/2010 (OJ No L277, 20.10. 2010, p. 4).
If the manufacture is carried out in the United Kingdom the manufacturer must hold a manufacturing authorisation for that type of product granted by the Secretary of State.
For provisions on breaking open packages see regulation 7(3).
Published by the World Health Organization at: www.who.int/medicines/en.
1980 c. 43; sections 51 and 52 have been substituted by the Courts Act 2003 (c.39), section 47.
Other offences are set out at the end of Schedules 1, 2, 3, 4 and 5.
OJ No L 92, 7.4.1990, p. 42.
OJ No L 228, 17.8.1991, p. 70.
OJ No L311, 28.11.2001, p. 1; last amended by Regulation (EC) No 596/2009 of the European Parliament and of the Council (OJ No L188, 18.7.2009, p. 14).
OJ No L334, 12.12.2008, p. 7.
OJ No L152, 16.6.2009, p. 11.
OJ No L229, 1.9.2009, p. 1, last amended by Commission Regulation (EU) No 939/2010 (OJ L277, 21.10.2010, p. 14).
OJ No L293, 11.11.2010, p.72; corrected at OJ L293, 11.11.2010, p. 72.
OJ No L 211, 28.11.2001, p. 1 as last amended by Regulation (EC) No 470/2009 of the European Parliament and of the Council (OJ No L152, 16.6.2009, p. 11). Annex I was inserted by Commission Directive 2009/9/EC (OJ No L 44, 14.2.2009, p. 10).
OJ No L334, 12.12.2008, p. 7.
ISBN 9287145873.
OJ No L 228, 17.8.91, p. 70.
S. I. 2001/3998; relevant amending instruments are S. I. 2003/1432 and 2005/1653.
Published at: http://www.vmd.defra.gov.uk/registers/sqpregister.aspx.
OJ No C 63, 1.3.94, p. 4.
The number of days of the withdrawal period is calculated by dividing 500 by the mean temperature of the water in degrees Celsius.
OJ No L42, 13.2.2013, p. 1.
Published at http://www.rcvs.org.uk/advice-and-guidance/code-of-professional-conduct-for-veterinary-surgeons/.
OJ No L 92, 7.4.1990, p. 42.
OJ No L 35, 8.2.2005, p. 1.
OJ No L86, 6.4.1979, p. 30.
OJ No L268, 18.10.2003, p. 29. Regulation (EC) no 1831/2003 was last amended by Article 29 of Regulation (EC) No 767/2009 (OJ No L229, 1.9.2009, p. 1.)
OJ No L 92, 7.4.90, p. 42.