ANNEX IU.K.SCOPE OF THE FIRST COMMUNITY METHODS OF ANALYSIS DIRECTIVE FOR EDIBLE CASEINS AND CASEINATES
General Provisions
Determination of moisture in:
acid caseins using method 1, Annex II
rennet caseins using method 1, Annex II
caseinates using method 1, Annex II
Determination of protein content in:
acid caseins using method 2, Annex II
rennet caseins using method 2, Annex II
caseinates using method 2, Annex II
Determination of titratable acidity in:
acid caseins using method 3, Annex II
Determination of ash (including P2O5) in:
acid caseins using method 4, Annex II
rennet caseins using method 5, Annex II
Determination of pH in:
caseinates using method 6, Annex II
ANNEX IIU.K.METHODS OF ANALYSIS RELATING TO THE COMPOSITION OF EDIBLE CASEINS AND CASEINATES
GENERAL PROVISIONS U.K.
1.PREPARATION OF THE ANALYSIS SAMPLEU.K.
1.1. General U.K.
The mass of the sample presented to the laboratory for analysis shall be at least 200 grams.
1.2. Preparation of the sample for analysis in the laboratory U.K.
1.2.1.Thoroughly mix and break down any lumps, etc., in the laboratory sample by repeatedly shaking and inverting the container (if necessary, after having transferred all of the laboratory sample to an air-tight container of sufficient capacity (twice Volume of sample) to allow this operation to be carried out).U.K.
1.2.2.Transfer a representative portion of the sample, i.e. about 50 grams of the thoroughly mixed laboratory sample (1.2.1) to the test sieve (3.3).U.K.
1.2.3.If the 50 gram portion completely or almost completely passes (at least 95 % by weight) through the sieve (3.3), use for the determination the sample as prepared in 1.2.1.U.K.
1.2.4.Otherwise, grind the 50 gram portion, using the grinding device (3.4), until it satisfies the sieving criterion (1.2.3). Immediately transfer all the sieved sample to an air-tight container of sufficient capacity (twice volume of sample) and mix thoroughly by repeated shaking and inverting. During these operations, take precautions to avoid any change in the moisture content of the product.U.K.
1.2.5.After the test sample has been prepared, any determination should be proceeded with as soon as possible.U.K.
1.3. Containers U.K.
The sample shall always be kept in an air-tight and moisture-tight container.
2.REAGENTSU.K.
2.1. Water U.K.
2.1.1.Wherever mention is made to water for solution, dilution or washing purposes, distilled water, or de-mineralized water of at least equivalent purity, shall be used.U.K.
2.1.2.Wherever reference is made to ‘solution’ or ‘dilution’ without further indication, ‘solution in water’ or ‘dilution with water’ is meant.U.K.
2.2. Chemicals U.K.
All chemicals used shall be of recognized analytical reagent quality except where otherwise specified.
3.EQUIPMENTU.K.
3.1. Lists of equipment U.K.
The lists of equipment contain only those items with a specialized use and items to a particular specification.
3.2. Analytical balance U.K.
Analytical balance means a balance capable of weighing to at least 0,1 mg.
3.3. Test sieve U.K.
The test ieves to be used are to be fitted with a lid, to be of diameter 200 mm, to be constructed of wire cloth with a nominal aperture size of 500 urn. The aperture tolerances and wire diameters to be permitted are as given in ISO 3310/1. (Test sieves — Technical requirements and testing — Part 1: Metal wire cloth. ISO 3310/1 — 1975).
The sieves are to be fitted with a receiver.
3.4. Grinding device U.K.
For grinding the laboratory sample if necessary (see 1.2.4), without development of undue heat and without loss or absorption of moisture, a hammer mill shall not be used.
4.EXPRESSION OF RESULTSU.K.
4.1. Results U.K.
The result stated in the analytical report is to be the mean value obtained from two determinations which satisfy the repeatability criterion for that method.
4.2. Calculation of percentage U.K.
Except where otherwise specified, the result shall be calculated as a percentage by mass of the sample.
5.TEST REPORTU.K.
The test report shall identify the method of analysis used as well as the results obtained. In addition, it shall mention all details of procedure not specified in the method of analysis or which are optional, as well as any circumstances that may have influenced the results obtained. The test report shall give all the information necessary for the complete identification of the sample.
METHOD 1U.K.
DETERMINATION OF MOISTURE CONTENT U.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method determines the moisture content in:
acid caseins
rennet caseins
caseinates
2.DEFINITIONU.K.
The moisture content of caseins and caseinates: the loss of mass as determined by the method specified.
3.PRINCIPLEU.K.
The residual mass of a test portion is determined after drying at atmospheric pressure in an oven at 102 oC ± 1 oC to constant mass. The loss of mass is calculated as a percentage by mass of the sample.
4.APPARATUSU.K.
4.1. Analytical balance U.K.
4.2. Dishes, flat-bottomed and of material non-corrodible under the conditions of test e.g. nickel, aluminium, stainless steel or glass. The dishes must have lids which fit tightly but which can readily be removed. Suitable dimensions are: diameter 60 to 80 mm and depth about 25 mm.U.K.
4.3. Atmospheric pressure drying oven, well ventilated, thermostatically controlled with temperature regulation (at 102 oC ± 1 oC). The temperature should be uniform throughout the oven.U.K.
4.4. Desiccator, containing freshly activated silica gel with a water content indicator or an equivalent desiccant.U.K.
4.5. Suitable device for handling dishes, e.g. laboratory tongs.U.K.
5.PROCEDUREU.K.
5.1. Preparation of the test sample U.K.
As described in Section 1.2 of the General Provisions.
5.2. Preparation of the dish U.K.
5.2.1.Heat the uncovered dish and its lid (4.2) in the oven (4.3), controlled at 102 oC ± 1 oC, for at least one hour.U.K.
5.2.2.Place the lid on the dish, transfer the covered dish to the desiccator (4.4), allow to cool to the temperature of the balance room and weigh to the nearest 0,1 mg (m0).U.K.
5.3. Test portion U.K.
Place 3 to 5 grams of the test sample (5.1) into the dish, cover with the lid and weigh to the nearest 0,1 mg (m1).
5.4. Determination U.K.
5.4.1.Uncover the dish and place it with its lid in the oven (4.3), controlled at 102 oC ± 1 oC, for four hours.U.K.
5.4.2.Replace the lid on the dish, transfer to the desiccator, allow to cool to the temperature of the balance room and weigh to the nearest 0,1 mg.U.K.
5.4.3.Uncover the dish and heat it again, with its lid, in the oven for one hour. Then repeat operation 5.4.2.U.K.
5.4.4.If the mass obtained in 5.4.3 is less than the mass obtained in 5.4.2 by more than 1 mg, repeat operation 5.4.3.U.K.
If an increase in mass occurs, use the lowest recorded mass in the calculation (6.1).
Let the final weight recorded be m2g. The total drying time should not normally exceed six hours.
6.EXPRESSION OF RESULTSU.K.
6.1. Method of calculation U.K.
The loss of mass on drying of the sample, expressed as a percentage by mass, is given by:

×
=
mass, in g of the dish and its lid after process 5.2;
=
mass, in g of the dish, its lid and the test portion before drying (process 5.3);
=
mass, in g of the dish, its lid and the test portion after drying (process 5.4.3 or 5.4.4).
Calculate the loss on drying to the nearest 0,01 %.
6.2. Repeatability U.K.
The difference in results between two determinations carried out simultaneously or in rapid succession on the same sample, by the same analyst, under the same conditions, shall not exceed 0,1 g of moisture per 100 grams of product.
This repeatability interval should be achieved in 95 % of the times that the method is carried out.
METHOD 2U.K.
DETERMINATION OF PROTEIN CONTENT U.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method determines the protein content of:
acid caseins,
rennet caseins,
caseinates,
except those containing ammonium caseinate or other ammonium or nitrogenous non-protein compounds.
2.DEFINITIONU.K.
The protein content: the nitrogen content as determined by the method specified and then multiplied by 6,38 and expressed as a percentage by mass.
3.PRINCIPLEU.K.
A test portion is digested with a mixture of potassium sulphate and sulphuric acid, in the presence of copper (II) sulphate as catalyst, to convert organic nitrogen to ammoniacal nitrogen. The ammonia is distilled and absorbed into boric acid solution and then titrated with standard hydrochloric acid solution. The nitrogen content is converted to protein content by multiplying by 6,38.
4.REAGENTSU.K.
4.1. Sulphuric acid, concentrated, S2O 1,84 g/ml.U.K.
4.2. Potassium sulphate, anhydrous (K2SO4).U.K.
4.3. Copper (II) sulphate pentahydrate (CuSO45H2O).U.K.
4.4. Sucrose (C12H22O11).U.K.
4.5. Boric acid, 40-g/l solution.U.K.
4.6. Sodium hydroxide, concentrated aqueous solution 30 % (m/m), carbonate free.U.K.
4.7. Hydrochloric acid, 0,1 mol/1.U.K.
4.8. Mixed indicator. Mix equal volumes of a 2 g/1 solution of methyl red in at least 95 % (V/V) ethanol and a 1 g/1 solution of methylene blue in at least 95 % (V/V) ethanol.U.K.
5.APPARATUSU.K.
5.1. Analytical balance U.K.
5.2. Kjeldahl flask, 500 ml capacity.U.K.
5.3. Digestion apparatus to hold the Kjeldahl flask (5.2) in an inclined position and with a heating device which will not heat the part of the flask above the surface of the liquid contents.U.K.
5.4. Condenser with straight inner tube.U.K.
5.5. Outlet tube with safety bulb connected to the lower end of the condenser (5.4) by a ground glass joint or a rubber tube. If rubber tubing is used, the glass ends must be near one another.U.K.
5.6. Splash-head connected to the Kjeldahl flask (5.2) and to the condenser (5.4) by soft, close-fitting rubber or other appropriate stoppers.U.K.
5.7. Conical flask, 500 ml capacity.U.K.
5.8. Graduated cylinders, 50 ml and 100 ml capacity.U.K.
5.9. Burette, 50 ml capacity, graduated in 0,1 ml.U.K.
5.10. Boiling aids: U.K.
5.10.1.For the digestion: small pieces of hard porcelain, or glass beads.U.K.
5.10.2.For the distillation; freshly calcined pieces of pumice.U.K.
6.PROCEDUREU.K.
6.1. Preparation of the test sample U.K.
As described in Section 1.2 of the General Provisions.
6.2. Test for presence of ammoniacal nitrogen U.K.
If the presence of ammonium caseinate or other ammonium compounds is suspected, carry out the following test. Add to 1 gram of sample in a small conical flask 10 ml of water and 100 mg of magnesium oxide. Rinse down any magnesium oxide adhering to the walls and close the flask with a cork stopper, inserting a piece of moistened red litmus paper between the stopper and the neck of the flask. Mix the contents of the flask carefully and heat the flask in a water bath at 60 to 65 o C. If the litmus paper colours blue within 15 minutes ammonia is present, and the method is not applicable (see Section 1).
6.3. Blank test U.K.
At the same time as the determination of the nitrogen content of the sample perform a blank determination using 0,5 grams of the sucrose (4.4) instead of the test portion, using the same apparatus, the same quantities of all reagents and the same procedure as described in 6.5. If the titration in the blank determination exceeds 0,5 ml of 0,1 mol/1 acid, the reagents shall be checked and the impure reagent or reagents purified or replaced.
6.4. Test portion U.K.
Transfer to the Kjeldahl flask (5.2) 0,3 to 0,4 grams of the test sample (6.1), weighed to the nearest 0,1 mg.
6.5. Determination U.K.
6.5.1.Transfer to the flask a few pieces of porcelain or a few glass beads (5.10.1) and about 10 grams of the anhydrous potassium sulphate (4.2).U.K.
Add 0,2 g of the copper (II) sulphate (4.3) and wash down the neck of the flask with a little water. Add 20 ml of the concentrated sulphuric acid (4.1). Mix the contents of the flask.
Heat gently on the digestion apparatus (5.3) until any frothing has ceased, boil gently until the solution is clear and a pale green-blue colour persists. During heating, swirl the flask occasionally.
Continue the boiling, regulating the heating so as to condense the vapours in the middle of the flask neck. Continue the heating for 90 minutes avoiding local overheating.
Allow to cool to room temperature. Carefully add about 200 ml of water and a few pieces of pumice (5.10.2). Mix and cool again.
6.5.2.Transfer into the conical flask (5.7) 50 ml of the boric acid solution (4.5) and four drops of the indicator (4.8). Mix. Place the conical flask under the condenser (5.4) so that the tip of the outlet tube (5.5) is immersed in the boric acid solution. Using a graduated cylinder (5.8), add to the Kjeldahl flask 80 ml of the sodium hydroxide solution (4.6). During this operation, hold the flask in an inclined position so that the sodium hydroxide solution runs down the side of the flask to form a bottom layer.U.K.
Immediately connect the Kjeldahl flask to the condenser by means of the splash-head (5.6).
Gently rotate the Kjeldahl flask to mix its contents. Boil gently at first, avoiding any frothing. Continue the distillation so that 150 ml of distillate are collected in approximately 30 minutes. The distillate should have a temperature below 25 o C. About two minutes before the end of the distillation, lower the conical flask so that the tip of the outlet tube is no longer immersed in the acid solution, and rinse the tip with a little water. Stop heating, remove the outlet tube and rinse its outer and inner walls with a little water, collecting the washings in the conical flask.
6.5.3.Titrate the distillate in the conical flask, using the standard volumetric hydrochloric acid solution (4.7).U.K.
7.EXPRESSION OF RESULTSU.K.
7.1. Formula and method of calculation U.K.
The protein content of the sample, expressed as a percentage by mass, is given by:
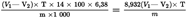
where:
is the volume, in millilitres, of the standard volumetric hydrochloric acid solution (4.7) used in the determination (6.5);
is the volume, in millilitres, of the standard volumetric hydrochloric acid solution (4.7) used in the blank test (6.3);
is the strength of the standard volumetric hydrochloric acid solution (4.7) in mol/1;
is the mass, in grams, of the test portion.
Calculate the protein content to the nearest 0,1 %.
7.2. Repeatability U.K.
The difference between the results of two determinations carried out simultaneously or in rapid succession on the same sample, by the same analyst under the same conditions shall not exceed 0,5 grams of protein per 100 grams of product.
This repeatability interval should be achieved in 95 % of the times that the method is correctly carried out.
METHOD 3U.K.
DETERMINATION OF TITRATABLE ACIDITY U.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
The method determines the titratable acidity of:
acid caseins.
2.DEFINITIONU.K.
The titratable acidity of acid caseins: the volume in millilitres, of a 0,1 mol/1 standard sodium hydroxide solution required to neutralize an aqueous extract of 1 gram of the product.
3.PRINCIPLEU.K.
An aqueous extract of the sample at 60 o C is obtained and filtered. The filtrate is titrated against standard sodium hydroxide using phenolphtalein indicator.
4.REAGENTSU.K.
Any water used in the method procedure or in the preparation of reagents shall be freed from carbon dioxide by boiling for 10 minutes before use.
4.1. Sodium hydroxide solution: 0,1 Mol/1.U.K.
4.2. Phenolphtalein indicator solution, 10 g/1 in ethanol (95 % V/V) neutralized to the indicator.U.K.
5.APPARATUSU.K.
5.1. Analytical balance U.K.
5.2. Conical flask, 500 ml capacity, with ground neck and fitted with a ground glass stopper.U.K.
5.3. One-mark pipette, 100 ml capacity.U.K.
5.4. Pipette, suitable for measuring 0,5 ml of indicator solution (4.2).U.K.
5.5. Conical flask, 250 ml capacity.U.K.
5.6. Measuring cylinder, 250 ml capacity.U.K.
5.7. Burette, graduated in 0,1 ml.U.K.
5.8. Water bath, capable of being controlled at a temperature of 60 oC ± 2 oC.U.K.
5.9. Appropriate filter U.K.
6.PROCEDUREU.K.
6.1. Preparation of the test sample U.K.
As described in Section 1.2 of the General Provisions.
6.2. Test portion U.K.
Weigh about 10 grams of the test sample (6.1) to the nearest 10 mg and transfer it to the conical flask (5.2).
6.3. Determination U.K.
Using the 250 ml measuring cylinder (5.6), add 200 ml of freshly boiled and cooled water, previously heated to 60 o C. Stopper the flask, mix by swirling and place in the water bath at 60 o C (5.8) for 30 minutes. Shake the flask at intervals of about 10 minutes.
Filter, and cool the filtrate to about 20 oC. The filtrate must be clear.
Transfer 100 ml of the cooled filtrate into the conical flask (5.5), using the pipette (5.3). Add 0,5 ml of the phenolphtalein indicator solution (4.2), using the pipette (5.4). Titrate with the standard volumetric sodium hydroxide solution (4.1), until the appearance of a faint pink colour, persisting for at least 30 seconds. Determine and record the volume used to the nearest 0,01 ml.
7.EXPRESSION OF RESULTSU.K.
7.1. Formula and method of calculation U.K.
The titratable acidity of the acid casein is given by:

where:
is the volume, in millilitres, of the standard volumetric sodium hydroxide solution (4.1) used;
is the strength of the standard volumetric sodium hydroxide solution (4.1) in mol/1;
is the mass, in grams, of the test portion.
Calculate the titratable acidity to two decimal places.
7.2. Repeatability U.K.
The difference in results between two determinations carried out simultaneously or in rapid succession on the same sample, by the same analyst, under the same conditions, shall not exceed 0,02 ml of 0,1 mol/1 sodium hydroxide per 1 gram of product.
This repeatability interval should be achieved in 95 % of the times that the method is correctly carried out.
METHOD 4U.K.
DETERMINATION OF ASH (including P2O5) U.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
The method determines the ash (including P2O5) content of:
acid caseins
2.DEFINITIONU.K.
The ash (including P2O5) content: the content of ash as determined by the method specified.
3.PRINCIPLEU.K.
A portion of the sample is incinerated at 825 o C ± 25 o C in the presence of magnesium acetate to bind all phosphorus of organic origin. The final ash is calculated after the weighing of the residue and subtraction of the mass of ash originating from the magnesium acetate.
4.REAGENTSU.K.
4.1. Magnesium acetate tetrahydrate solution, 120 g/1. Dissolve 120 grams of magnesium acetate tetrahydrate [Mg (CH3 CO2)2 4 H2 O] in water and make up one litre with water.U.K.
5.APPARATUSU.K.
5.1. Analytical balance U.K.
5.2. One-mark pipette, 5 ml.U.K.
5.3. Silica or platinum dishes, about 70 mm diameter and 25 to 50 mm deep.U.K.
5.4. Drying oven, capable of being controlled at 102 oC ± 1 oC.U.K.
5.5. Electrical furnace, capable of being controlled at 825 oC ± 25 oC.U.K.
5.6. Boiling water bath U.K.
5.7. Desiccator containing freshly activated silica gel with a water content indicator or an equivalent desiccant.U.K.
6.PROCEDUREU.K.
6.1. Preparation of the test sample U.K.
As described in Section 1.2 of the General Provisions.
6.2. Preparation of the dishes U.K.
Heat two dishes (A,B) (5.3) in the electrical furnace (5.5), controlled at 825 o C ± 25 o C, for 30 minutes. Allow the dishes to cool somewhat and then place in the desiccator (5.7) to the temperature of the balance room and weigh to the nearest 0,1 mg.
6.3. Test portion U.K.
Weigh, to the nearest 0,1 mg approximately 3 grams of the test sample (6.1), directly into one of the prepared dishes (A).
6.4. Determination U.K.
Using the pipette (5.2), add to the dish (A) exactly 5 ml of the magnesium acetate solution (4.1) so as to wet all of the test portion, and allow to stand for 20 minutes.
To the other prepared dish (B), add with the pipette (5.2) exactly 5 ml of the magnesium acetate solution (4.1).
Evaporate the contents of both dishes (A and B) to dryness on the boiling water bath (5.6).
Place both dishes in the oven (5.4), controlled at 102 oC ± 1 oC, for 30 minutes.
Heat dish A with its contents on a low flame, a hot plate or under an I/R lamp, until the test portion is completely charred, taking care that it does not burst into flame.
Transfer both dishes (A and B) to the electrical furnace (5.5), controlled at 825 oC ± 25 oC, and heat for at least one hour until all carbon has disappeared from dish A. Allow both dishes to cool somewhat and then place in the desiccator (5.7) to the temperature of the balance room and weigh to the nearest 0,1 mg.
Repeat the operations of heating for approximately 30 minutes, in the electrical furnace (5.5), cooling and weighing, until the mass remains constant to within 1 mg or begins to increase. Record the minimum mass.
7.EXPRESSION OF RESULTSU.K.
7.1. Method of calculation U.K.
The content of ash, including P2O5, in the sample, as a percentage by mass, is given by:

where:
is the mass, in grams, of the test portion;
is the mass, in grams, of dish A and residue;
is the mass, in grams, of the prepared dish A;
is the mass, in grams, of dish B and residue;
is the mass, in grams, of the prepared dish B.
Calculate the final result to the nearest 0,01 %.
7.2. Repeatability U.K.
The difference in results between the determinations carried out simultaneously or in rapid succession on the same sample, by the same analyst, under the same conditions, shall not exceed 0,1 grams per 100 grams of product.
The repeatability interval should be achieved in 95 % of the times that the method is correctly carried out.
METHOD 5U.K.
DETERMINATION OF ASH (including P2O5) U.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method determines the ash (including P2O 5) content of:
rennet casein
2.DEFINITIONU.K.
The ash (including P2O5) content: the content of ash as determined by the method specified.
3.PRINCIPLEU.K.
A portion of the sample is incinerated at 825 oC ± 25 oC to constant mass. The residue is determined by weighing and calculated as a percentage by mass of the sample.
4.APPARATUSU.K.
4.1. Analytical balance U.K.
4.2. Silica or platinum dish, about 70 mm diameter and 25 to 50 mm deep.U.K.
4.3. Electrical furnace with air circulation, capable of being controlled at 825 oC ± 25 oC.U.K.
4.4. Desiccator, containing freshly activated silica gel with a water content indicator or an equivalent desiccant.U.K.
5.PROCEDUREU.K.
5.1. Preparation of the test sample U.K.
As described in Section 1.2 of the General Provisions.
5.2. Preparation of the dish U.K.
Heat the dish (4.2) in the electrical furnace (4.3), controlled at 825 oC ± 25 oC, for 30 minutes. Allow the dish to cool somewhat and then place in the desiccator (4.4) to the temperature of the balance room and weigh to the nearest 0,1 mg.
5.3. Test portion U.K.
Weigh, to the nearest 0,1 mg approximately 3 grams of the test sample (5.1) directly into the prepared dish.
5.4. Determination U.K.
Heat the dish with its contents on a low flame, a hot plate or an I/R lamp until the test portion is completely charred, taking care that it does not burst into flame.
Transfer the dish to the electrical furnace (4.3), controlled at 825 oC ± 25 oC, and heat for at least one hour until all carbon has disappeared from the dish. Allow the dish to cool somewhat and then place in the desiccator (4.4) to the temperature of the balance room and weigh to the nearest 0,1 mg.
Repeat the operations of heating for approximately 30 minutes, in the electrical furnace (4.3), cooling and weighing, until the mass remains constant to within 1 mg or begins to increase. Record the minimum mass.
6.EXPRESSION OF RESULTSU.K.
6.1. Method of calculation and formula U.K.
The ash content of the sample, including P2O5, as a percentage per mass, is given by:

where:
is the mass, in grams, of the test portion;
is the mass, in grams, of the dish and residue;
is the mass, in grams, of the prepared dish.
Calculate the final result to the nearest 0,01 %.
6.2. Repeatability U.K.
The difference between the results of two determinations carried out simultaneously or in rapid succession on the same sample, by the same analyst, under the same conditions, shall not exceed 0,15 grams of ash per 100 grams of product.
This repeatability interval should be achieved in 95 % of the times that the method is correctly carried out.
METHOD 6U.K.
DETERMINATION OF pH U.K.
1.SCOPE AND FIELD OF APPLICATIONU.K.
This method determines the pH of:
caseinates.
2.DEFINITIONU.K.
The pH of caseinates: the pH, at 20 oC, of an aqueous solution of caseinates, as determined by the method specified.
3.PRINCIPLEU.K.
The electrometric determination of pH of an aqueous solution of caseinate, using a pH meter.
4.REAGENTSU.K.
Any water used in the preparation of the reagents or in the Procedure (6) shall be recently distilled water that has been protected from carbon dioxide absorption.
4.1. Buffer solutions, for calibration of the pH meter (5.2)U.K.
Two standard buffer solutions with pH values at 20 oC which are known to the second decimal place and will bracket the pH value of the sample under test, for example phtalate buffer solution of pH approximately 4 and a borax buffer solution of pH approximately 9.
5.APPARATUSU.K.
5.1. Balance, accuracy 0,1 grams.U.K.
5.2. pH meter, minimum sensitivity 0,05 pH unit, with a suitably calibrated elecrode, e.g. glass electrode and a calomel or other reference electrode.U.K.
5.3. Thermometer, accuracy 0,5 oC.U.K.
5.4. Conical flask, capacity 100 ml, fitted with a ground glass stopper.U.K.
5.5. Beaker, capacity 50 ml.U.K.
5.6. Mixer U.K.
5.7. Beaker, for the mixer (5.6) of at least 250 ml capacity.U.K.
6.PROCEDUREU.K.
6.1. Preparation of the test sample U.K.
As described in Section 1.2 of the General Provisions.
6.2. Determination U.K.
6.2.1. Calibration of pH meter U.K.
Adjust the temperature of the buffer solutions (4.1) to 20 oC and calibrate the pH meter in accordance with the manufacturer's instructions.
NOTES U.K.
1.The calibration should be carried out while the flasks are standing for 20 mintues (see 6.2.2).U.K.
2.If a series of samples is being tested, check the calibration of the pH meter with one or more of the standard buffer solutions at least every 30 minutes.U.K.
6.2.2. Preparation of the test solution U.K.
Transfer to the beaker (5.7) 95 ml of water, add 5,0 grams of the test sample (6.1), and mix using the mixer (5.6) for 30 seconds.
Allow to stand for 20 minutes at about 20 oC, covered with a watch glass.
6.2.3. Measurement of pH U.K.
6.2.3.1.Pour about 20 ml of the solution into the beaker (5.5) and immediately determine the pH of this liquid, using the pH meter (5.2) after having rinsed the glass electrode carefully with water.U.K.
6.2.3.2.Measure the pH.U.K.
7.EXPRESSION OF RESULTSU.K.
7.1. Recording of pH U.K.
Record, as the pH of the aqueous solution of caseinate, the value read from the dial of the pH meter to at least two decimal places.
7.2. Repeatability U.K.
The difference between the results of two determinations carried out simultaneously or in rapid succession on the same sample, by the same analyst, under the same conditions, shall not exceed 0,05 pH unit.
This repeatability interval should be achieved in 95 % of the times that the method is correctly carried out.