- Latest available (Revised)
- Point in Time (20/10/2019)
- Original (As adopted by EU)
Commission Regulation (EEC) No 2568/91Show full title
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
You are here:

What Version
Advanced Features
- Show Geographical Extent(e.g. England, Wales, Scotland and Northern Ireland)
- Show Timeline of Changes
More Resources
Revised version PDFs
- Revised 20/10/20193.52 MB
- Revised 04/12/20163.60 MB
- Revised 11/10/20163.55 MB
- Revised 04/08/20163.60 MB
- Revised 16/10/20153.42 MB
- Revised 01/01/20153.68 MB
- Revised 01/03/20143.69 MB
- Revised 01/01/20142.84 MB
- Revised 09/08/20122.49 MB
- Revised 01/04/20112.46 MB
- Revised 01/10/20081.42 MB
- Revised 01/01/20081.42 MB
- Revised 01/11/20035.95 MB
- Revised 22/05/20020.48 MB
- Revised 01/11/20010.66 MB
- Revised 01/07/20010.66 MB
- Revised 01/06/19990.65 MB
- Revised 01/11/19980.65 MB
- Revised 10/02/19980.65 MB
- Revised 31/10/19950.59 MB
- Revised 28/05/19950.59 MB
- Revised 01/11/19940.55 MB
- Revised 19/02/19940.55 MB
- Revised 05/02/19940.55 MB
- Revised 01/05/19930.55 MB
- Revised 21/03/19930.56 MB
- Revised 20/02/19930.56 MB
- Revised 01/11/19920.55 MB
- Revised 21/07/19920.55 MB
- Revised 03/07/19920.54 MB
- Revised 05/06/19920.54 MB
- Revised 21/12/19910.52 MB
- Revised 06/09/19910.52 MB
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


This item of legislation originated from the EU
Legislation.gov.uk publishes the UK version. EUR-Lex publishes the EU version. The EU Exit Web Archive holds a snapshot of EUR-Lex’s version from IP completion day (31 December 2020 11.00 p.m.).
Changes over time for: ANNEX XVII
Status:
Point in time view as at 20/10/2019.
Changes to legislation:
There are outstanding changes by UK legislation not yet made to Commission Regulation (EEC) No 2568/91. Any changes that have already been made to the legislation appear in the content and are referenced with annotations.
Changes to Legislation
Changes and effects yet to be applied by the editorial team are only applicable when viewing the latest version or prospective version of legislation. They are therefore not accessible when viewing legislation as at a specific point in time. To view the ‘Changes to Legislation’ information for this provision return to the latest version view using the options provided in the ‘What Version’ box above.
[F1ANNEX XVII U.K. METHOD FOR THE DETERMINATION OF STIGMASTADIENES IN VEGETABLE OILS
Textual Amendments
F1 Inserted by Commission Regulation (EC) No 656/95 of 28 March 1995 amending Regulation (EEC) No 2568/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis and Council Regulation (EEC) No 2658/87 on the tariff and statistical nomenclature and on the Common Customs Tariff.
1. PURPOSE U.K.
Determination of stigmastadienes in vegetable oils containing low concentrations of these hydrocarbons, particularly in virgin olive oil and crude olive-residue oil.
2. SCOPE U.K.
The standard may be applied to all vegetable oils although measurements are reliable only where the content of these hydrocarbons lies between 0,01 and 4,0 mg/kg. The method is particularly suited to detecting the presence of refined vegetable oils (olive, olive residue, sunflower, palm, etc.) in virgin olive oil since refined oils contained stigmastadienes and virgin oils do not.
3. PRINCIPLE U.K.
Isolation of unsaponifiable matter. Separation of steroidal hydrocarbon fraction by column chromatography on silica gel and analysis by capillary gas chromatography.
4. APPARATUS U.K.
4.1. 250 ml flasks suitable for use with a reflux condenser. U.K.
4.2. Separating funnels of 500 ml capacity. U.K.
4.3. 100 ml round-bottom flasks. U.K.
4.4. Rotary evaporator. U.K.
4.5. Glass chromatography column (1,5 to 2,0 cm internal diameter by 50 cm length) with Teflon tap and a plug of glass wool fibre or sintered glass disc at the bottom. To prepare silica gel column, pour hexane into the chromatography column to a depth of approximately 5 cm and then fill with a slurry of silica gel in hexane (15 g in 40 ml) with the help of hexane portions. Allow to settle and finish settling by applying slight vibration. Add anhydrous sodium sulphate to a height of approximately 0,5 cm, finally elute the excess hexane. U.K.
4.6. Gas chromatograph with flame ionization detector, split or cold on-column injector and oven programmable to within ± 1 °C. U.K.
4.7. Fused silica capillary column for gas chromatography (0,25 or 0,32 mm internal diameter by 25 m length) coated with 5 %-phenylmethylsilicone phase, 0,25 mm film thickness. U.K.
Note 1: U.K.
Other columns of similar or lower polarity can be used. U.K.
4.8. Integrator-recorder with possibility of valley-valley integration mode. U.K.
4.9. 5 to 10 ml microsyringe for gas chromatography with cemented needle. U.K.
4.10. Electrical heating mantle or hot place. U.K.
5. REAGENTS U.K.
All reagents should be of analytical grade unless otherwise specified. The water used should be distilled water, or water of at least equivalent purity.
[F25.1. Hexane or mixture of alkanes of b.p. interval 65 to 70 °C, distilled with rectifying column. Hexane may be replaced by iso-octane (2,2,4-trimethyl pentane in chromatography grade), provided that comparable precision values are achieved. The residue after evaporation of 100 ml of solvent may be controlled. Solvents with higher boiling point than n-hexane take longer to evaporate. However, they are preferred due to the toxicity of hexane.] U.K.
Textual Amendments
5.2. 96 v/v ethanol. U.K.
5.3. Anhydrous sodium sulphate. U.K.
5.4. Alcoholic potassium hydroxide solution at 10 %. Add 10 ml of water to 50 g potassium hydroxide, stir, and then dissolve the mixture in ethanol to 500 ml. U.K.
Note 3: U.K.
Alcoholic potash turns brown on standing. It should be prepared freshly each day and kept in well stoppered dark glass bottles. U.K.
5.5. Silica gel 60 for column chromatography, 70 to 230 mesh, (Merck, reference 7734 or similar). U.K.
Note 4: U.K.
Usually, silica gel can be used directly from the container without any treatment. However, some batches of silica gel may show low activity resulting in bad chromatographic separations. Under this circumstance, the silica gel should be treated in the following way: Activate the silica gel by heating for a minimum of four hours at 550 °C. After heating, place the silica gel in a desiccator while the gel is cooling and then transfer the silica gel to a stoppered flask. Add 2 % of water and shake until no lumps can be seen and the powder flows freely. U.K.
If batches of silica gel result in chromatograms with interfering peaks, the silica gel should be treated as above. An alternative could be the use of extra pure silica gel 60 (Merck, reference 7754).
5.6. Stock solution (200 ppm) of cholesta-3,5-diene (Sigma, 99 % purity) in hexane (10 mg in 50 ml). U.K.
5.7. Standard solution of cholesta-3,5-diene hexane at concentration of 20 ppm, obtained by dilution of above solution. U.K.
Note 5: U.K.
The solutions 5.6 and 5.7 are stable for a period of at least four months if kept at less than 4 °C. U.K.
5.8. Solution of n-nonacosane in hexane at concentration of approximately 100 ppm. U.K.
5.9. Carrier gas for chromatography: helium or hydrogen of 99,9990 % purity. U.K.
5.10. Auxiliary gases for flame ionization detector: hydrogen of 99,9990 % purity and purified air. U.K.
6. PROCEDURE U.K.
6.1. Preparation of unsaponifiable matter U.K.
6.1.1. Weigh 20 ± 0,1 g of oil into a 250-ml flask (4.1), add 1 ml of the standard solution of cholesta-3,5-diene (20μg) and 75 ml of alcoholic potash at 10 %, fit reflux condenser, and heat to slight boiling for 30 minutes, Remove the flask containing the sample from the heat and allow the solution to cool slightly (do not allow to cool completely as the sample will set). Add 100 ml of water and transfer the solution to a separating funnel (4.2) with the aid of 100 ml of hexane. Shake the mixture vigorously for 30 seconds and allow the separate. U.K.
Note 6: U.K.
If an emulsion is produced which does not rapidly disappear, add small quantities of ethanol. U.K.
6.1.2. Transfer the aqueous phase beneath to a second separating funnel and extract again with 100 ml of hexane. Once more run off the lower phase and wash the hexane extracts (combined in another separating funnel) three times with 100 ml each time of a mixture of ethanol-water (1: 1) until neutral pH is reached. U.K.
6.1.3. Pass the hexane solution through anhydrous sodium sulphate (50 g), wash with 20 ml hexane and evaporate in a rotary evaporator at 30 °C under reduced pressure until dryness. U.K.
6.2. Separation of steroidal hydrocarbon fraction U.K.
6.2.1. Take the residue to the fractioning column with the aid of two 1-ml portions of hexane, run the sample onto the column by allowing the solution level to drop to the top of the sodium sulphate and start the chromatographic elution with hexane at a flow rate of 1 ml/min approximately. Discard the first 25 to 30 ml of eluate and then collect the following 40 ml fraction. After collection, transfer this fraction to a 100-ml round bottomed flask (4.3). U.K.
Note 7: U.K.
The first fraction contains saturated hydrocarbons (Figure 1 a) and the second fraction the steroidal ones. Further elution provides squalene and related compounds. To achieve a good separation between saturated and steroidal hydrocarbons, the optimization of fraction volumes is required. For this, the volume of the first fraction should be adjusted so that when the second fraction is analysed the peaks representing the saturated hydrocarbons are low (see Figure 1 c); if they do not appear but the intensity of the standard peak is low, the volume should be reduced. Anyway, a complete separation between the components of the first and second fractions is unnecessary; as there is no overlapping of peaks during GC analysis if GC conditions are ajusted as indicated in 6.3.1. The optimization of the volume of the second fraction if generally not needed as a good separation exists with the further components. Nevertheless, the presence of a large peak at approximately 1,5 minutes lower retention time than the standard is due to squalene, and it is indicative of a bad separation. U.K.
6.2.2. Evaporate the second fraction in a rotary evaporator at 30 °C under reduced pressure until dryness, and immediately dissolve the residue in 0,2 ml of hexane. Keep the solution in the refrigerator until analysis. U.K.
Note 8: U.K.
Residues 6.1.3 and 6.2.2 should not be kept dry and at room temperature. As soon as they are obtained, the solvent should be added and the solutions should be kept in the refrigerator. U.K.
6.3. Gas chromatography U.K.
6.3.1. Working conditions for split injection: U.K.
injector temperature: 300 °C,
detector temperature: 320 °C,
integrator-recorder: the parameters for integration should be fixed so as to give a correct assessment of the areas. Valley-valley integration mode is recommended,
sensitivity: about 16 times the minimum attenuation,
amount of solution injected: 1μl,
oven programming temperatures: initial 235 °C for six minutes and then rising at 2 °C/minute up to 285 °C,
injector with 1: 15 flow divider,
carrier: helium or hydrogen at about 120 kPa pressure.
These conditions may be adjusted in accordance with the characteristics of the chromatograph and the column to give chromatograms meeting the following requirements: internal standard peak within approximately five minutes of the time given in 6.3.2; the internal standard peak should be at least 80 % of the full scale.
The gas chromatographic system must be checked injecting a mixture of the stock solution of cholestadiene (5.6) and n-nonacosane solution (5.8). The cholesta-3,5-diene peak must appear before the n-nonacosane (Figure 1c); if it does not occur two actions can be undertaken: reduce the oven temperature and/or use a less polar column.
6.3.2. Peak identification U.K.
The internal standard peak appears at approximately 19 minutes and the 3,5-stigmastadiene at a relative retention time of approximately 1,29 (see Figure 1b). The 3,5-stigmastadiene occurs with small quantities of an isomer, and usually, both elute together as a single chromatographic peak. Nevertheless, if the column is too polar or shows a high resolving power, the isomer can appear as a small peak before and close to that of stigmasta-3,5-diene (Figure 2). In order to ensure that the stigmastadienes are eluted as one peak, it is advisable to replace the column by one which is either less polar or has a wider internal diameter.
Note 9: U.K.
Stigmastadienes for reference can be obtained from the analysis of a refined vegetable oil by using less amount of sample (1 to 2 g). Stigmastadienes originate a prominent and easily identifiable peak. U.K.
6.3.3. Quantitative analysis U.K.
The stigmastadienes content is determined according to the formula:

| where: | A s = area of stigmastadienes peak (if the peak is resolved into two isomers, sum of areas of the two peaks), A c = area of internal standard (cholestadiene), M c = mass of standard added, in micrograms, M o = mass of oil taken, in grams. |
Detection limit: about 0,01 mg/kg.
[F3Note 10: When stigmastadienes appear in concentrations of more than 4 mg/kg, if quantifying is required, the method of the International Olive Council for determination of sterenes in refined oil must be applied.]
Textual Amendments
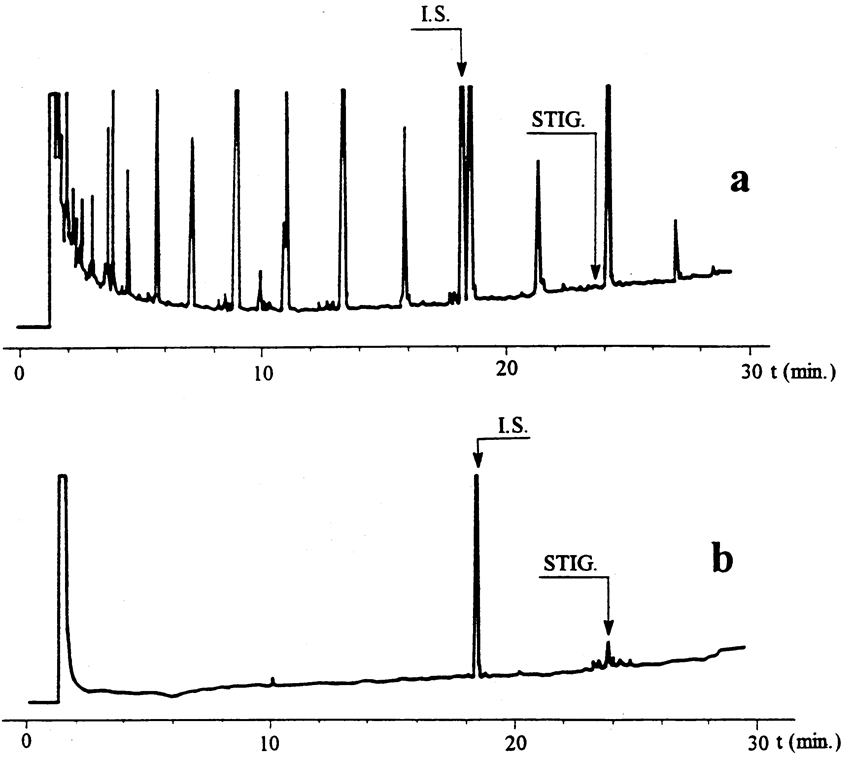
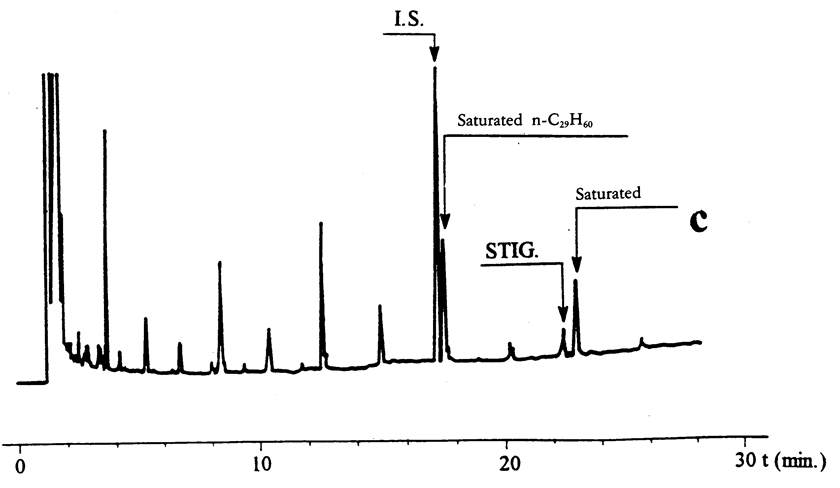
Figure 1 U.K.
Gas chromatograms obtained from olive oil samples analysed on a fused silica capillary column (0,25 mm internal diameter by 25 m) coated with 5 %-phenylmethylsilicone, 0,25 μm film thickness. U.K.
(a)
First fraction (30 ml) from a virgin oil, spiked with standard.
(b)
Second fraction (40 ml) from an olive oil containing 0,10 mg/kg of stigmastadienes.
(c)
Second fraction (40 ml) containing a small proportion of the first fraction.
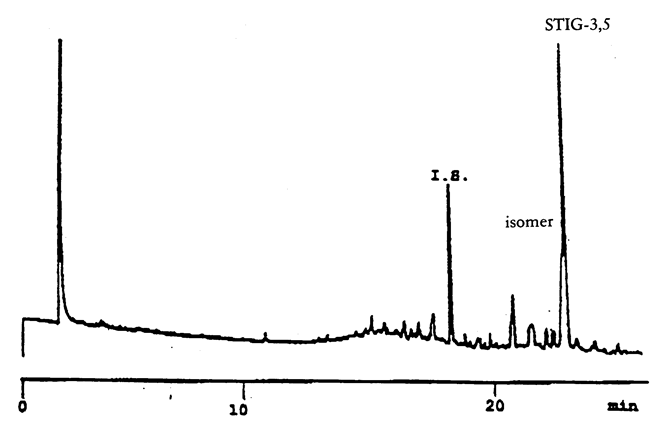
Gas chromatogram obtained from a refined olive oil sample analysed on DB-5 column showing the isomer of 3,5-stigmastadiene.]
Options/Help
Print Options
PrintThe Whole Regulation
PrintThis Annex only
You have chosen to open the Whole Regulation
The Whole Regulation you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
The Schedules you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As adopted by EU): The original version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Point in Time: This becomes available after navigating to view revised legislation as it stood at a certain point in time via Advanced Features > Show Timeline of Changes or via a point in time advanced search.
See additional information alongside the content
Geographical Extent: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Show Timeline of Changes: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Opening Options
Different options to open legislation in order to view more content on screen at once
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the EU Official Journal
- lists of changes made by and/or affecting this legislation item
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
Timeline of Changes
This timeline shows the different versions taken from EUR-Lex before exit day and during the implementation period as well as any subsequent versions created after the implementation period as a result of changes made by UK legislation.
The dates for the EU versions are taken from the document dates on EUR-Lex and may not always coincide with when the changes came into force for the document.
For any versions created after the implementation period as a result of changes made by UK legislation the date will coincide with the earliest date on which the change (e.g an insertion, a repeal or a substitution) that was applied came into force. For further information see our guide to revised legislation on Understanding Legislation.
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.