- Latest available (Revised)
- Original (As adopted by EU)
Commission Regulation (EEC) No 2568/91Show full title
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
You are here:

What Version
Advanced Features
- Show Geographical Extent(e.g. England, Wales, Scotland and Northern Ireland)
- Show Timeline of Changes
More Resources
Revised version PDFs
- Revised 20/10/20193.52 MB
- Revised 04/12/20163.60 MB
- Revised 11/10/20163.55 MB
- Revised 04/08/20163.60 MB
- Revised 16/10/20153.42 MB
- Revised 01/01/20153.68 MB
- Revised 01/03/20143.69 MB
- Revised 01/01/20142.84 MB
- Revised 09/08/20122.49 MB
- Revised 01/04/20112.46 MB
- Revised 01/10/20081.42 MB
- Revised 01/01/20081.42 MB
- Revised 01/11/20035.95 MB
- Revised 22/05/20020.48 MB
- Revised 01/11/20010.66 MB
- Revised 01/07/20010.66 MB
- Revised 01/06/19990.65 MB
- Revised 01/11/19980.65 MB
- Revised 10/02/19980.65 MB
- Revised 31/10/19950.59 MB
- Revised 28/05/19950.59 MB
- Revised 01/11/19940.55 MB
- Revised 19/02/19940.55 MB
- Revised 05/02/19940.55 MB
- Revised 01/05/19930.55 MB
- Revised 21/03/19930.56 MB
- Revised 20/02/19930.56 MB
- Revised 01/11/19920.55 MB
- Revised 21/07/19920.55 MB
- Revised 03/07/19920.54 MB
- Revised 05/06/19920.54 MB
- Revised 21/12/19910.52 MB
- Revised 06/09/19910.52 MB
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


This item of legislation originated from the EU
Legislation.gov.uk publishes the UK version. EUR-Lex publishes the EU version. The EU Exit Web Archive holds a snapshot of EUR-Lex’s version from IP completion day (31 December 2020 11.00 p.m.).
Changes over time for: Division 5.
Changes to legislation:
There are outstanding changes not yet made to Commission Regulation (EEC) No 2568/91. Any changes that have already been made to the legislation appear in the content and are referenced with annotations.
Changes to Legislation
Revised legislation carried on this site may not be fully up to date. Changes and effects are recorded by our editorial team in lists which can be found in the ‘Changes to Legislation’ area. Where those effects have yet to be applied to the text of the legislation by the editorial team they are also listed alongside the legislation in the affected provisions. Use the ‘more’ link to open the changes and effects relevant to the provision you are viewing.
Changes and effects yet to be applied to the whole legislation item and associated provisions
- Signature words omitted by S.I. 2019/1422 reg. 6(10)
- Art. 1(8) inserted by S.I. 2019/1422 reg. 6(2)(b)
- Art. 1(8)(a)(ii)(bb) omitted in earlier amending provision S.I. 2019/1422, reg. 6(2)(b) by S.I. 2020/1453 reg. 14(16)(a)(i)
- Art. 1(8)(b) words substituted in earlier amending provision S.I. 2019/1422, reg. 6(2)(b) by S.I. 2020/1453 reg. 14(16)(a)(ii)
- Art. 1(8)(c)(ii) omitted in earlier amending provision S.I. 2019/1422, reg. 6(2)(b) by S.I. 2020/1453 reg. 14(16)(a)(iii)
- Annex 1a para. 1.1 words substituted by S.I. 2019/1422 reg. 6(11)(a)
- Annex 1a para. 1.2 words substituted by S.I. 2019/1422 reg. 6(11)(b)
- Art. 2a(3)(e) words substituted by S.I. 2019/1422 reg. 6(4)(c) (This amendment not applied to legislation.gov.uk. Reg. 6(4)(c) substituted immediately before IP completion day by S.I. 2020/1453, regs. 1(2)(b), 14(16)(c)(ii))
- Art. 2a(3)(e) words substituted by S.I. 2019/1422, reg. 6(4)(c) (as substituted) by S.I. 2020/1453 reg. 14(16)(c)(ii)
- Art. 3 words substituted by S.I. 2019/1422 reg. 6(5)(a)
- Art. 3 words substituted by S.I. 2019/1422 reg. 6(5)(b)
[F15. PROCEDURE U.K.
5.1. Preparation of the chromatography column U.K.
Suspend 15 g of silica gel (point 4.1) in n-hexane (point 4.2) and introduce into the column (point 3.2). Allow to settle spontaneously. Complete settling with the aid of an electric shaker to make the chromatographic bed more homogeneous. Percolate 30 ml of n-hexane to remove any impurities. Weigh exactly about 500 mg of the sample into the 25-ml flask (point 3.1), using the analytical balance (point 3.8), and add a suitable amount of internal standard (point 4.5), depending on the assumed wax content, e.g. add 0,1 mg of lauryl arachidate in the case of olive oil, 0,25-0,50 mg in the case of olive-pomace oil and 0,05 mg of methyl heptadecanoate for olive oils (point 4.6).
Transfer the prepared sample to the chromatography column with the aid of two 2-ml portions of n-hexane (point 4.2).
Allow the solvent to flow to 1 mm above the upper level of the absorbent. Percolate a further of n-hexane/ethyl ether (99:1) and collect 220 ml at a flow of about 15 drops every 10 seconds. ( This fraction contains the methyl and ethyl esters and waxes ). ( Note 4 ) ( Note 5 ).
Note 4: The n-hexane/ethyl ether (99:1) mixture should be freshly prepared every day U.K.
Note 5: 100 μl of Sudan I dye at 1 % in the elution mixture can be added to the sample solution to check visually that the waxes are eluted properly. U.K.
The retention time of the dye lies in between that of the waxes and triacylglycerols. Hence, when the dye reaches the bottom of the chromatography column, elution has to be suspended because all the waxes have been eluted.
Evaporate the resultant fractions in a rotary evaporator until the solvent is almost removed. Remove the last 2 ml under a weak current of nitrogen. Collect the fraction containing the methyl and ethyl esters is diluted with 2-4 ml of n-heptane or iso-octane.
5.2. Gas chromatography analysis U.K.
5.2.1. Preliminary procedure U.K.
Fit the column to the gas chromatograph (point 3.3), connecting the inlet port to the on-column system and the outlet port to the detector. Check the gas chromatography apparatus (operation of gas loops, efficiency of detector and recorder system, etc.).
If the column is being used for the first time, it is advisable to condition it. Run a light flow of gas through the column, then switch on the gas chromatography apparatus. Gradually heat until a temperature of 350 °C is reached after approximately 4 h.
Maintain this temperature for at least 2 h, then regulate the apparatus to the operating conditions (regulate gas flow, light flame, connect to electronic recorder (point 3.3.4), regulate oven temperature for column, regulate detector, etc.). Record the signal at a sensitivity at least twice as high as that required for the analysis. The base line should be linear, with no peaks of any kind, and must not have any drift.
Negative straight-line drift indicates that the column connections are not correct while positive drift indicates that the column has not been properly conditioned.
5.2.2. Choice of operating conditions for waxes and methyl and ethyl esters (Note 6). U.K.
The operating conditions are generally as follows:


— Detector temperature
:
350 °C.
— Amount injected
:
1 μl of n-heptane solution (2-4 ml).
— Carrier gas
:
helium or hydrogen at the optimal linear speed for the gas chosen (see Appendix A).
— Instrument sensitivity
:
suitable for fulfilling the above conditions.
Note 6: Due to the high final temperature, positive drift is allowed but may not exceed more than 10 % of the full-scale value. U.K.
These conditions may be modified to suit the characteristics of the column and the gas chromatograph in order to separate all the waxes and fatty acid methyl and ethyl esters and to obtain satisfactory peak separation (see Figures 2, 3 and 4) and a retention time of 18 ± 3 minutes for the lauryl arachidate internal standard. The most representative peak of the waxes must be over 60 % of the full-scale value while the methyl heptadecanoate internal standard for the methyl and ethyl esters must reach the full-scale value.
The peak integration parameters should be determined in such a way as to obtain a correct evaluation of the peak areas considered.
5.3. Performance of the analysis U.K.
Take up 10 μl of the solution with the aid of the 10 μl micro-syringe, drawing back the plunger until the needle is empty. Introduce the needle into the injection system and inject quickly after 1–2 s. After about 5 s, gently extract the needle.
Perform the recording until the waxes or stigmastadienes are completely eluted, depending on the fraction being analysed.
The base line must always meet the required conditions.
5.4. Peak identification U.K.
Identify the peaks from the retention times by comparing them with mixtures of waxes with known retention times, analysed under the same conditions. The alkyl esters are identified from mixtures of methyl and ethyl esters of the chief fatty acids in olive oils (palmitic and oleic).
Figure 1 provides a chromatogram of the waxes in a virgin olive oil. Figures 2 and 3 show the chromatograms of two retail extra virgin olive oils, one with methyl and ethyl esters and the other without them. Figure 4 gives the chromatograms for a top-quality extra virgin olive oil and the same oil spiked with 20 % deodorised oil.
5.5. Quantitative analysis of the waxes U.K.
Determine the area of the peaks corresponding to the lauryl arachidate internal standard and the aliphatic esters from C 40 to C 46 with the aid of the integrator.
Determine the total waxes content by adding each individual wax, in mg/kg of fat, as follows:

where:
A x
=
area corresponding to the peak for the individual ester, in computer counts
A s
=
area corresponding to the peak for the lauryl arachidate internal standard, in computer counts
m s
=
mass of the lauryl arachidate internal standard added, in milligrams;
m
=
mass of the sample taken for determination, in grams.
5.5.1. Quantitative analysis of the methyl and ethyl esters U.K.
With the aid of the integrator, determine the areas of the peaks corresponding to the methyl heptadecanoate internal standard, the methyl esters of the C 16 and C 18 fatty acids and the ethyl esters of the C 16 and C 18 fatty acids.
Determine the content of each alkyl ester, in mg/kg of fat, as follows:
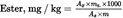
where:
A x
=
area corresponding to the peak for the individual C 16 and C 18 ester, in computer counts
A s
=
area corresponding to the peak for the methyl heptadecanoate internal standard, in computer counts
m s
=
mass of the methyl heptadecanoate internal standard added, in milligrams;
m
=
mass of the sample taken for determination, in grams.]
Options/Help
Print Options
PrintThe Whole Regulation
PrintThe Whole Annex
PrintThis Division only
You have chosen to open the Whole Regulation
The Whole Regulation you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
The Schedules you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As adopted by EU): The original version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
See additional information alongside the content
Geographical Extent: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Show Timeline of Changes: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Opening Options
Different options to open legislation in order to view more content on screen at once
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the EU Official Journal
- lists of changes made by and/or affecting this legislation item
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
Timeline of Changes
This timeline shows the different versions taken from EUR-Lex before exit day and during the implementation period as well as any subsequent versions created after the implementation period as a result of changes made by UK legislation.
The dates for the EU versions are taken from the document dates on EUR-Lex and may not always coincide with when the changes came into force for the document.
For any versions created after the implementation period as a result of changes made by UK legislation the date will coincide with the earliest date on which the change (e.g an insertion, a repeal or a substitution) that was applied came into force. For further information see our guide to revised legislation on Understanding Legislation.
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.