
ANNEX IIU.K.CRITERIA FOR SAMPLE PREPARATION AND FOR METHODS OF ANALYSIS USED FOR THE OFFICIAL CONTROL OF THE LEVELS OF MYCOTOXINS IN FOODSTUFFS
1.INTRODUCTIONU.K.
1.1.PrecautionsU.K.
As the distribution of mycotoxins is generally non-homogeneous, samples shall be prepared, and especially homogenised, with extreme care.
The complete sample as received by the laboratory shall be homogenized, in case the homogenisation is performed by the laboratory.
For the analysis of aflatoxins, daylight should be excluded as much as possible during the procedure, since aflatoxin gradually breaks down under the influence of ultra-violet light.
1.2.Calculation of proportion of shell/kernel of whole nutsU.K.
The limits fixed for aflatoxins in Regulation (EC) No 466/2001 apply to the edible part. The level of aflatoxins in the edible part can be determined by:
samples of nuts ‘in shell’ can be shelled and the level of aflatoxins is determined in the edible part.
the nuts ‘in shell’ can be taken through the sample preparation procedure. The method of sampling and analysis shall estimate the weight of nut kernel in the aggregate sample. The weight of nut kernel in the aggregate sample shall be estimated after establishing a suitable factor for the proportion of nut shell to nut kernel in whole nuts. This proportion is used to ascertain the amount of kernel in the bulk sample taken through the sample preparation and method of analysis.
Approximately 100 whole nuts shall be taken at random separately from the lot or shall be put aside from each aggregate sample. The ratio may, for each laboratory sample, be obtained by weighing the whole nuts, shelling and re-weighing the shell and kernel portions.
However, the proportion of shell to kernel may be established by the laboratory from a number of samples and so can be assumed for future analytical work. But if a particular laboratory sample is found to be in contravention of any limit, the proportion shall be determined for that sample using the approximately 100 nuts that have been set aside.
2.TREATMENT OF THE SAMPLE AS RECEIVED IN THE LABORATORYU.K.
Each laboratory sample shall be finely grinded and mixed thoroughly using a process that has been demonstrated to achieve complete homogenisation.
In case the maximum level applies to the dry matter, the dry matter content of the product shall be determined on a part of the homogenised sample, using a method that has been demonstrated to determine accurately the dry matter content.
3.REPLICATE SAMPLESU.K.
The replicate samples for enforcement, trade (defence) and reference (referee) purposes shall be taken from the homogenised material unless such procedure conflicts with [F1the rules applicable in each constituent territory of Great Britain] as regards the rights of the food business operator.
Textual Amendments
F1Words in Annex 2 para. 3 substituted (31.12.2020) by The Contaminants in Food (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/639), regs. 1, 17 (as substituted by S.I. 2020/1504, regs. 1(2), 8(6)); 2020 c. 1, Sch. 5 para. 1(1)
4.METHOD OF ANALYSIS TO BE USED BY THE LABORATORY AND LABORATORY CONTROL REQUIREMENTSU.K.
4.1.DefinitionsU.K.
A number of the most commonly used definitions that the laboratory shall be required to use are the following:
=
Repeatability, the value below which the absolute difference between two single test results obtained under repeatability conditions, namely same sample, same operator, same apparatus, same laboratory, and short interval of time may be expected to lie within a specific probability (typically 95 %) and hence r = 2,8 × sr.
=
Standard deviation, calculated from results generated under repeatability conditions.
=
Relative standard deviation, calculated from results generated under repeatability conditions [(sr / 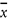
=
Reproducibility, the value below which the absolute difference between single test results obtained under reproducibility conditions, namely on identical material obtained by operators in different laboratories, using the standardised test method may be expected to lie within a certain probability (typically 95 %); R = 2,8 × sR.
=
Standard deviation, calculated from results under reproducibility conditions.
=
Relative standard deviation calculated from results generated under reproducibility conditions [(sR / 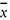
[F24.2. General requirements U.K.
Confirmatory methods of analysis used for food control purposes shall comply with the provisions of items 1 and 2 of Annex III to Regulation (EC) No 882/2004.
Textual Amendments
4.3. Specific requirements U.K.
4.3.1. Specific requirements for confirmatory methods U.K.
4.3.1.1. Performance criteria U.K.
It is recommended that fully validated confirmatory methods (i.e. methods validated by collaborative trials for relevant matrices) are used where appropriate and available. Other suitable validated confirmatory methods (e.g. methods validated in-house on relevant matrices belonging to the commodity group of interest) may also be used provided they fulfil the performance criteria set out in the following tables.
Where possible, the validation of in-house validated methods shall include a certified reference material.
Performance criteria for aflatoxins
| Note: | |||
| |||
| [X1Criterion | Concentration Range | Recommended Value | Maximum permitted Value |
|---|---|---|---|
| Blanks | All | Negligible | — |
| Recovery — Aflatoxin M 1 | 0,01-0,05 μg/kg | 60 to 120 % | |
| > 0,05 μg/kg | 70 to 110 % | ||
| Recovery — Aflatoxins B 1 , B 2 , G 1 , G 2 | < 1,0 μg/kg | 50 to 120 % | |
| 1-10 μg/kg | 70 to 110 % | ||
| > 10 μg/kg | 80 to 110 % | ||
| Reproducibility RSD R | All | As derived from Horwitz Equation (*)(**) | 2 × value derived from Horwitz Equation (*)(**) |
| Repeatability RSD r may be calculated as 0,66 times Reproducibility RSD R at the concentration of interest.] | |||
Performance criteria for ochratoxin A
| Level μg/kg | Ochratoxin A | ||
|---|---|---|---|
| RSD r % | RSD R % | Recovery % | |
| < 1 | ≤ 40 | ≤ 60 | 50 to 120 |
| ≥ 1 | ≤ 20 | ≤ 30 | 70 to 110 |
Performance criteria for patulin
| Level μg/kg | Patulin | ||
|---|---|---|---|
| RSD r % | RSD R % | Recovery % | |
| < 20 | ≤ 30 | ≤ 40 | 50 to 120 |
| 20-50 | ≤ 20 | ≤ 30 | 70 to 105 |
| > 50 | ≤ 15 | ≤ 25 | 75 to 105 |
Performance criteria for deoxynivalenol
| Level μg/kg | Deoxynivalenol | ||
|---|---|---|---|
| RSD r % | RSD R % | Recovery % | |
| > 100-≤ 500 | ≤ 20 | ≤ 40 | 60 to 110 |
| > 500 | ≤ 20 | ≤ 40 | 70 to 120 |
Performance criteria for zearalenone
| Level μg/kg | Zearalenone | ||
|---|---|---|---|
| RSD r % | RSD R % | Recovery % | |
| ≤ 50 | ≤ 40 | ≤ 50 | 60 to 120 |
| > 50 | ≤ 25 | ≤ 40 | 70 to 120 |
Performance criteria for Fumonisin B 1 and B 2 individually
| Level μg/kg | Fumonisin B 1 and B 2 individually | ||
|---|---|---|---|
| RSD r % | RSD R % | Recovery % | |
| ≤ 500 | ≤ 30 | ≤ 60 | 60 to 120 |
| > 500 | ≤ 20 | ≤ 30 | 70 to 110 |
Performance criteria for T-2 and HT-2 toxin individually
| Level μg/kg | T-2 and HT-2 toxin individually | ||
|---|---|---|---|
| RSD r % | RSD R % | Recovery % | |
| 15-250 | ≤ 30 | ≤ 50 | 60 to 130 |
| > 250 | ≤ 25 | ≤ 40 | 60 to 130 |
Performance criteria for citrinin
| Level μg/kg | Citrinin | |||
|---|---|---|---|---|
| RSD r % | Recommended RSD R % | Maximum allowed RSD R % | Recovery % | |
| All | 0,66 × RSD R | As derived from Horwitz Equation (*) (**) | 2 × value derived from Horwitz Equation (*) (**) | 70 to 120 |
Notes to the performance criteria for the mycotoxins:
The detection limits of the methods used are not stated as the precision values given at the concentrations of interest.
The precision values are calculated from the Horwitz equation, in particular the original Horwitz equation (for concentrations 1,2 × 10 –7 ≤ C ≤ 0,138) (*) and the modified Horwitz equation (for concentrations C < 1,2 × 10 –7 ) (**).
(*)Horwitz equation for concentrations 1,2 × 10 –7 ≤ C ≤ 0,138:
RSD R = 2 (1-0.5logC)
(ref: W. Horwitz, L.R. Kamps, K.W. Boyer, J.Assoc.Off.Analy.Chem.,1980, 63, 1344)
(**)Modified Horwitz equation (*) for concentrations C < 1,2 × 10 –7 :
RSD R = 22 %
(ref: M. Thompson, Analyst, 2000, 125, p. 385-386)
Where:
RSD R is the relative standard deviation calculated from results generated under reproducibility conditions [(sR/) × 100]
C is the concentration ratio (i.e. 1 = 100g/100g, 0,001 = 1 000 mg/kg)
This is a generalised precision equation which has been found to be independent of analyte and matrix but solely dependent on concentration for most routine methods of analysis.
Editorial Information
X1 Substituted by Corrigendum to Commission Regulation (EU) No 519/2014 of 16 May 2014 amending Regulation (EC) No 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis (Official Journal of the European Union L 147 of 17 May 2014).
4.3.1.2. ‘ Fitness-for-purpose ’ approach U.K.
For in-house validated methods, as an alternative, a ‘fitness-for-purpose’ approach (1) may be used to assess their suitability for official control. Methods suitable for official control must produce results with a standard measurement uncertainty (u) less than the maximum standard measurement uncertainty calculated using the formula below:

where:
Uf is the maximum standard measurement uncertainty (μg/kg)
LOD is the limit of detection of the method (μg/kg)
α is a constant, numeric factor to be used depending on the value of C. The values to be used are set out in Table hereafter.
C is the concentration of interest (μg/kg)
If the analytical method provides results with uncertainty measurements less than the maximum standard uncertainty the method shall be considered being equally suitable to one which meets the performance criteria given in point 4.3.1.1.
Table
Numeric values to be used for α as constant in formula set out in this point, depending on the concentration of interest
| C (μg/kg) | α |
|---|---|
| ≤ 50 | 0,2 |
| 51-500 | 0,18 |
| 501- 1 000 | 0,15 |
| 1 001 - 10 000 | 0,12 |
| > 10 000 | 0,1 |
4.3.2. Specific requirements for semi-quantitative screening methods U.K.
4.3.2.1. Scope U.K.
The scope applies to bioanalytical methods based on immuno-recognition or receptor binding (such as ELISA, dip-sticks, lateral flow devices, immuno-sensors) and physicochemical methods based on chromatography or direct detection by mass spectrometry (e.g. ambient MS). Other methods (e.g. thin layer chromatography) are not excluded provided the signals generated relate directly to the mycotoxins of interest and allow that the principle described hereunder is applicable.
The specific requirements apply to methods of which the result of the measurement is a numerical value, for example a (relative) response from a dip-stick reader, a signal from LC-MS, etc., and that normal statistics apply.
The requirements do not apply to methods that do not give numerical values (e.g. only a line that is present or absent), which require different validation approaches. Specific requirements for these methods are provided in point 4.3.3.
This document describes procedures for the validation of screening methods by means of an inter-laboratory validation, the verification of the performance of a method validated by means of an inter-laboratory exercise and the single-laboratory validation of a screening method.
4.3.2.2. Terminology U.K.
Screening target concentration (STC): the concentration of interest for detection of the mycotoxin in a sample. When the aim is to test compliance with regulatory limits, the STC is equal to the applicable maximum level. For other purposes or in case no maximum level has been established, the STC is predefined by the laboratory.
Screening method: means method used for selection of those samples with levels of mycotoxins that exceed the screening target concentration (STC), with a given certainty. For the purpose of mycotoxin screening, a certainty of 95 % is considered fit-for-purpose. The result of the screening analysis is either ‘ negative ’ or ‘ suspect ’ . Screening methods shall allow a cost-effective high sample-throughput, thus increasing the chance to discover new incidents with high exposure and health risks to consumers. These methods shall be based on bio-analytical, LC-MS or HPLC methods. Results from samples exceeding the cut-off value shall be verified by a full re-analysis from the original sample by a confirmatory method.
‘ Negative sample ’ means the mycotoxin content in the sample is < STC with a certainty of 95 % (i.e. there is a 5 % chance that samples will be incorrectly reported as negative).
‘ False negative sample ’ means the mycotoxin content in the sample is > STC but it has been identified as negative.
‘ Suspect sample ’ (screen positive) means the sample exceeds the cut-off level (see below) and may contain the mycotoxin at a level higher than the STC. Any suspect result triggers a confirmatory analysis for unambiguous identification and quantification of the mycotoxin.
‘ False suspect sample ’ is a negative sample that has been identified as suspect.
‘ Confirmatory methods ’ means methods that provide full or complementary information enabling the mycotoxin to be identified and quantified unequivocally at the level of interest.
Cut-off level: the response, signal, or concentration, obtained with the screening method, above which the sample is classified as ‘ suspect ’ . The cut-off is determined during the validation and takes the variability of the measurement into account.
Negative control (blank matrix) sample: a sample known to be free (2) of the mycotoxin to be screened for, e.g. by previous determination using a confirmatory method of sufficient sensitivity. If no blank samples can be obtained, then material with the lowest obtainable level might be used as long as the level allows the conclusion that the screening method is fit for purpose.
Positive control sample: sample containing the mycotoxin at the screening target concentration, e.g. a certified reference material, a material of known content (e.g. test material of proficiency tests) or otherwise sufficiently characterised by a confirmatory method. In the absence of any of the above, a blend of samples with different levels of contamination or a spiked sample prepared within laboratory and sufficiently characterised can be used, provided it can be proven that the contamination level has been verified.
4.3.2.3. Validation procedure U.K.
The aim of the validation is to demonstrate the fitness of purpose of the screening method. This is done by determination of the cut-off value and determination of the false negative and false suspect rate. In these two parameters performance characteristics such as sensitivity, selectivity, and precision are embedded.
Screening methods can be validated by inter-laboratory or by single laboratory validation. If inter-laboratory validation data is already available for a certain mycotoxin/matrix/STC combination, a verification of method performance is sufficient in a laboratory implementing the method.
4.3.2.3.1. Initial validation by single laboratory validation U.K.
Mycotoxins:
The validation shall be performed for every individual mycotoxin in the scope. In case of bio-analytical methods that give a combined response for a certain mycotoxin group (e.g. aflatoxins B 1 , B 2 , G 1 & G 2 ; fumonisins B 1 & B 2 ), applicability must be demonstrated and limitations of the test mentioned in the scope of the method. Undesired cross-reactivity (e.g. DON-3-glycoside, 3- or 15-acetyl-DON for immuno-based methods for DON) is not considered to increase the false negative rate of the target mycotoxins, but may increase the false suspect rate. This unwanted increasing will be diminished by confirmatory analysis for unambiguous identification and quantification of the mycotoxins.
Matrices:
An initial validation should be performed for each commodity, or, when the method is known to be applicable to multiple commodities, for each commodity group. In the latter case, one representative and relevant commodity is selected from that group (see table A).
Sample set:
The minimum number of different samples required for validation is 20 homogeneous negative control samples and 20 homogeneous positive control samples that contain the mycotoxin at the STC, analysed under intermediate precision (RSD Ri ) conditions spread over 5 different days. Optionally, additional sets of 20 samples containing the mycotoxin at other levels can be added to the validation set to gain insight to what extent the method can distinguish between different mycotoxin concentrations.
Concentration:
For each STC to be used in routine application, a validation has to be performed.
4.3.2.3.2. Initial validation through collaborative trials U.K.
Validation through collaborative trials shall be done in accordance with an internationally recognised protocol on collaborative trials (e.g. ISO 5725:1994 or the IUPAC International Harmonised Protocol) which requires inclusion of valid data from at least eight different laboratories. Other than that, the only difference compared to single laboratory validations is that the ≥ 20 samples per commodity/level can be evenly divided over the participating laboratories, with a minimum of two samples per laboratory.
4.3.2.4. Determination of cut-off level and rate of false suspected results of blank samples U.K.
The (relative) responses for the negative control and positive control samples are taken as basis for the calculation of the required parameters.
Screening methods with a response proportional with the mycotoxin concentration
For screening methods with a response proportional with the mycotoxin concentration the following applies:
Cut-off = R STC – t-value 0,05 * SD STC
mean response of the positive control samples (at STC)
one tailed t-value for a rate of false negative results of 5 % (see table B)
standard deviation
Screening methods with a response inversely proportional with the mycotoxin concentration
Similarly, for screening methods with a response inversely proportional with the mycotoxin concentration, the cut-off is determined as:
Cut-off = R STC + t-value 0,05 * SD STC
By using this specific t-value for establishing the cut-off value, the rate of false negative results is by default set at 5 %.
Fitness for purpose assessment
Results from the negative control samples are used to estimate the corresponding rate of false suspect results. The t-value is calculated corresponding to the event that a result of a negative control sample is above the cut off value, thus erroneously classified as suspect.
=
(cut off – mean blank )/SD blank
for screening methods with a response proportional with the mycotoxin concentration
or
=
( mean blank – cut off)/SD blank
for screening methods with a response inversely proportional with the mycotoxin concentration
From the obtained t-value, based on the degrees of freedom calculated from the number of experiments, the probability of false suspect samples for a one tailed distribution can either be calculated (e.g.. spread sheet function ‘ TDIST ’ ) or taken from a table for t-distribution.
The corresponding value of the one tailed t-distribution specifies the rate of false suspect results.
This concept is described in detail with an example in Analytical and Bioanalytical Chemistry DOI 10.1007/s00216 -013-6922-1.
4.3.2.5. Extension of the scope of the method U.K.
4.3.2.5.1. Extension of scope to other mycotoxins: U.K.
When new mycotoxins are added to the scope of an existing screening method, a full validation is required to demonstrate the suitability of the method.
4.3.2.5.2. Extension to other commodities: U.K.
If the screening method is known or expected to be applicable to other commodities, the validity to these other commodities shall be verified. As long as the new commodity belongs to a commodity group (see Table A) for which an initial validation has already been performed, a limited additional validation is sufficient. For this, a minimum of 10 homogeneous negative control and 10 homogeneous positive control (at STC) samples shall be analysed under intermediate precision conditions. The positive control samples shall all be above the cut-off value. In case this criterion is not met, a full validation is required.
4.3.2.6. Verification of methods already validated through collaborative trials U.K.
For screening methods that have already been successfully validated through a collaborative laboratory trial, the method performance shall be verified. For this a minimum of 6 negative control and 6 positive control (at STC) samples shall be analysed. The positive control samples shall all be above the cut-off value. In case this criterion is not met, the laboratory has to perform a root-cause analysis to identify why it cannot meet the specification as obtained in the collaborative trial. Only after taking corrective action it shall re-verify the method performance in its laboratory. In case the laboratory is not capable to verify the results from the collaborative trial, it will need to establish its own cut-off in a complete single laboratory validation.
4.3.2.7. Continuous method verification/on-going method validation U.K.
After initial validation, additional validation data are acquired by including at least two positive control samples in each batch of samples screened. One positive control sample is a known sample (e.g. one used during initial validation), the other is a different commodity from the same commodity group (in case only one commodity is analysed, a different sample of that commodity is used instead). Inclusion of a negative control sample is optional. The results obtained for the two positive control samples are added to the existing validation set.
At least once a year the cut-off value is re-established and the validity of the method is re-assessed. The continuous method verification serves several purposes:
quality control for the batch of samples screened
providing information on robustness of the method at conditions in the laboratory that applies the method
justification of applicability of the method to different commodities
allowing to adjust cut-off values in case of gradual drifts over time.
4.3.2.8. Validation report U.K.
The validation report shall contain:
A statement on the STC
A statement on the obtained cut-off.
Note: The cut-off must have the same number of significant figures as the STC. Numerical values used to calculate the cut-off need at least one more significant figure than the STC. U.K.
A statement on calculated false suspected rate
A statement on how the false suspected rate was generated.
Note: The statement on the calculated false suspected rate indicates if the method is fit-for-purpose as it indicates the number of blank (or low level contamination) samples that will be subject to verification. U.K.
Table A
Commodity groups for the validation of screening methods
| a If a buffer is used to stabilise the pH changes in the extraction step, then this commodity group can be merged into one commodity group ‘ High water content ’ . | ||
| b ‘ Difficult or unique commodities ’ should only be fully validated if they are frequently analysed. If they are only analysed occasionally, validation may be reduced to just checking the reporting levels using spiked blank extracts. | ||
| Commodity groups | Commodity categories | Typical representative commodities included in the category |
|---|---|---|
| High water content | Fruit Juices | Apple juice, grape juice |
| Alcoholic beverages | Wine, beer, cider | |
| Root and tuber vegetables | Fresh ginger | |
| Cereal or fruit based purees | Purees intended for infants and small children | |
| High oil content | Tree nuts | Walnut, hazelnut, chestnut |
| Oil seeds and products thereof | Oilseed rape, sunflower, cotton-seed, soybeans, peanuts, sesame etc. | |
| Oily fruits and products thereof | Oils and pastes (e.g. peanut butter, tahina) | |
| High starch and/or protein content and low water and fat content | Cereal grain and products thereof | Wheat, rye, barley, maize, rice, oats Wholemeal bread, white bread, crackers, breakfast cereals, pasta |
| Dietary products | Dried powders for the preparation of food for infants and small children | |
| High acid content and high water content a | Citrus products | |
| ‘Difficult or unique commodities’b | Cocoa beans and products thereof, copra and products thereof, coffee, tea Spices, liquorice | |
| High sugar low water content | Dried fruits | Figs, raisins, currants, sultanas |
| Milk and milk products | Milk | Cow, goat and buffalo milk |
| Cheese | Cow, goat cheese | |
| Dairy products (e.g. milk powder) | Yogurt, cream | |
Table B
One tailed t-value for a false negative rate of 5 %
| Degrees of Freedom | Number of replicates | t-value (5 %) |
|---|---|---|
| 10 | 11 | 1,812 |
| 11 | 12 | 1,796 |
| 12 | 13 | 1,782 |
| 13 | 14 | 1,771 |
| 14 | 15 | 1,761 |
| 15 | 16 | 1,753 |
| 16 | 17 | 1,746 |
| 17 | 18 | 1,74 |
| 18 | 19 | 1,734 |
| 19 | 20 | 1,729 |
| 20 | 21 | 1,725 |
| 21 | 22 | 1,721 |
| 22 | 23 | 1,717 |
| 23 | 24 | 1,714 |
| 24 | 25 | 1,711 |
| 25 | 26 | 1,708 |
| 26 | 27 | 1,706 |
| 27 | 28 | 1,703 |
| 28 | 29 | 1,701 |
| 29 | 30 | 1,699 |
| 30 | 31 | 1,697 |
| 40 | 41 | 1,684 |
| 60 | 61 | 1,671 |
| 120 | 121 | 1,658 |
| ∞ | ∞ | 1,645 |
4.3.3. Requirements for qualitative screening methods (methods that do not give numerical values) U.K.
The development of validation guidelines for binary test methods is currently subject of various standardization bodies (e.g. AOAC, ISO). Very recently AOAC has drafted a guideline on this matter. This document can be regarded as the current state of the art in its field. Therefore methods that give binary results (e.g. visual inspection of dip-stick tests) should be validated according to this guideline
http://www.aoac.org/imis15_prod/AOAC_Docs/ISPAM/Qual_Chem_Guideline_Final_Approved_031412.pdf
4.4. Estimation of measurement uncertainty, recovery calculation and reporting of results (3) U.K.
4.4.1. Confirmatory methods U.K.
The analytical result must be reported as follows:
Corrected for recovery, the level of recovery being indicated. The correction for recovery is not necessary in case the recovery rate is between 90-110 %.
As x +/– U whereby x is the analytical result and U is the expanded measurement uncertainty, using a coverage factor of 2 which gives a level of confidence of approximately 95 %.
For food of animal origin, the taking into account of the measurement uncertainty can also be done by establishing the decision limit (CCα) in accordance with Commission Decision 2002/657/EC (4) (point 3.1.2.5 of Annex I — the case of substances with established permitted limit).
However if the result of the analysis is significantly (> 50 %) lower than the maximum level or much higher than the maximum level (i.e. more than 5 times the maximum level), and on the condition that the appropriate quality procedures are applied and the analysis serves only the purpose of checking compliance with legal provisions, the analytical result might be reported without correction for recovery and the reporting of the recovery rate and measurement uncertainty might be omitted in these cases.
The present interpretation rules of the analytical result in view of acceptance or rejection of the lot apply to the analytical result obtained on the sample for official control. In case of analysis for defence or referee purposes, the national rules apply.
4.4.2. Screening methods U.K.
The result of the screening shall be expressed as compliant or suspected to be non-compliant.
‘ Suspected to be non-compliant ’ means the sample exceeds the cut-off level and may contain the mycotoxin at a level higher than the STC. Any suspect result triggers a confirmatory analysis for unambiguous identification and quantification of the mycotoxin.
‘ Compliant ’ means that the mycotoxin content in the sample is < STC with a certainty of 95 % (i.e. there is a 5 % chance that samples will be incorrectly reported as negative). The analytical result is reported as ‘ < level of STC ’ with the level of STC specified.]
4.5.Laboratory quality standardsU.K.
Laboratory must comply with the provisions of Article 12 of Regulation (EC) No 882/2004 on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules(5).
[F2Ref: M. Thompson and R. Wood, Accred. Qual. Assur., 2006, 10, p. 471-478.]
[F2Samples are considered free of analyte if the amount present in the sample does not exceed more than 1/5 th of the STC. If the level can be quantified with a confirmatory method, the level must be taken into consideration for the validation assessment.]
[F2More details on procedures for the estimation of measurement uncertainty and on procedures for assessing recovery can be found in the report ‘ Report on the relationship between analytical results, measurement uncertainty, recovery factors and the provisions of EU food and feed legislation ’ — http://ec.europa.eu/food/food/chemicalsafety/contaminants/report-sampling_analysis_2004_en.pdf]
[F2Commission Decision 2002/657/EC of 14 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results ( OJ L 221, 17.8.2002, p. 8 ).]
See also the transitional arrangements provided for in article 18 of Commission Regulation (EC) No 2076/2005 of 5 December 2005 laying down transitional arrangements for the implementation of Regulation (EC) No 853/2004, 854/2004 and 882/2004 of the European Parliament and of the Council and amending Regulations (EC) No 853/2004 and 854/2004 (OJ L 338, 22.12.2005, p. 83).
Textual Amendments