Commission Regulation (EC) No 273/2008
of 5 March 2008
laying down detailed rules for the application of Council Regulation (EC) No 1255/1999 as regards methods for the analysis and quality evaluation of milk and milk products (repealed)
THE COMMISSION OF THE EUROPEAN COMMUNITIES,
Having regard to the Treaty establishing the European Community,
Having regard to Council Regulation (EC) No 1255/1999 of 17 May 1999 on the common organisation of the market in milk and milk products(1), and in particular Articles 10 and 15 and Articles 26(3), 29(1) and 31(4) thereof,
Whereas:
(1) Commission Regulation (EC) No 213/2001(2) lays down detailed rules for the application of Council Regulation (EC) No 1255/1999 as regards methods for the analysis and quality evaluation of milk and milk products. In the light of technical developments in the field of analytical methodology further substantial changes need to be made. In the interest of clarity and efficiency and given the number and technical nature of the amendments, Regulation (EC) No 213/2001 should be repealed and replaced by a new regulation.
(2) The composition and quality requirements for milk and milk products laid down under the arrangements provided for in Regulation (EC) No 1255/1999 must be verified to ensure that they are strictly complied with.
(3) The reference methods for such verifications are often methods published by international organisations such as the European Committee for Standardization (CEN), the International Dairy Federation (IDF), the International Organisation for Standardisation (ISO) and the Scientific Association dedicated to the analytical Excellence (AOAC International), which are regularly updated by those organisations. In some cases a Community reference method is laid down, while in other cases no reference method is specified in the Community rules. In order to ensure that reference methods are uniformly applied, a list of reference methods should be drawn up and provision should be made for the Commission to adapt the list where necessary.
(4) The use of routine methods should not be ruled out. Minimal conditions for using them should therefore be specified.
(5) Common procedures should also be established to ensure uniform practice in evaluating the results of analyses, in sensory evaluation of the products concerned and in re-examining results which have been disputed.
(6) For some analyses, there are currently no internationally accepted reference methods which have been validated and thus no information is available on the between-laboratory-variation of analytical results. Community methods should therefore be laid down, which have been validated according to internationally established rules and should be applied as reference methods.
(7) Commission Regulation (EC) No 1898/2005(3) lays down detailed rules for implementing Council Regulation (EC) No 1255/1999 as regards measures for the disposal of cream, butter and concentrated butter on the Community market and provides for the tracing of cream, butter and concentrated butter in certain circumstances in order to ensure the correct end use of these products. Tracing is important for the proper functioning of the scheme. In order to ensure that operators participating in it receive equal treatment, common methods should be established for determining some of these tracers.
(8) Under Article 9 of Regulation (EC) No 1255/1999, private storage aid may be granted for cheeses made from ewes’ milk. A special refund for the same products can be granted under Article 31 of that Regulation. Cheeses made from ewes’ milk, goats’ milk, buffalos’ milk and mixtures of ewes’, goats’ and buffalos’ milk may be imported into the Community under preferential arrangements from certain third countries. In view of the above, appropriate checks are needed to ensure that no cow’s milk has been incorporated in the products concerned. A Community reference method should therefore be established for detecting cow’s milk, without prejudice to the use of routine methods, provided they comply with certain criteria.
(9) Under Commission Regulation (EEC) No 2921/90 of 10 October 1990 on aid for the production of casein and caseinates from skimmed milk(4), the absence of coliforms must be detected. The internationally accepted reference methods for detecting coliforms in milk and milk products is ISO 4831. A Community reference method for detecting coliforms has been established based on the abovementioned standard.
(10) Council Regulation (EEC) No 2658/87 of 23 July 1987 on the tariff and statistical nomenclature and on the Common Customs Tariff(5) provides for different rates of customs duty for compound feedingstuffs falling within tariff heading No 2309, depending on their milk-product content. To ensure that the rules in question are uniformly applied, a generally recognised method for analysing lactose content should be laid down for compulsory use in all Member States.
(11) Under Regulation (EC) No 1255/1999, butter and skimmed-milk powder intended for intervention or, in the case of skimmed-milk powder, for use as animal feed, must meet certain quality requirements. Reference methods should be laid down to verify that those requirements are met.
(12) Some methods are introduced for the first time in this Regulation. A sufficient period from the time of entry into force of this Regulation should be provided in order to allow laboratories to correctly introduce and use these new methods. Whenever a reference method referred to in Annex I is revised and published by the Standards Developing Organisation, the laboratories should be allowed six months to update their analytical procedures to conform to the new standard.
(13) The measures provided for in this Regulation are in accordance with the opinion of the Management Committee for Milk and Milk products,
HAS ADOPTED THIS REGULATION:
CHAPTER IGENERAL PROVISIONS
Article 1Subject matter and scope
1.This Regulation lays down certain reference methods for the chemical, physical and microbiological analysis and sensory evaluation of milk and milk products to be used under the arrangements provided for in the common organisation of the market in milk and milk products established by Regulation (EC) No 1255/1999 and the rules for applying those methods.
2.The list of the reference methods applicable to analyses as referred to in paragraph 1 is laid down in Annex I to this Regulation.
3.The Commission shall update the list in accordance with the procedure laid down in Article 42 of Regulation (EC) No 1255/1999.
Article 2Routine methods
Routine methods may be used for analyses required by the Community rules provided that they are properly calibrated and regularly checked against the reference method. Results shall be compared taking into account the constant bias, the repeatability and the reproducibility.
In cases of dispute, the result obtained by the reference method shall be decisive.
Member States shall inform the Commission on the use of routine methods in the analysis referred to in Article 1.
CHAPTER IIMETHODS OF ANALYSIS
Article 3Evaluation of compliance of a consignment with a legal limit
Except for the analysis of tracers, Annex II to this Regulation shall apply in order to define compliance with legal compositional requirements.
Article 4Sensory evaluation
1.For milk and milk products other than butter for public storage, the reference method to be used by the Member States for sensory evaluation shall be either IDF standard 99C:1997 or other comparable methods which they shall notify to the Commission.
The procedures described in Annex III shall be applied to check the performance of assessors and the reliability of results in sensory analyses.
2.For butter for public storage, the procedures described in Annex III shall be applied to check the performance of assessors and the reliability of results in sensory analyses.
The procedure laid down in Annex IV shall be applied as a reference method for sensory evaluation.
Article 5Tracers
1.The method of analysis laid down in Annex V shall be used as the reference method for determining the content of enanthic acid triglyceride in butter, butter-oil and cream.
2.The method of analysis laid down in Annex VI shall be used as the reference method for determining vanillin in concentrated butter, butter and cream.
3.The method of analysis laid down in Annex VII shall be used as the reference method for determining the ethyl ester of beta-apo-8' carotenic acid content of concentrated butter and butter.
4.The analysis method laid down in Annex VIII shall be used as the reference method for determining the β-sitosterol or stigmasterol content of butter and concentrated butter.
5.Concentrated butter, butter and cream are considered to be traced in conformity with the relevant Community rules if the results obtained are in accordance with the specifications of points 10 and 11 of Annex V and point 8 of Annexes VI, VII and VIII.
Article 6Detection of cows’ milk casein
1.The reference method of analysis laid down in Annex IX shall be used to ensure that cheese made exclusively from ewes’ milk, goats’ milk or buffalos’ milk or from a mixture of ewes’, goats’ and buffalos’ milk does not contain cows’ milk casein.
Cows’ milk casein is considered to be present if the cows’ milk casein content of the analysed sample is equal to or higher than the content of the reference sample containing 1 % cows’ milk as laid down in Annex IX.
2.Routine methods for detecting cows’ milk casein in cheeses referred to in paragraph 1 may be used provided that:
(a)the detection limit is maximum 0,5 %; and
(b)there are no false-positive results; and
(c)cows’ milk casein is detectable with the required sensitivity even after long ripening periods, as may occur in usual commercial conditions.
If any of the above mentioned requirements is not met, the reference methods laid down in Annex IX shall be used.
Article 7Detection of coliforms
Coliforms in butter, skimmed-milk powder, casein and caseinates shall be detected in accordance with the reference method laid down in Annex X.
Article 8Determination of the lactose content
The lactose content of products falling within CN code 2309 shall be determined in accordance with the reference method laid down in Annex XI.
Article 9Detection of rennet whey
1.Rennet whey in skimmed-milk powder intended for public storage shall be detected in accordance with the reference method laid down in Annex XII.
2.Rennet whey in skimmed-milk powder and mixtures intended for use as animal feed shall be detected in accordance with the reference method laid down in Annex XII. In case of detection of rennet whey, Annex XIII should be implemented.
Article 10Detection of buttermilk
Buttermilk in skimmed-milk powder shall be detected in accordance with the reference method laid down in Annex XIV.
Article 11Detection of antimicrobiotic residues
Antimicrobial residues in skimmed-milk powder shall be detected in accordance with the reference method laid down in Annex XV.
Article 12Determination of skimmed-milk powder content
Skimmed-milk powder content in compound feedingstuffs shall be determined in accordance with the reference method laid down in Annex XVI.
Article 13Detection of starch
Starch in skimmed-milk powder, denatured milk powder and compound feedingstuffs shall be detected in accordance with the reference method laid down in Annex XVII.
Article 14Determination of moisture content in dried cream
Moisture content in dried cream shall be determined in accordance with the reference method laid down in Annex XVIII.
Article 15Determination of moisture content of acid buttermilk powder
Moisture content in acid buttermilk powder intended for use in feedingstuffs shall be determined in accordance with the reference method laid down in Annex XIX.
Article 16Determination of milk fat purity
Milk fat purity shall be determined in accordance with the reference method laid down in Annex XX.
CHAPTER IIIGENERAL AND FINAL PROVISIONS
Article 17Quality assurance
Analyses shall be performed in laboratories having an analytical quality assurance system including internal quality control procedures. Not accredited laboratories shall participate in proficiency testing schemes at least once per year and their results shall not deviate by more than 2σR (reproducibility standard deviation of the reference method) from the consensus value. A detailed description of the systems used shall be available for consultation in the laboratory.
Laboratories that are accredited in accordance with the standards referred to in Article 12 of Regulation (EC) No 882/2004 of the European Parliament and of the Council of 29 April 2004 on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules(6) shall be exempted from the obligation to participate in proficiency testing.
Article 18Sampling and disputes over the results of analysis
1.Sampling shall be carried out in accordance with the relevant regulation for the product under consideration. If no sampling provisions are presented, then the provision given in ISO 707 | IDF 50, Milk and milk products — Guidance of sampling, shall be used.
2.Laboratory reports of the results of the analysis must contain sufficient information for an evaluation of the results to be made in accordance with Annex II and Annex XXI.
3.Duplicate samples must be taken for analyses required under Community rules.
4.The procedure described in Annex XXI shall be used in cases where the results of an analysis are not accepted by the operator.
5.If the manufacturer can prove, within five working days of sampling, that the sampling procedure was not carried out correctly, sampling must be repeated where possible. If sampling cannot be repeated, the consignment must be accepted.
Article 19Transition period
Evaluation of compliance according to Annex II to this Regulation shall be performed within 12 months after its entry into force. Member States will immediately report to the Commission where necessary if any major problem is encountered during this period with the statistical control procedure.
Article 20Repeals
Regulation (EC) No 213/2001 is repealed.
References to the repealed Regulation shall be construed as references to this Regulation and shall be read in accordance with the correlation table in Annex XXII.
Article 21Entry into force
This Regulation shall enter into force on the third day following that of its publication in the Official Journal of the European Union.
It shall apply from 31 March 2008.
This Regulation shall be binding in its entirety and directly applicable in all Member States.
Done at Brussels, 5 March 2008.
For the Commission
Mariann Fischer Boel
Member of the Commission
ANNEX I(Article 1)
LIST OF REFERENCE METHODS
Index Min. = minimum, Max. = maximum, Annex = Annex to quoted Regulation, SNF = solids non fat, PV = peroxide value, A = appearance, F = flavour, C = consistency, TBC = total bacterial count, Therm = thermophilic bacterial count, MS = Member State, IDF = International Dairy Federation, ISO = International Standards Organisation, IUPAC = International Union of Pure and Applied Chemistry, ADPI = American Dairy Products Institute, SCM = sweetened condensed milk, EMC = evaporated milk or cream.
PART A
| a Without prejudice to the requirements in the specific Regulation | |||||
| b The minimum protein content would be 34 % per 1 September 2009. | |||||
| Commission Regulation | Product | Parameter | Limita | Reference method | Remark |
|---|---|---|---|---|---|
| Regulation (EC) No 2771/1999 — Public storage | Unsalted butter | Fat | Min. 82 % m/m | ISO 17189:2003|IDF 194:2003 | |
| Water | Up to 16 % m/m | ISO 3727-1:2001|IDF 80-1:2001 | |||
| SNF | Up to 2 % m/m | ISO 3727-2:2001|IDF 80-2:2001 | |||
| Fat acidity | 1,2 mmole/100 g of fat | ISO 1740:2004|IDF 6:2004 | |||
| PV (max.) | 0,3 meq. oxygen/1 000 g fat | ISO 3976:2006|IDF 74:2006 | Note 1 | ||
| Coliforms | Not detectable in 1 g | Annex X | Note 3 | ||
| Non-milk fat | Not detectable by triglyceride analysis | Annex XX | |||
| Sterol tracers | Not detectable, β-sitosterol ≤ 40 mg/kg | Annex VIII | |||
| Other tracers | |||||
| Not detectable | Annex VI | |||
| ≤ 6 mg/kg | Annex VII | |||
| Not detectable | Annex V | |||
| Sensory characteristics | At least 4 out of 5 points for A, F and C | Annex IV | |||
| Water dispersion | At least 4 points | ISO 7586:1985 — IDF 112A:1989 | |||
| Regulation (EC) No 2771/1999 — Private storage | Unsalted butter | Fat | Min. 82 % m/m | ISO 17189:2003|IDF 194:2003 | |
| Water | Up to 16 % m/m | ISO 3727-1:2001|IDF 80-1:2001 | |||
| SNF | Up to 2 % m/m | ISO 3727-2:2001|IDF 80-2:2001 | |||
| Regulation (EC) No 2771/1999 — Private storage | Salted butter | Fat | Min. 80 % m/m | ISO 17189:2003|IDF 194:2003 | |
| Water | Up to 16 % m/m | ISO 3727-1:2001|IDF 80-1:2001 | |||
| SNF (excluding salt) | Up to 2 % m/m | ISO 3727-2:2001|IDF 80-2:2001 | |||
| Salt | Up to 2 % m/m | ISO 15648:2004|IDF 179:2004 | |||
| Regulation (EC) No 1898/2005 chapter II | Unsalted butter | Fat | Min. 82 % m/m | ISO 17189:2003|IDF 194:2003 | |
| Non-milk fat | Annex XX | ||||
| Water | Up to 16 % m/m | ISO 3727-1 2001|IDF 80-1:2001 | |||
| SNF | Up to 2 % m/m | ISO 3727-2:2001|IDF 80-2:2001 | |||
| Tracers: | |||||
| See Annex VIII | Annex VIII | |||
| See Annex VI | Annex VI | |||
| See Annex VII | Annex VII | |||
| See Annex V | Annex V | |||
| Regulation (EC) No 1898/2005 chapter II | Salted butter | Fat | Min. 80 % m/m | ISO 17189:2003|IDF 194:2003 | |
| Non-milk fat | Annex XX | ||||
| Water | Up to 16 % m/m | ISO 3727-1:2001|IDF 80-1:2001 | |||
| SNF (excluding salt) | Up to 2 % m/m | ISO 3727-2:2001|IDF 80-2:2001 | |||
| Salt | Up to 2 % m/m | ISO 15648:2004|IDF 179:2004 | |||
| Tracers: | |||||
| See Annex VIII | Annex VIII | |||
| See Annex VI | Annex VI | |||
| See Annex VII | Annex VII | |||
| See Annex V | Annex V | |||
| Regulation (EC) No 1898/2005 chapter II | Concentrated butter | Fat | Min. 99,8 % m/m | IDF 24:1964 | |
| Water and SNF | Up to 0,2 % m/m | ISO 5536:2002|IDF 23:2002 (moisture) IDF 24:1964 (SNF) | |||
| Fat acidity | 1,2 mmole/100 g of fat | ISO 1740:2004|IDF 6:2004 | |||
| PV (max.) | 0,5 meq. oxygen/1 000 g fat | ISO 3976:2006|IDF 74:2006 | Note 1 | ||
| Non-milk fat | Absence | Annex XX | |||
| Flavour | Clean | ||||
| Smell | Absence of foreign odours | ||||
| Other | Absence of neutralising agents, anti-oxidants and preservatives | ||||
| Tracers: | |||||
| See Annex VIII | Annex VIII | |||
| See Annex VI | Annex VI | |||
| See Annex VII | Annex VII | |||
| See Annex V | Annex V | |||
| Regulation (EC) No 1898/2005 chapter II | Cream | Fat | Minimum 35 % m/m | ISO 2450:1999|IDF 16 C:1987 | |
| Non-milk fat | Annex XX | ||||
| Tracers: | |||||
| See Annex VIII | Note 2 | |||
| See Annex VI | Annex VI | |||
| See Annex VII | Note 2 | |||
| See Annex V | Annex V | |||
| Regulation (EC) No 1898/2005 chapter III | Concentrated butter | Fat | Min. 96 % m/m | Note 2 | |
| Non-milk fat | Annex XX | ||||
| SNF | Up to 2 % m/m | Note 2 | |||
| Tracers: | |||||
| 15 g/100 kg of concentrated butter | Annex VIII | |||
| 17 g/100 kg of concentrated butter | Annex VIII | |||
| 10,34 kg/t of concentrated butter | Annex V | |||
|
| Note 2 | |||
|
| Note 2 | |||
| lecithin (E 322) | Up to 0,5 % m/m | Note 2 | |||
| NaC1 | Up to 0,75 % m/m | ISO 15648:2004|IDF 179:2004 | |||
| Fat acidity | 1,2 mmole/100 g of fat | ISO 1740:2004|IDF 6:2004 | |||
| PV (max.) | Up to 0,5 meq. oxygen/1 000 g fat | ISO 3976:2006|IDF 74:2006 | Note 1 | ||
| Flavour | Clean | ||||
| Smell | Absence of foreign odours | ||||
| Other | Absence of neutralising agents, anti-oxidants and preservatives | ||||
| Regulation (EC) No 1898/2005 chapter IV | Unsalted butter | Fat | Min. 82 % m/m | ISO 17189:2003|IDF 194:2003 | |
| Water | Up to 16 % m/m | ISO 3727-1:2001|IDF 80-1:2001 | |||
| SNF | Up to 2 % m/m | ISO 3727-2:2001|IDF 80-2:2001 | |||
| Regulation (EC) No 1898/2005 chapter IV | Salted butter | Fat | Min. 80 % m/m | ISO 17189:2003|IDF 194:2003 | |
| Water | Up to 16 % m/m | ISO 3727-1:2001|IDF 80-1:2001 | |||
| SNF (excluding salt) | Up to 2 % m/m | ISO 3727-2:2001|IDF 80-2:2001 | |||
| Salt | Up to 2 % m/m | ISO 15648:2004|IDF 179:2004 | |||
| Article 9 and Title II of Regulation (EC) No 1255/1999 | Cheese made from ewes’ and/or goats’ milk | Cows’ milk | < 1 % m/m | Annex IX | |
| Regulation (EEC) No 2921/90 | Annex I — Acid casein | Water | Up to 12,00 % m/m | ISO 5550:2006|IDF 78:2006 | |
| Fat | Up to 1,75 % m/m | ISO 5543:2004|IDF127:2004 | |||
| Free acidity | Up to 0,30 ml of 0,1 N NaOH solution/g | ISO 5547:1978|IDF 91:1979 | |||
| Regulation (EEC) No 2921/90 | Annex I — Rennet- casein | Water | Up to 12,00 % m/m | ISO 5550:2006|IDF 78:2006 | |
| Fat | Up to 1,00 % m/m | ISO 5543:2004|IDF 127:2004 | |||
| Ash | Min. 7,50 % m/m | ISO 5545:1978|IDF 90:1979 | |||
| Regulation (EEC) No 2921/90 | Annex I — Caseinates | Water | Up to 6,00 % m/m | ISO 5550:2006|IDF 78:2006 | |
| Milk protein | Min. 88,00 % m/m | ISO 5549:1978|IDF 92:1979 | |||
| Fat and ash | Up to 6,00 % m/m | ISO 5543:2004|IDF 127:2004 | |||
| Fixed ash | ISO 5544:1978|IDF 89:1979 | ||||
| Ash | ISO 5545:1978|IDF 90:1979 | ||||
| Regulation (EEC) No 2921/90 | Annex II — Acid casein | Water | Up to 10,00 % m/m | ISO 5550:2006|IDF 78:2006 | |
| Fat | Up to 1,50 % m/m | ISO 5543:2004|IDF 127:2004 | |||
| Free acidity | Up to 0,20 ml of 0,1 N NaOH solution/g | ISO 5547:1978|IDF 91:1979 | |||
| TBC (max.) | 30 000/ g | ISO 4833:2003 | Note 3 | ||
| Coliforms | Absence in 0,1 g | Annex X | Note 3 | ||
| Therm. (max.) | 5 000/ g | ISO 4833:2003 | Notes 3 and 4 | ||
| Regulation (EEC) No 2921/90 | Annex II — Rennet-casein | Water | Up to 8,00 % m/m | ISO 5550:2006|IDF 78:2006 | |
| Fat | Up to 1,00 % m/m | ISO 5543:2004|IDF 127:2004 | |||
| Ash | Min. 7,50 % m/m | ISO 5545:1978|IDF 90:1979 | |||
| TBC (max.) | 30 000/ g | ISO 4833:2003 | Note 3 | ||
| Coliforms | Absence in 0,1 g | Annex X | Note 3 | ||
| Therm. (max.) | 5 000/ g | ISO 4833:2003 | Notes 3 and 4 | ||
| Regulation (EEC) No 2921/90 | Annex II — Caseinates | Water | Up to 6,00 % m/m | ISO 5550:2006|IDF 78:2006 | |
| Milk protein | Min. 88,00 % m/m | ISO 5549:1978|IDF 92:1979 | |||
| Fat and ash | Up to 6,00 % m/m | ISO 5543:2004|IDF 127:2004 ISO 5544:1978|IDF 89:1979 or ISO 5545:1978|IDF 90:1979 | |||
| TBC (max.) | 30 000/ g | ISO 4833:2003 | Note 3 | ||
| Coliforms | Absence in 0,1 g | Annex X | Note 3 | ||
| Therm. (max.) | 5 000/ g | ISO 4833:2003 | Notes 3 and 4 | ||
| Regulation (EEC) No 2921/90 | Annex III — Caseinates | Water | Up to 6,00 % m/m | ISO 5550:2006|IDF 78:2006 | |
| Milk protein | Min. 85,00 % m/m | ISO 5549:1978|IDF 92:1979 | |||
| Fat | Up to 1,50 % m/m | ISO 5543:2004|IDF 127:2004 | |||
| Lactose | Up to 1,00 % m/m | ISO 5548:2004|IDF 106:2004 | |||
| Ash | Up to 6,50 % m/m | ISO 5544:1978|IDF 89:1979 or ISO 5545:1978|IDF 90:1979 | |||
| TBC (max.) | 30 000/ g | ISO 4833:2003 | Note 3 | ||
| Coliforms | Absence in 0,1 g | Annex X | Note 3 | ||
| Therm. (max.) | 5 000/ g | ISO 4833:2003 | Notes 3 and 4 | ||
| Regulation (EC) No 2799/1999 | Compound feedingstuffs and skimmed-milk powder (SMP) (for use in feedingstuffs) | Water (acid buttermilk powder) | Up to 5 % m/m | Annex XIX | |
| Protein | 31,4 % m/m (min.) of the non-fat dry matter | ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 | |||
| Water (SMP) | Up to 5 % m/m | ISO 5537:2004|IDF 26:2004 | |||
| Fats (SMP) | Up to 11 % m/m | ISO 1736:2000|IDF 9C:1987 | |||
| Rennet whey (SMP) | Absence | Annex XIII | Note 6 | ||
| Starch (SMP) | Absence | Annex XVII | |||
| Water (mixtures) | Up to 5 % m/m of the non-fat matter | ISO 5537:2004|IDF 26:2004 | |||
| Fat (mixtures) | Commission Directive 84/4/EEC (OJ L 15, 18.1.1984, p. 29) | ||||
| Rennet whey (mixtures) | Absence | Annex XIII | |||
| SMP content (of end product) | Min. 50 % m/m | Annex XVI | |||
| Fat (in end product) | Min. 2,5 % m/m or 5 % m/m | Commission Directive 84/4/EEC (OJ L 15, 18.1.1984, p. 29) | Note 7 | ||
| Starch (in end product) | Min. 2 % m/m | Annex XVII | Note 8 | ||
| Copper (in end product) | 25 ppm | Commission Directive 78/633/EEC (OJ L 206, 26.7.1987, p. 43) | |||
| Regulation (EC) No 214/2001 | SMP (spray) | Fat | Up to 1,0 % m/m | ISO 1736:2000|IDF 9C:1987 | |
| Protein | 31,4 %b m/m (min.) of the non-fat dry matter | ISO 8968-1/2:2001|IDF 20-1/2:2001 | |||
| Water | Up to 3,5 % m/m | ISO 5537:2004|IDF 26:2004 | |||
| Acidity | Up to 19,5 ml, 0,1 N NaOH, 10 g solids non-fat | ISO 6091:1980|IDF 86:1981 | |||
| Lactates | Up to 150 mg/100 g solids non-fat | ISO 8069:2005|IDF 69:2005 | |||
| Phosphatase | Negative | ISO 11816-1:2006|IDF 155-1:2006 | |||
| Insolubility index | Up to 0,5 ml at 24 °C | ISO 8156:2005|IDF 129:2005 | |||
| Scorched particles | Disc A or B (15,0 mg) | ADPI (1990) | |||
| TBC | 40 000/ g | ISO 4833:2003 | Note 3 | ||
| Coliforms | Negative/0,1 g | Annex X | Note 3 | ||
| Buttermilk | Negative | Annex XIV | |||
| Rennet whey | Negative | Annex XII | |||
| Acid whey | Negative | Note 2 | |||
| Anti-microbial agents | Annex XV | ||||
PART B
The reference methods listed in Part B may be used for analysing products covered by any of the Regulations listed in column 1.
| Commission Regulation | Product | CN code | Parameter | Limit | Reference method | Remark |
|---|---|---|---|---|---|---|
| Regulation (EEC) No 2658/87 Regulation (EC) No 2535/2001 Regulation (EC) No 1282/2006 | Milk and cream, not concentrated nor containing added sugar or other sweetening matter | 0401 | Fat (≤ 6 % m/m) | The limits are those specified in the description of the CN code for the specific product, further specified, where applicable, in Commission Regulation (EEC) No 3846/87 (OJ L 366, 24.12.1987, p. 1), Part 9 of the export nomenclature or Regulation (EC) No 2535/2001 (OJ L 341, 22.12.2001, p. 29) | ISO 1211:2001|IDF 1D:1996 | |
| Fat (> 6 % m/m) | ISO 2450:1999|IDF 16C:1987 | |||||
| Milk and cream, concentrated or containing added sugar or other sweetening matter | 0402 | Fat (liquid form) | ISO 1737:1999|IDF 13C:1987 | |||
| Fat (solid form) | ISO 1736:2000|IDF 9C:1987 | |||||
| Protein | ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 | |||||
| Sucrose (normal content) | ISO 2911:2004|IDF 35:2004 | |||||
| Sucrose (low content) | Note 2 | |||||
| Solids (SCM) | ISO 6734:1989|IDF 15B:1991 | |||||
| Solids (EMC) | ISO 6731:1989|IDF 21B:1987 | |||||
| Water (milk powder) | ISO 5537:2004|IDF 26:2004 | |||||
| Water (cream powder) | Annex XVIII | |||||
| Buttermilk, fermented or acidified milk and cream, concentrated or not concentrated, containing added sugar or other sweetening matter | 0403 | Fat | ISO 1211:2001|IDF 1D:1996 ISO 1736:2000|IDF 9C:1987 ISO 2450:1999|IDF 16 C:1987 ISO 7208:1999|IDF 22B:1987 ISO 8262-3:2005|IDF 124-3:2005 | |||
| Protein | ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 | |||||
| Sucrose (normal content) | ISO 2911:2004|IDF 35:2004 | |||||
| Sucrose (low content) | Note 2 | |||||
| Water (acid buttermilk powder) | Annex XIX | |||||
| Water (sweet buttermilk powder) | ISO 5537:2004|IDF26:2004 | |||||
| Solids (other products) | Methods approved by the competent authority | |||||
| Whey, whether or not concentrated or containing added sugar or other sweetening matter; products consisting of natural milk constituents | 0404 | Fat | ISO 1736:2000|IDF 9C:1987 ISO 2450:1999|IDF 16C:1987 ISO 7208:1999|IDF 22B:1987 | |||
| Protein | ISO 8968-1|2|3:2001|IDF 20-1|2|3:2001 | |||||
| Sucrose (normal content) | ISO 2911:2004|IDF 35:2004 | |||||
| Sucrose (low content) | Note 2 | |||||
| 0404 90 | Protein | ISO 8968 1/2 2001|IDF 20-1/2:2001 | ||||
| Water | IDF 21B:1987 | |||||
| Solids | ISO 6734:1989|IDF 15B:1991 | |||||
| (Concentrated products) | ISO 6731:1989|IDF 21B:1987 | |||||
| Butter and other fats derived from milk; dairy spreads | 0405 | Fat (if ≤ 85 % m/m) | ISO 17189:2003|IDF 194:2003 | |||
| Butter | Water | ISO 3727-1:2001|IDF 80-1:2001 | ||||
| SNF | ISO 3727-2:2001|IDF 80-2:2001 | |||||
| NaCl | ISO 15648:2004|IDF 179:2004 | |||||
| Fat (if > 99 % m/m) | IDF 24:1964 | |||||
| Butteroil | Water (if fat < 99 % m/m) | ISO 5536:2002|IDF 23:2002 | ||||
| Cheese and curd | 0406 | Fat | ISO 1735:2004|IDF 5:2004 | |||
| Solids | ISO 5534:2004|IDF 4:2004 | |||||
| Solids (Ricotta) | ISO 2920:2004|IDF 58:2004 | |||||
| NaCl | ISO 5943:2006|IDF 88:2006 | |||||
| Lactose | ISO 5765-1/2:2002|IDF 79-1/2:2002 | |||||
| Regulation (EEC) No 2658/87 | Compound feedingstuffs | 2309 | Lactose | Annex XI |
Notes to list of European Union reference methods:
Note 1: Milk fat isolation as described in ISO 1740:1991 (protection from light).
Note 2: No reference method has been established. Methods approved by the competent authority.
Note 3: Sample to be prepared according to ISO 8261:2001|IDF 122:2001.
Note 4: Incubation for 48 hours at a temperature of 55 °C, care should be taken to prevent the culture medium from drying out.
Note 5: % m/m SNF = % m/m solids — % m/m fat.
Note 6: Commission Directive 84/4/EEC.
Note 7: Commission Regulation (EC) No 2799/1999 (OJ L 340, 31.12.1999, p. 3-27).
Note 8: Commission Directive 78/633/EEC.
ANNEX II(Article 3)
EVALUATION OF COMPLIANCE OF A CONSIGNMENT WITH THE LEGAL LIMIT
1.PRINCIPLE
In cases where detailed sampling procedures are given by the relevant legislation, these procedures are followed. In all other cases a sample of at least 3 sample units taken randomly from the consignment submitted to control is used. A composite sample may be prepared. The result obtained is compared with the legal limits by calculation of a 95 % confidence interval as 2 x standard deviation, where the relevant standard deviation depends on whether (1) the method is validated through international collaboration with values for σr and σR or (2) in the case of in-house validation, an internal reproducibility has been calculated. This confidence interval will then equate to the measurement uncertainty of the result.
2.THE METHOD IS VALIDATED THROUGH INTERNATIONAL COLLABORATION
In this case the repeatability standard deviation σr and the reproducibility standard deviation σR have been established and the laboratory can demonstrate compliance with the performance characteristics of the validated method.
Calculate the arithmetic mean 
Calculate the expanded uncertainty (k = 2) of 

If the final result x of measurement is calculated using a formula of the form x = y 1 + y 2, x = y 1 – y 2, x = y 1 · y 2 or x = y 1 / y 2 the usual procedures for combining standard deviations in such cases must be followed.
The consignment is judged to be not in compliance with the upper legal limit UL if

otherwise it is judged to be in compliance with UL.
The consignment is judged to be not in compliance with the lower legal limit LL if

otherwise it is judged to be in compliance with LL.
3.IN-HOUSE VALIDATION WITH CALCULATION OF INTERNAL REPRODUCIBILITY STANDARD DEVIATION
In cases where methods not specified in this Regulation are used and precision measures have not been established an in-house validation must be carried out. Internal repeatability standard deviation sir and the internal reproducibility standard deviation siR have to be used instead of σr and σ R , resp., in the formulae for the computation of the expanded uncertainty U.
The decision rules are as under (1). However, if the consignment is judged to be not in compliance with the legal limit the measurements must be repeated with the method specified in this Regulation and the decision reached according to (1).
ANNEX III(Article 4)
EVALUATION OF ASSESSORS AND THE RELIABILITY OF RESULTS IN SENSORY ANALYSES
The following procedures are applicable if scoring methods are used (IDF Standard 99C:1997).
A.DETERMINATION OF THE ‘REPEATABILITY INDEX’
At least ten samples will be analysed as blind duplicates by an assessor within a period of 12 months. This will usually happen in several sessions. The results for individual product characteristics are evaluated using the following formula:

where:
:
repeatability index
:
score for the first evaluation of sample xi
:
score for the second evaluation of sample xi
:
number of samples
The samples to be evaluated should reflect a broad quality range. wI should not exceed 1,5 (5-point scales).
B.DETERMINING THE ‘DEVIATION INDEX’
This index should be used to check whether an assessor uses the same scale for quality evaluation as an experienced group of assessors. The scores obtained by the assessor are compared with the average of the scores obtained by the assessor group.
The following formula is used for the evaluation of results:

where:
:
see section (A)


:
average score of the assessor group for the first and second evaluation respectively of sample xi
:
number of samples (at least 10 per 12 months).
The samples to be evaluated should reflect a broad quality range. DI should not exceed 1,5 (5-point scales).
Member States must notify any difficulties encountered when applying this procedure.
Where individual assessors are found to exceed the 1,5 limit for Deviation or Repeatability indices, the Official authority expert/s must perform one or more random ‘Re-performance’ checks on samples graded by them over the next few weeks, or perform one or more ‘Accompanied’ checks with those assessors. Close monitoring is necessary to decide whether to retain their services. Findings should be documented and retained as proof of follow up action.
C.COMPARISON OF THE RESULTS OBTAINED IN DIFFERENT REGIONS OF A MEMBER STATE AND IN DIFFERENT MEMBER STATES
Where applicable, a test must be organised at least once per year to compare the results obtained by assessors from different regions. If significant differences are observed, the necessary steps should be taken to identify the reasons and arrive at comparable results.
Member States may organise tests to compare the results obtained by their own assessors and by assessors from neighbouring Member States. Significant differences should lead to an in-depth investigation with the aim of arriving at comparable results.
Member States should notify the Commission of the results of these comparisons.
ANNEX IV(Article 4)
SENSORY EVALUATION OF BUTTER
1.SCOPE
The purpose of this procedure for sensory evaluation of butter is to provide a uniform method applicable in all Member States.
Refer to the current IDF International Standard for Milk and Milk Products, IDF 99 — Parts 1, 2, 3 on Sensory Evaluation, for further detail.
2.DEFINITIONS
‘Sensory evaluation’ (assessment) means the examination of the attributes of a product by the sense organs.
‘Panel’ means a group of selected assessors working, during the assessment, without intercommunication, and without influencing one another.
‘Assessor’ is defined as someone chosen for his/her ability to perform a sensory test. This type of assessor may have limited experience.
‘Expert Assessor’ is defined as someone with a high degree of sensory sensitivity and experience of sensory methodology, who is able to make consistent and reliable sensory assessments of various products. This type of assessor will have a good long term sensory memory.
‘Scoring’ means sensory evaluation by a panel, using a numerical scale. A nomenclature of defects must be used.
‘Grading’ means a quality classification which is performed on the basis of scoring.
‘Control documents’: documents used to record the individual scores for each attribute and the final grade of the product. (This document may also be used to record chemical composition.)
3.TEST ROOM
Refer to ISO 8589 and ISO/DIS 22935-2 | IDF 99-2 par 7 for more details.
Precautions must be taken in order that the assessors in the test room are not influenced by external factors.
The test room must be free from foreign odours and easy to clean. The walls must be of a light colour and non reflective.
The test room and its lighting must be such that the properties of the products to be scored are not affected.
The room must be equipped with appropriate thermostatic control to enable a constant temperature of butter to be maintained. Butter should have a temperature of 12 °C (±2 °C) at the time of grading.
4.SELECTION OF ASSESSORS
An assessor must be familiar with butter products and be competent to carry out sensory grading. His/her competence should be monitored on a regular basis (at least once a year) by the competent authority.
4.1.ISO/DIS 22935-1 | IDF 99-1 par 4 (recruitment) and par 5.1 should be consulted for details on general requirements and screening tests which may be used prior to official use of a new assessor.
It is essential that training is ongoing and general sessions should be held on a regular basis. Refer to ISO 8586-1 for information on panel training.
4.2.Initial training should cover the following:
general theory and practical importance of sensory evaluation,
methods, scales and description of sensory impressions,
detection and recognition of sensory attributes and specific sensory terms,
background training on the manufacture of butter,
validated references and samples to help the assessor to identify specific flavours and flavour intensity within the product.
5.REQUIREMENTS FOR THE PANEL
The number of assessors in the panel should be uneven, the minimum number being three. The majority must be employees of the competent authority or authorised persons not employed by the dairy industry.
A panel Leader shall be responsible for the entire procedure and may participate in the panel.
A number of factors must be taken into account before evaluation in order to obtain optimal performances from the subjects:
subjects must not be suffering from an illness which could affect their performance. In such a case, the assessor concerned should be replaced on the panel by another,
subjects must be on time to take part in the evaluation and make sure that they have enough time to make their evaluation,
subjects must not use strong-smelling substances like perfume, after-shave lotion, deodorant, etc. and should avoid eating strong-flavoured (e.g. highly spiced) food, etc.,
subjects may not smoke, eat or drink anything other than water during the half hour before the evaluation.
6.PERFORMANCE
All Assessors should participate in regular sensory evaluation panels to maintain their competence. The frequency will depend on the volume and throughput of butter, and where possible, should be at least one panel per month.
Senior Assessors should also participate in a number of panels each year, and where possible, at least once per quarter.
7.SAMPLING AND PREPARATION OF THE SAMPLE
It is essential that the identity of the samples is concealed during the assessment so that any possible bias is avoided. The samples should be coded.
This should be organized prior to the evaluation. Requirement for temperature of butter during its transportation to the test room should be set (6 °C ± 2 °C).
When the sensory evaluation is carried out at a cold store, the sample is taken using a butter trier. If the sensory evaluation is carried out at another location other than the cold store, then at least a 500 g sample should be taken. During the evaluation, the butter should have the temperature of 12 °C (±2 °C) (refer: in ISO/DIS 22935-2 | IDF 99-2 the evaluation temperature of butter is 14 °C ± 2 °C). Large deviations should be avoided at all cost.
8.ASSESSMENT OF THE VALUE OF EACH ATTRIBUTE
8.1.The sensory evaluation is to be carried out in relation to the following three attributes: appearance, consistency and flavour:
‘Appearance’ involves the following features: colour, visible purity, absence of physical contamination, absence of mould growth and uniformity of water dispersion. Water dispersion is tested according to IDF-Standard 112A/1989.
‘Consistency’ involves the following features: Body, texture and firmness. Spreadability may be monitored using physical means should an Individual Member State so wish in order to satisfy customer requirements. The Commission may decide to harmonise methodology in the future.
‘Body’ is the term which refers to the cohesiveness of the product as it is being consumed. It is normally associated with firmness and spreadability, and should be uniform throughout the product. It is closely related to texture and is the ability of the product to stand up under its own weight. It is indicated by resistance during cutting and can be measured mechanically and by mouthfeel and fingerfeel.
‘Flavour’ is the characteristic as perceived in the mouth, predominantly by the taste buds of the tongue.
‘Aroma’ is the characteristic as perceived by the nose and sense of smell.
A significant deviation from the recommended temperature prevents a reliable evaluation of consistency and flavour. The temperature is of paramount importance.
Grading of butter must be deferred if the temperature is outside the recommended band.
8.2.Each attribute has to be sensory evaluated separately. The scoring has to be done according to table 1.
8.3.It may be desirable for the assessors to score together, before starting the assessment, one or more reference samples for appearance, consistency and flavour, in order to achieve uniformity.
8.4.Scoring for acceptance is as follows:
Refer to part 7 — Nomenclature, and description of criteria applicable to points, when scoring.
| Maximum | Required | |
|---|---|---|
| Appearance | 5 | 4 |
| Consistency | 5 | 4 |
| Flavour/aroma | 5 | 4 |
Where the required score is not obtained, a description of the defect has to be given.
The score given by each assessor for each attribute must be recorded in the control document.
The product is accepted or rejected on the basis of a majority decision.
Cases where differences between the individual scoring for each attribute are wider than adjacent points should not occur frequently (not more than once per 20 samples). Otherwise the competence of the panel should be checked by the panel leader.
9.SUPERVISION
A panel leader who must be an official employee of the competent authority and may be a member of the panel must be generally responsible for the entire procedure. He/she must record the individual scores for each attribute in the control document and certify whether the product is accepted or rejected.
10.NOMENCLATURE
Refer to the appended table 2.
11.REFERENCE
FIL-IDF 99C:1997 Sensory evaluation of dairy products by scoring — Reference method
ISO/DIS 22935 | IDF 99 International Standard for Milk and Milk Products — Sensory analysis — Parts 1-3
ISO 8586-1 Sensory analysis — General guidance for selection, training and monitoring of assessors — Part 1
ISO 8589 Sensory analysis — General guidance for the design of test rooms
FIL-IDF 112A:1989 Butter — Determination of water dispersion value
Table 1
Butter scoring
| a Table 2. | ||||||||
| b The defects mentioned under ‘good’ are only very small deviations from the ideal type. | ||||||||
| Appearance | Consistency | Flavour + aroma | ||||||
|---|---|---|---|---|---|---|---|---|
| Points | Noa | Remarks | Points (quality class) | Noa | Remarks | Points (quality class) | Noa | Remarks |
| 5 | Very good ideal type highest quality (equal dry) | 5 | Very good ideal type highest quality (well spreadable) | 5 | Very good ideal type highest quality (absolutely pure finest aroma) | |||
| 4 | Good b no evident defects | 4 | 17 18 | Good b hard soft | 4 | Good b no evident defects | ||
| 3 | 1 2 3 4 5 6 7 8 | Fair (slight defects) loose (free), moisture not uniform, two coloured streaky mottled, marbled speckled oil separation overcoloured weak, open texture | 3 | 14 15 16 17 18 | Fair (slight defects) short, brittle, crumbly pasty, doughy, greasy sticky hard soft | 3 | 21 22 25 27 33 34 35 | Fair (slight defects) unclean foreign flavour acid cooked flavour, scorched flavour feed flavour coarse, bitter oversalted |
| 2 | 1 3 4 5 6 10 11 12 | Poor (evident defects) loose (free) moisture streaky mottled, marbled speckled oil separation foreign matter mouldy undissolved salt | 2 | 14 15 16 17 18 | Poor (evident defects) short, brittle, crumbly pasty, doughy, greasy sticky hard soft | 2 | 21 22 23 25 32 33 34 35 36 38 | Poor (evident defects) unclean foreign flavour stale acid oxidized flavour, metallic flavour feed flavour coarse, bitter oversalted musty-flat, putrid chemical flavour |
| 1 | 1 3 4 5 6 7 9 10 11 12 | Very poor (strong defects) loose (free) moisture streaky mottled, marbled speckled oil separation overcoloured granular foreign matter mouldy undissolved salt | 1 | 14 15 16 17 18 | Very poor (strong defects) short, brittle, crumbly pasty, doughy, greasy sticky hard soft | 1 | 22 24 25 26 28 29 30 31 32 34 35 36 37 38 | Very poor (strong defects) foreign flavour cheesy, lactic cheese flavour acid yeasty mould flavour rancid oily, fishy tallowy oxidized flavour, metallic flavour coarse, bitter oversalted musty-flat, putrid malty chemical flavour |
Table 2
Table of butter defects
| a This designation should be used as seldom as possible and only when the defect cannot be described more accurately. |
I.Appearance |
1.loose (free), moisture |
2.not uniform, two coloured |
3.streaky |
4.mottled, marbled |
5.speckled |
6.oil separation |
7.overcoloured |
8.weak (open texture) |
9.granular |
10.foreign matter |
11.mouldy |
12.undissolved salt |
II.Consistency |
14.short, brittle, crumbly |
15.pasty, doughy, greasy |
16.sticky |
17.hard |
18.soft |
III.Flavour and aroma |
20.without flavour |
21.uncleana |
22.foreign flavour |
23.stale |
24.cheesy, lactic cheese flavour |
25.acid |
26.yeasty |
27. (a)cooked flavour |
(b)scorched flavour |
28.mouldy flavour |
29.rancid |
30.oily, fishy |
31.tallowy |
32. (a)oxidized flavour |
(b)metallic flavour |
33.feed flavour |
34.coarse, bitter |
35.oversalted |
36.musty-flat, putrid |
37.malty |
38.chemical flavour |
ANNEX V(Article 5)
DETERMINATION OF THE CONTENT OF ENANTHIC ACID TRIGLYCERIDE IN BUTTER, BUTTER-OIL AND CREAM BY GAS CHROMATOGRAPHIC ANALYSIS OF TRIGLYCERIDES
1.SCOPE
This method lays down a method for the determination of the content of the triglyceride of enanthic acid in butter-oil, butter and cream.
2.TERMS AND DEFINITION
Enanthic acid content: content of the triglyceride of enanthic acid determined by the procedure specified in this method.
Note: The enanthic acid content is expressed in kg per ton of product for butter-oil and butter, and it is expressed in kg per ton of milk fat for cream.
3.PRINCIPLE
Milk fat is extracted from the different products according to ISO 14156 | IDF 172:2001. The quantitative determination of the content of the triglyceride of enanthic acid in the extracted fat is determined by capillary gas chromatography (GC). The result obtained for the sample is evaluated by reference to the triglyceride of caproic acid as internal standard.
Note: Tributyrin has also been found to be a satisfactory internal standard.
4.REAGENTS
Use only reagents of recognized analytical grade.
4.1.n-Hexane
4.2.Standard triglyceride of caproic acid, at least 99 % pure
4.3.Standard triglyceride of enanthic acid, at least 99 % pure
4.4.Anhydrous sodium sulfate (Na2SO4).
5.APPARATUS
Usual laboratory equipment and particularly the following:
5.1.Analytical balance precise at 1 mg
5.2.Volumetric flasks, of capacities 10 ml and 20 ml
5.3.Tubes for centrifuge, of capacity 30 ml
5.4.Rotary evaporator
5.5.Oven, capable of being maintained at a temperature of 50 °C ± 5 °C
5.6.Filter paper, medium porosity, of diameter about 15 cm
5.7.
Gas chromatography equipment
5.7.1.Gas chromatograph equipped with a split/splitless or on-column injector and a flame ionization detector (FID)
5.7.2.
GC column, with a stationary phase which has successfully employed to perform triglyceride separation (100 % dimethylpolysiloxane or 5 % phenyl-95 % methylpolysiloxane). Select the stationary phase, the column length (between 4 m and 15 m), the internal diameter (between 0,22 mm and 0,50 mm) and the film thickness (0,12 μm or more) taking into account the laboratory experience and the injection system applied. In any case the selected column shall produce both a complete separation between the solvent peak and the triglyceride of caproic acid and a baseline resolution between triglyceride of caproic and enanthic acid peaks. Examples of applicable conditions are listed below.
5.7.2.1.Example of applicable conditions using a split injector:
Carrier gas: helium
Column head pressure: 100 KPa
Column: 12 m length, 0,5 mm internal diameter, 0,1 μm film thickness fused silica column
Stationary phase: 100 % dimethylpolysiloxane or 5 % phenyl-95 % dimethylpolysiloxane (for ex. HT5)
Column temperature: initial temperature of 130 °C, maintained for 1 min, raised at a rate of 20 °C/min up to 260 °C and then raised at a rate of 30 °C/min up to 360 °C; maintain 10 mn at 360 °C
Detector temperature: 370 °C
Injector temperature: 350 °C
Split ratio 1:30
Amount of sample injected: 1 μl.
5.7.2.2.Example of applicable conditions using an on-column injector:
Carrier gas: hydrogen (constant flow system)
Column head pressure: 89 kPa
Column: 4 m length, 0,32 mm internal diameter, 0,25 μm film thickness, fused silica column
Stationary phase: 5 % phenyl, 95 % dimethylpolysiloxane
Column temperature: initial temperature of 60 °C, maintained for 2 min, raised at a rate of 35 °C/min up to 340 °C, maintained at this temperature for 5 min
Detector temperature: 350 °C
Amount of sample injected: 1 μl
5.8.Injection syringe, of capacity 5 μl.
6.SAMPLING
It is important that the laboratory receives a sample which is truly representative and has not been damaged or changed during transport or storage.
Sampling is not part of the method specified in this International Standard. A recommended sampling method is given in IDF: standard 50C:1995 or ISO 707-1997 — Milk and milk products — Methods of sampling.
7.PROCEDURE
7.1.Preparation of the test sample and test portion
Proceed according to ISO 14156 | IDF 172:2001
7.1.1.Butter-oil, Butter
7.1.1.1.Melt 50 g to 100 g of test sample in the oven (5.5)
7.1.1.2.Place 0,5 g to 1,0 g of anhydrous sodium sulfate (5.4) in a folded filter paper
7.1.1.3.Filter the fat through the filter paper containing anhydrous sodium sulfate collecting the filtrate in a beaker maintained in the oven (5.5). When decanting the melted butter onto the filter paper, take care that no serum is transferred
7.1.2.Cream
7.1.2.1.Bring the test sample to a temperature of 20 °C ± 2 °C
7.1.2.2.Mix or stir the sample thoroughly
7.1.2.3.Dilute a suitable amount of test sample so as to obtain 100 ml of test portion with a mass fraction of fat of approximately 4 %
7.1.2.4.Proceed as with raw milk and homogenized milk (see ISO 14156 | IDF 172:2001, §8.3) to extract the fat from the cream
7.1.2.5.Weigh in a 10 ml volumetric flask (5.2), to the nearest 1 mg, 1 g of the extracted fat. Add 1 ml of the solution 7.2.2. Complete to 10 ml with n-hexane (4.1) and homogenise
7.1.2.6.Introduce 1 ml of the solution 7.1.1.2 in a 10 ml volumetric flask (5.2) and dilute to 10 ml with n-hexane (4.1)
7.2.Preparation of the calibration standards
7.2.1.Dissolve 100 mg of the triglyceride of enanthic acid (4.3) in 10 ml of n-hexane (4.1)
7.2.2.Dissolve 100 mg of the triglyceride of caproic acid (4.2) in 10 ml of n-hexane (4.1)
7.2.3.Introduce 1 ml of the solution 7.2.2 in a 10 ml volumetric flask (5.2). Complete to 10ml with n-hexane (4.1)
7.2.4.Introduce 1 ml of the solution 7.2.1 and 1 ml of the solution 7.2.2 in a 10 ml volumetric flask (5.2). Complete to 10 ml with n-hexane (4.1)
7.2.5.Introduce 1 ml of the solution 7.2.4 in a 10 ml volumetric flask (5.2) and complete to 10 ml with n-hexane (4.1)
7.3.Chromatographic determination
7.3.1.Inject 1 μl of the standard solution 7.2.5 twice
7.3.2.Inject 1 μl of each sample solution
Note: If the on column injector system is adopted an increased dilution should be applied both to the standard and sample solutions.
7.3.3.Repeat the operation 7.3.1 every 3 samples in order to bracket samples between duplicate standard injections. Results are based upon the mean average response factors from the standard chromatograms.
8.CALCULATION OF RESULTS
For each chromatogram, integrate the area of the peaks associated with the triglycerides of enanthic acid and caproic acid.
Follow those instructions for each bracketed sequence i.e. for a set of bracketed samples, the standard injected twice immediately before them is STD1 and the standard injected twice immediately after them is STD2.
8.1.Calibration
8.1.1.Calculate the response factor for each duplicate of STD1, Rf1(a) and Rf1(b)
Rf1 (a) or (b) = (Peak area for caproic acid triglyceride/Peak area for enanthic acid triglyceride) × 100
Calculate the mean average response factor, Rf1
Rf1 = (Rf1(a) + Rf1(b)) / 2
8.1.2.Similarly, calculate the mean average response factor STD2, Rf2
8.1.3.Calculate the mean average response factor, Rf
Rf = (Rf1 + Rf2) /2
8.2.Test samples
For each sample chromatogram obtained between STD1 and STD2, calculate the enanthic acid content, C (kg/t):
C = (Peak area for enanthic acid triglyceride × Rf × 100)/(Peak area for caproic acid triglyceride × Wt × 1 000)
where:
Wt = weight of fat taken (g),
100 = dilution volume for sample,
1 000 = conversion factor (for μg/g to kg/t)
For butter samples, take the fat content of butter into account and calculate a corrected concentration value, Cbutter (kg/t of butter)
Cbutter = Cfat × F
where F is the fat content of butter.
9.PRECISION
Details of an interlaboratory test on butter in accordance with ISO 5725-1 and ISO 5725-2 on the precision method are shown in (12.).
The values for repeatability and reproducibility limit are expressed for the 95 % probability level and may not be applicable to concentration ranges and matrices other than those given.
9.1.Repeatability
The absolute differences between two individual single test results, obtained with the same method on identical test material in the same laboratory by the same operator using the same equipment within a short interval of time, will in not more than 5 % of cases be greater than 0,35 kg/t.
9.2.Reproducibility
The absolute differences between two individual single test results, obtained with the same method on identical test material in different laboratories with different operators using different equipment will in not more than 5 % of cases be greater than 0,66 kg/t.
10.TOLERANCE LIMITS: LOWER LIMITS (CASE OF INSUFFICIENT QUANTITIES)
10.1. Three samples must be taken from the traced product in order to check on the correct tracing of the product
10.2.Butter and concentrated butter
10.2.1.The incorporation rate is 11 kg of at least 95 % pure enanthic acid triglyceride per tonne of butter, i.e. 10,45 kg/t
10.2.2.The results of three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
9,51 kg/t (95 % of the minimum incorporation rate of 95 % pure enanthic acid triglyceride, single determination),
6,89 kg/t (70 % of the minimum incorporation rate of 95 % pure enanthic acid triglyceride, single determination),
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation respectively between 9,51 kg/t and 6,89 kg/t.
10.3.Cream
10.3.1.The incorporation is 10 kg of at least 95 % pure enanthic acid triglyceride per tonne of milk fat, i.e. 9,50 kg/t traced milk fat
10.3.2.The results of the three samples obtained from the analysis of the product are used to check the rate of the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
8,60 kg/t (95 % of the minimum incorporation rate of 95 % pure enanthic acid triglyceride, single determination),
6,23 kg/t (70 % of the minimum incorporation rate of 95 % pure enanthic acid triglyceride, single determination),
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation respectively between 8,60 kg/t and 6,23 kg/t.
11.TOLERANCE LIMITS: UPPER LIMITS (CASE OF EXCEEDING QUANTITY BY MORE THAN 20 %)
11.1. Three samples must be taken from the traced product in order to check on the correct tracing of the product
11.2.Butter and concentrated butter
11.2.1.The results of three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the mean of these results is compared with the following limits:
Upper limit is 12,96 kg/t
11.3.Cream
11.3.1.The results of three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the mean of these results is compared with the following limits:
Upper limit is 11,82 kg/t.
12.ADDITIONAL INFORMATION: STATISTICAL ANALYSIS OF RESULTS ON THE DETERMINATION OF TRIENANTOATE IN BUTTERFAT BY TRIGLYCERIDE ANALYSIS
Four collaborative trials have been carried out to determine the trienantoate content in traced butter.
Nine laboratories participated to the 1st ring test and no specifications were provided about the analytical methods to use:
10 laboratories participated to the 2nd ring test and 4 different methods were applied:
Quantification of methylheptanoate by using n-nonane or methylnonanoate as internal standard
Quantification of trienantoate by using tricaproate as internal standard
Quantification of methylheptanoate by using a calibration sample/mixture
Quantification of methylheptanoate by using a calibration mixture.
Moreover, if FAME were analysed, two different methylation procedures were used (De Francesco and Christopherson & Glass).
Due to the results obtained, two methods were chosen to perform the 3rd ring test:
Quantification of methylheptanoate by using n-nonane or methylnonanoate as internal standard
Quantification of trienantoate by using tricaproate as internal standard.
The results of 7 labs showed that the FAME method produced a higher variability and consequently it was decided to use only the determination of trienantoate as triglyceride following the procedure of the q Quantification of trienantoate by using tricaproate as internal standard. Moreover the triglyceride analysis has to be carried out by capillary column.
In the 4th ring test four samples (A, B, C, D) were circulated and nine laboratories provided results (Tables 1-2).
Two laboratories (DE and UE) analysed the samples by using FAME method.
Due to the reduced number of laboratories, the Statistical calculation has been performed both on the complete set (Figures 1-2) of data including FAME results and on the data obtained from TG analysis.
Tests for outliers:
sample A. Dixon, Cochran and Grubbs tests at levels 1 and 5 %, showed one laboratory outlier.
sample B. Grubbs test at level 5 % showed one laboratory outlier.
sample C. Dixon and Grubbs tests at levels 1 and 5 %, showed one laboratory outlier.
sample D. Dixon and Grubbs tests at levels 1 end 5 %, showed one laboratory outlier.
The outlier has been excluded from the calculation.
It is worth noting that the results obtained by FAME method were never considered as outliers by the tests applied.
Precision parameters
Tables 1 and 2 report the results of all the laboratories and the precision parameters calculated on an acceptable number (8) of labs but, unfortunately not deriving from the same analytical method.
Tables 3 and 4 report the results deriving only from TG method and the corresponding precision parameters. The acceptance of these parameters is subjected to the acceptance of the low number of laboratories (6).
Figures 2 and 3 show the trend of Sr and SR calculated on the 4 samples of the 2 data set described above.
Table 5 reports the Sr and SR values together with the corresponding pooled values and overall r and R parameters.
Finally the Critical Difference at 95 % of probability level has been calculated.
Table 1
Statistical Results of TG + FAME* methods
| Sample A | R1 | R2 | Mean | N. of labs retained after eliminating outliers | 8 | |
| RENNES | FR1 | 11,0 | 11,1 | 11,1 | N. of outliers | 1 |
| RIKILT | NL | 11,2 | 11,2 | 11,2 | Outliers | DК |
| ZPLA | DE* | 11,6 | 11,8 | 11,7 | Mean value | 11,3 |
| ADAS | GB | 11,4 | 11,2 | 11,3 | True value | 11,0 |
| CNEVA | FR2 | 11,4 | 11,4 | 11,4 | Repeatability standard deviation (Sr) | 0,09 |
| LODI | IT | 11,1 | 11,3 | 11,2 | Repeatability relative sd (RSDr%) | 0,80 |
| EELA | FI | 11,3 | 11,2 | 11,3 | Repeatability r (95 %) | 0,26 |
| ISPRA | UE* | 11,0 | 11,0 | 11,0 | Relative Repeatability r % | 2,24 |
| D.V.F.A. | DK | 13,3 | 11,8 | 12,6 | Reproducibility standard deviation (SR) | 0,23 |
| Reproducibility relative sd (RSDR%) | 2,04 | |||||
| Reproducibility R (95 %) | 0,84 | |||||
| Relative Reproducibility R % | 5,71 | |||||
| Sample B | R1 | R2 | Mean | N. of labs retained after eliminating outliers | 8 | |
| RENNES | FR1 | 12,7 | 12,8 | 12,8 | N, of outliers | 1 |
| RIKILT | NL | 13,5 | 13,3 | 13,4 | Outliers | DK |
| ZPLA | DE* | 14,0 | 13,8 | 13,9 | Mean value | 13,4 |
| ADAS | GB | 13,4 | 13,5 | 13,5 | True value | 13,5 |
| CNEVA | FR2 | 13,3 | 13,4 | 13,4 | Repeatability standard deviation (Sr) | 0,14 |
| LODI | IT | 13,9 | 13,5 | 13,7 | Repeatability relative sd (RSDr%) | 1,04 |
| EELA | FI | 13,4 | 13,2 | 13,3 | Repeatability r (95 %) | 0,40 |
| ISPRA | UE* | 13,2 | 13,3 | 13,3 | Relative Repeatability r % | 2,91 |
| D.V.F.A. | DK | 14,1 | 14,8 | 14,5 | Reproducibility standard deviation (SR) | 0,35 |
| Reproducibility relative sd (RSDR%) | 2,61 | |||||
| Reproducibility R (95 %) | 0,99 | |||||
| Relative reproducibility R % | 7,31 |
Table 2
Statistical Results of TG + FAME* methods
| Sample C | R1 | R2 | Mean | N. of labs retained after eliminating outliers | 8 | |
| RENNES | FR1 | 8,9 | 9,2 | 9,1 | N. of outliers | 1 |
| RIKILT | NL | 9,2 | 9,3 | 9,3 | Outliers | DK |
| ZPLA | DE* | 9,2 | 9,4 | 9,3 | Mean value | 9,3 |
| ADAS | GB | 9,5 | 9,3 | 9,4 | True value | 9,3 |
| CNEVA | FR2 | 9,4 | 9,4 | 9,4 | Repeatability standard deviation (Sr) | 0,14 |
| LODI | IT | 9,2 | 9,5 | 9,4 | Repeatability relative sd (RSDr%) | 1,50 |
| EELA | FI | 9,4 | 9,6 | 9,5 | Repeatability r (95 %) | 0,40 |
| ISPRA | UE* | 9,4 | 9,3 | 9,4 | Relative Repeatability r % | 4,20 |
| D.V.F.A. | DK | 10,7 | 10,9 | 10,8 | Reproducibility standard deviation (SR) | 0,17 |
| Reproducibility relative sd (RSDR%) | 1,82 | |||||
| Reproducibility R (95 %) | 0,47 | |||||
| Relative Reproducibility R % | 5,10 | |||||
| Sample D | R1 | R2 | Mean | N. of labs retained after eliminating outliers | 8 | |
| RENNES | R1 | 1,6 | 1,6 | 1,6 | N. of outliers | 1 |
| RIKILT | NL | 2,1 | 2,1 | 2,1 | Outliers | DK |
| ZPLA | DE* | 2,3 | 2,3 | 2,3 | Mean value | 2,1 |
| ADAS | GB | 2,1 | 2,2 | 2,2 | True value | 2,1 |
| CNEVA | FR2 | 2,1 | 2,1 | 2,1 | Repeatability standard deviation (Sr) | 0,08 |
| LODI | IT | 2,2 | 1,9 | 2,1 | Repeatability relative sd (RSDr%) | 3,81 |
| EELA | FI | 2,3 | 2,3 | 2,3 | Repeatability r (95 %) | 0,22 |
| ISPRA | UE* | 2,3 | 2,3 | 2,3 | Relative Repeatability r % | 10,67 |
| D.V.F.A. | DK | 3,4 | 2,9 | 3,2 | Reproducibility standard deviation (SR) | 0,24 |
| Reproducibility relative sd (RSDR%) | 11,43 | |||||
| Reproducibility R (95 %) | 0,67 | |||||
| Relative Reproducibility R % | 32,00 |
Table 3
Statistical Results of TG + FAME* methods
| Sample A | R1 | R2 | Mean | N. of labs retained after eliminating outliers | 6 | |
| RENNES | FR1 | 11,0 | 11,1 | 11,1 | N. of outliers | 1 |
| RIKILT | NL | 11,2 | 11,2 | 11,2 | Outliers | DК |
| ADAS | GB | 11,4 | 11,2 | 11,3 | Mean value | 11,2 |
| CNEVA | FR2 | 11,4 | 11,4 | 11,4 | True value | 11,0 |
| LODI | IT | 11,1 | 11,3 | 11,2 | Repeatability standard deviation (Sr) | 0,09 |
| EELA | FI | 11,3 | 11,2 | 11,3 | Repeatability relative sd (RSDr%) | 0,80 |
| D.V.F.A. | DK | 13,3 | 11,8 | 12,6 | Repeatability r (95 %) | 0,25 |
| Relative Repeatability r % | 2,24 | |||||
| Reproducibility standard deviation (SR) | 0,13 | |||||
| Reproducibility relative sd (RSDR%) | 1,16 | |||||
| Reproducibility R (95 %) | 0,36 | |||||
| Relative Reproducibility R % | 3,25 | |||||
| Sample B | R1 | R2 | Mean | N. of labs retained after eliminating outliers | 6 | |
| RENNES | FR1 | 12,7 | 12,8 | 12,8 | N. of outliers | 1 |
| RIKILT | NL | 13,5 | 13,3 | 13,4 | Outliers | DК |
| ADAS | GB | 13,4 | 13,5 | 13,5 | Mean value | 13,3 |
| CNEVA | FR2 | 13,3 | 13,4 | 13,4 | True value | 13,5 |
| LODI | IT | 13,9 | 13,5 | 13,7 | Repeatability standard deviation (Sr) | 0,15 |
| EELA | FI | 13,4 | 13,2 | 13,3 | Repeatability relative sd (RSDr%) | 1,13 |
| D.V.F.A. | DK | 14,1 | 14,8 | 14,5 | Repeatability r (95 %) | 0,42 |
| Relative Repeatability r % | 3,16 | |||||
| Reproducibility standard deviation (SR) | 0,33 | |||||
| Reproducibility relative sd (RSDR%) | 2,48 | |||||
| Reproducibility R (95 %) | 0,93 | |||||
| Relative Reproducibility R % | 6,94 |
Table 4
Statistical Results of TG method
| Sample C | R1 | R2 | Mean | N. of labs retained after eliminating outliers | 6 | |
| RENNES | FR1 | 8,9 | 9,2 | 9,1 | N. of outliers | 1 |
| RIKILT | NL | 9,2 | 9,3 | 9,3 | Outliers | DК |
| ADAS | GB | 9,5 | 9,3 | 9,4 | Mean value | 9,3 |
| CNEVA | FR2 | 9,4 | 9,4 | 9,4 | True value | 9,3 |
| LODI | IT | 9,2 | 9,5 | 9,4 | Repeatability standard deviation (Sr) | 0,15 |
| EELA | FI | 9,4 | 9,6 | 9,5 | Repeatability relative sd (RSDr%) | 1,61 |
| D.V.F.A. | DK | 10,7 | 10,9 | 10,8 | Repeatability r (95 %) | 0,42 |
| Relative Repeatability r % | 4,51 | |||||
| Reproducibility standard deviation (SR) | 0,19 | |||||
| Reproducibility relative sd (RSDR%) | 2,04 | |||||
| Reproducibility R (95 %) | 0,53 | |||||
| Relative Reproducibility R % | 5,71 | |||||
| Sample D | R1 | R2 | Mean | N, of labs retained after eliminating outliers | 6 | |
| RENNES | FR1 | 1,6 | 1,6 | 1,6 | N. of outliers | 1 |
| RIKILT | NL | 2,1 | 2,1 | 2,1 | Outliers | DK |
| Mean Value | 2,1 | |||||
| ADAS | GB | 2,1 | 2,2 | 2,2 | True value | 2,1 |
| CNEVA | FR2 | 2,1 | 2,1 | 2,1 | Repeatability standard deviation (Sr) | 0,09 |
| LODI | IT | 2,2 | 1,9 | 2,1 | Repeatability relative sd (RSDr%) | 4,29 |
| EELA | FI | 2,3 | 2,3 | 2,3 | Repeatability r (95 %) | 0,26 |
| D.V.F.A. | DK | 3,4 | 2,9 | 3,2 | Relative Repeatability r % | 12,01 |
| Reproducibility standard deviation (SR) | 0,25 | |||||
| Reproducibility relative sd (RSDR%] | 11,90 | |||||
| Reproducibility R (95 %) | 0,69 | |||||
| Relative Reproducibility R % | 33,32 |
Table 5
Repeatability and reproducibility (with FAME)
| CrD95 =0,40 Minimum purity stated for trienantoate = 95 % Minimum limit stated for trienantoate in butterfat = 11 kg/t Taking the Critical Difference for a 95 % probability level into consideration, the mean of the two results shall not be less than:
| ||||
| No of labs | Outlier | RepeatabilitySr (95 %) | ReproducibilitySR (95 %) | |
|---|---|---|---|---|
| Sample A | 8 | 1 | 0,09 | 0,23 |
| Sample Β | 8 | 1 | 0,14 | 0,35 |
| Sample C | 8 | 1 | 0,14 | 0,17 |
| Sample D | 8 | 1 | 0,08 | 0,24 |
| Pooled value | 0,116 | 0,256 | ||
| r | R | |||
| Pooled value* 2,8 | 0,324 | 0,716 | ||
Repeatability and reproducibility (without FAME)
| CrD95 = 0,36 Minimum purity stated for trienantoate = 95 % Minimum limit stated for trienantoate in butterfat = 11 kg/t Taking the Critical Difference for a 95 % probability level into consideration, the mean of the two results shall not be less than:
| ||||
| No of labs | Outlier | RepeatabilitySr (95 %) | ReproducibilitySR (95 %) | |
|---|---|---|---|---|
| Sample A | 6 | 1 | 0,09 | 0,13 |
| Sample B | 6 | 1 | 0,15 | 0,33 |
| Sample C | 6 | 1 | 0,15 | 0,19 |
| Sample D | 6 | 1 | 0,09 | 0,25 |
| Pooled value | 0,124 | 0,237 | ||
| r | R | |||
| Pooled value * 2,8 | 0,347 | 0,663 | ||
Figure 1(7)
Experimental results: Sample A

Experimental results: Sample B
Experimental results: Sample C

Experimental results: Sample D




ANNEX VI(Article 5)
DETERMINING THE VANILLIN CONTENT IN CONCENTRATED BUTTER, BUTTER OR CREAM BY HIGH-PERFORMANCE LIQUID CHROMATOGRAPHY
1.SCOPE AND FIELD OF APPLICATION
The method describes a procedure for the quantitative determination of vanillin in concentrated butter, butter or cream.
2.PRINCIPLE
Extraction of a known quantity of sample with a mixture of isopropanol/ethanol/acetonitrile (1:1:2). Precipitation of the majority of fat by cooling between -15 °C and -20 °C, followed by centrifugation.
After dilution with water, determination of the vanillin content by high-performance liquid chromatography (HPLC).
3.APPARATUS
Usual laboratory apparatus and, in particular, the following:
3.1.Freezer, operating in the temperature range -15 °C to -20 °C
3.2.Syringes, disposable of 2 ml capacity
3.3.Membrane microfilters of 0,45 μm pore size, resistant to a solution containing 5 % extraction solution (4.4)
3.4.Liquid chromatography system consisting of a pump (flow of 1,0 ml/min), an injector (20 μl injection, automatic or manual), an UV detector (operated at 306 nm, 0,01 Å full scale), a recorder or integrator and a column thermostat operating at 25 °C
3.5.Analytical column (250 mm × 4,6 mm ID) packed with LiChrospher RP 18 (Merck, 5 μm) or equivalent
3.6.Guard column (ca. 20 mm × 3 mm ID) dry-packed with LiChrospher RP 18 (5 to 10 μm) or equivalent
3.7.Centrifuge operating at 2 000 rpm.
4.REAGENTS
All reagents used must be of recognised analytical quality.
4.1.Isopropanol
4.2.Ethanol 96 % (v/v)
4.3.Acetonitrile
4.4.Extraction solution
Mix isopropanol (4.1), ethanol (4.2) and acetonitrile (4.3) in the ratio of 1:1:2 (v/v).
4.5.
Vanillin (4-hydroxy-3-methoxybenzaldehyde) ≥ 98 %
4.5.1.Vanillin stock solution (= 500 μg/ml)
Weigh accurately to 0,1 mg about 50 mg (CM mg) vanillin (4.5) in a 100 ml volumetric flask, add 25 ml extraction solution (4.4) and make up with water.
4.5.2.Vanillin standard solution (= 10 μg/ml)
Pipette 5 ml of the vanillin stock solution (4.5.1) into a volumetric flask of 250 ml and make up with water.
4.5.3. Methanol, HPLC quality
4.5.4. Acetic acid, glacial
4.5.5. Water, HPLC quality
4.5.6.HPLC mobile phase
Mix 300 ml methanol (4.5.3) with about 500 ml water (4.5.5) and 20 ml acetic acid (4.5.4) in a volumetric flask of 1 000 ml and make up with water (4.5.5). Filter through 0,45 μm filter (3.3).
5.PROCEDURE
5.1.Preparation of the test sample
5.1.1.Butter
Heat the sample until melting starts. Avoid local overheating at about 30 °C. The butter may not separate in two phases, in any case. When the sample becomes sufficiently plastic, homogenise it by shaking. Stir the butter for 15 s before taking a sample. Weigh, to the nearest 1 mg, about 5 g (SM g) of butter into a 100 ml volumetric flask.
5.1.2.Concentrated butter
Immediately before sampling place the container, with concentrated butter, into an oven at 40 to 50 °C until it is melted completely. Mix the sample by swirling or stirring, avoiding formation of air bubbles by too vigorous stirring. Weigh, to the nearest 1 mg, about 4 g (SM g) of concentrated butter into a 100 ml volumetric flask.
5.1.3.Cream
Heat the sample in a waterbath or incubator at a temperature of 35 to 40 °C. Distribute the fat homogeneously by swirling and, if necessary, by stirring. Cool the sample quickly to 20 ± 2 °C. The sample should look homogenous; otherwise the procedure should be repeated. Weigh, to the nearest 1 mg, about 10 g (SM g) of cream into a 100 ml volumetric flask.
5.2.Preparation of the test solution
Add about 75 ml extraction solution (4.4) to the test portion (5.1.1, 5.1.2 or 5.1.3), stir, or shake vigorously, for about 15 minutes and make up with extraction solution (4.4). Transfer about 10 ml of this extract to a test tube fitted with stopper. Place the test tube in the freezer (3.1) and allow it to stand for about 30 minutes. Centrifuge the cold extract for 5 minutes at approximately 2 000 rpm and decant immediately. Allow the decanted solution to adjust to room temperature. Pipette 5 ml of the decanted solution into a 100 ml volumetric flask and make up with water. Filter an aliquot through a membrane microfilter (3.3) using a syringue (3.2). The filtrate is ready for determination by HPLC.
5.3.Calibration
Pipette 5 ml of the vanillin standard solution (4.5.2) into a 100 ml volumetric flask. Add 5 ml extraction solution (4.4) and make up to the mark with water. This solution contains 0,5 μg/ml of vanillin.
5.4.Determination by HPLC
Allow the chromatographic system to stabilise for about 30 minutes. Inject the standard solution (5.3). Repeat this until the difference in peak area or peak height between two successive injections is less than 2 %. Under the conditions described the retention time of vanillin is about 9 minutes. Analyse the standard solution (5.3) in duplicate by injecting 20 μl. Inject 20 μl of the test solutions (5.2). Determine the area or height of the vanillin peak obtained. Repeat the duplicate injection of the standard solution (5.3) after 10 injections of test samples (5.2).
6.CALCULATION OF THE RESULTS
Calculate the average peak area (or height) (AC), of the vanillin peaks associated with the bracketing duplicate injections for each batch of test solutions (four areas or heights in total).
Calculate the response factor (R):

where CM is the mass of vanillin in mg (4.5.1).
The content (mg/kg) of vanillin I in the test sample is given by:

where:
=
peak area or height of the vanillin peak of the test sample
=
mass of test sample in g (5.1.1, 5.1.2 or 5.1.3).
Note: Where cream is analysed for vanillin, the tracer concentration has to be expressed as mg tracer/kg milk fat. This is done by multiplying C by 100/f. f is the fat content of the cream in percent (m/m).
=
factor which takes into account the dilutions of the standard and the test sample
=
correction factor for the fat content in first dilution of the test sample
Note: Instead of peak area, peak heights can be used (see 8.3).
7.ACCURACY OF THE METHOD
7.1.Repeatability (r)
The difference between the results of two determinations carried out within the shortest feasible time interval, by one operator using the same apparatus on identical test material, may not exceed 16 mg/kg.
7.2.Reproducibility (R)
The difference between the results of two determinations carried out by operators in different laboratories, using different apparatus on identical test material, may not exceed 27 mg/kg.
8.TOLERANCE LIMITS
8.1.Three samples must be taken from the traced product in order to check homogeneity
8.2.
Tracer obtained either from vanilla or from synthetic vanillin
8.2.1.The incorporation rate for 4-hydroxy-3-methoxybenzaldehyde is 250 g per tonne of concentrated butter or butter. Where cream is traced, the incorporation rate is 250 g per tonne of milk fat
8.2.2.The results for the three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
220,8 mg/kg (95 % of the minimum incorporation rate),
158,3 mg/kg (70 % of the minimum incorporation rate).
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation between 220,8 mg/kg and 158,3 mg/kg.
8.3.
Tracer obtained exclusively from vanilla beans or integral extracts thereof:
8.3.1.The incorporation rate for 4-hydroxy-3-methoxybenzaldehyde is 100 g per tonne of concentrated butter or butter. Where cream is traced, the incorporation rate is 100 g per tonne of milk fat.
8.3.2.The results for the three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
78,3 mg/kg (95 % of the minimum incorporation rate),
53,3 mg/kg (70 % of the minimum incorporation rate).
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation between 78,3 mg/kg and 53,3 mg/kg.
9.NOTES
9.1.Recovery of added vanillin at a level of 250 mg/kg butteroil varies from 97,0 to 103,8. The average content found was 99,9 % with a standard deviation of 2,7 %.
9.2.The standard solution contains 5 % extraction solution to compensate for peak broadening caused by the presence of 5 % of the extraction solution of the test samples. This enables quantification by peak height.
9.3.The analysis is based on a linear calibration line with a zero intercept.
9.4.By using appropriate dilutions of the standard solution (4.5.2), the linearity should be checked the first time the analysis is carried out and then at regular intervals and after changes in or repair of the HPLC equipment. Vanillin may be degraded to vanillin acid, divanillin and other compounds by activity of intrinsic enzymes in unpasteurised cream or products thereof.
ANNEX VII(Article 5)
DETERMINING THE ETHYL ESTER OF BETA-APO-8'-CAROTENIC ACID IN CONCENTRATED BUTTER AND BUTTER BY SPECTROMETRY
1.SCOPE AND FIELD OF APPLICATION
The method describes a procedure for the quantitative determination of the ethyl ester of beta-apo-8'-carotenic acid (apo-carotenic ester) in concentrated butter and butter. The apo-carotenic ester is the sum of all substances present in an extract of samples obtained under the conditions described in the method which absorb light at 440 nm.
2.PRINCIPLE
The butterfat is dissolved in light petroleum and the absorbance measured at 440 nm. The apo-carotenic ester content is determined by reference to an external standard.
3.APPARATUS
3.1.Pipettes — graduated, of capacity 0,25, 0,50, 0,75 and 1,0 ml
3.2.Spectrophotometer — suitable for use at 440 nm (and 447-449 nm) and equipped with cells of optical path length 1 cm
3.3.Volumetric flasks, 20 ml and 100 ml
3.4.Analytical balance, sensitivity 0,1 mg capable of weighing to the nearest 1 mg, with a readability of 0,1 mg
3.5.Oven, 45 °C ± 1 °C
3.6.Fast-filtering ashless filters.
4.REAGENTS
All reagents must be of recognised analytical grade.
4.1.
Apo-carotenic ester suspension (approximately 20 %)
4.1.1.Establish the content of the suspension as follows:
Warm the suspension between 45 °C and 50 °C and homogenize in the unopened original container. Weigh about 400 mg in a volumetric flask (100 ml), dissolve in 20 ml chloroform (4.4) and make up the volume with cyclohexane (4.5). Dilute 5 ml of this solution to 100 ml with cyclohexane (solution A). Dilute 5 ml of solution A to 100 ml with cyclohexane. Measure the absorbance at 447-449 nm (measure the maximum against cyclohexane as a blank using cells with 1 cm optical path length).
Apo-carotenic ester content P (%) = (Absmax × 40 000) / (Msusp × 2 550) or develop: (Absmax / 2 550) × (100 / 5) × (100 / 5) × (100 / Msusp)
=
absorbance of the measuring solution at the maximum
=
mass of suspension (g)
=
reference Abs (1 %, 1 cm) value
=
Purity (content) of the suspension (%)
Note: Apo-carotenic ester suspension is sensitive to air, heat and light. In the unopened, original container (sealed under nitrogen) and in a cool place it can be stored for about 12 months. After opening the contents should be used within a short period.
4.1.2.Apo-carotenic ester standard solution, approx. 0,2 mg/ml
Weigh to the nearest 1 mg about 0,100 g of apo-carotenic ester suspension (4.1.1) (W), dissolve in petroleum spirit (4.2), transfer quantitatively into a volumetric flask of capacity 100 ml, and make up to the mark with petroleum spirit.
This solution contains (W × P) / 10 mg/ml of apo-carotenic ester.
Note: The solution must be stored in a cool place in the dark. Discard unused solution after one month.
4.2.Petroleum spirit (40-60 °C)
4.3.Sodium sulphate, anhydrous, granular, previously dried at 102 °C for two hours
4.4.Chloroform
4.5.Cyclohexane
5.PROCEDURE
5.1.Preparation of the test sample
5.1.1.Concentrated butter
Melt the sample in an oven at approximately 45 °C.
5.1.2.Butter
Melt the sample in an oven at approximately 45 °C and filter a representative portion through a filter containing about 10 g of anhydrous sodium sulphate (4.3) in an environment shielded from strong natural and artificial light and maintained at 45 °C. Collect a suitable amount of butterfat.
5.2.Determination
Weigh, to the nearest 1 mg approximately 1 g of concentrated butter (or extracted butterfat (5.1.2)), (M). Transfer quantitatively to a 20 ml (V) volumetric flask using petroleum spirit (4.2), make up to the mark and mix thoroughly.
Transfer an aliquot to a 1 cm cell and measure the absorbance at 440 nm, against a petroleum spirit blank. Obtain the concentration of apo-carotenic ester in the solution by referring to the obtained standard curve (C μ/ml).
5.3.Calibration
Pipette 0, 0,25, 0,5, 0,75 and 1,0 ml of apo-carotenic ester standard solution (4.1.2) into five 100 ml volumetric flasks. Dilute to volume with petroleum spirit (4.2) and mix.
The approximate concentrations of the solutions range from 0 to 2 μg/ml and are calculated accurately by reference to the concentration of the standard solution (4.1.2) (W × P) / 10 mg/ml. Measure the absorbances at 440 nm against a petroleum spirit (4.2) blank.
Plot the values of absorbance on the y axis against apo-carotenic ester concentration on the x axis. Calculate the equation of the standard curve.
6.CALCULATION OF THE RESULTS
6.1.Apo-carotenic ester content, expressed as mg/kg product, is given by:
Concentrated butter: (C × V)/M
Butter: 0,82 (C × V)M
where:
=
apo-carotenic ester content, μg/ml, read from the calibration graph (5.3)
=
volume (ml) of the test solution (5.2)
=
mass (g) of the test portion (5.2)
=
a correction factor for the butterfat content of butter.
7.ACCURACY OF THE METHOD
7.1.Repeatability
7.1.1.Butter analysis
The difference between the results of two determinations carried out within the shortest feasible time interval, by one operator using the same apparatus on identical test material, may not exceed 1,4 mg/kg.
7.1.2.Concentrated butter analysis
The difference between the results of two determinations carried out within the shortest feasible time interval, by one operator using the same apparatus on identical test material, may not exceed 1,6 mg/kg.
7.2.Reproducibility
7.2.1.Butter analysis
The difference between the results of two determinations carried out by operators in different laboratories, using different apparatus on identical test material, may not exceed 4,7 mg/kg.
7.3.Concentrated butter analysis
The difference between the results of two determinations carried out by operators in different laboratories, using different apparatus on identical test material, may not exceed 5,3 mg/kg.
7.4.Source of precision data
The precision data were determined from an experiment conducted in 1995 involving 11 laboratories and 12 traced samples (six blind duplicates) for butter and 12 traced (six blind duplicates) for concentrated butter.
8.TOLERANCE LIMITS
8.1.Three samples must be taken from the traced product in order to check on the correct tracing of the product.
8.2.Butter
8.2.1.The incorporation rate for butter, taking into account background absorbance, is 22 mg/kg.
8.2.2.The results for the three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
17,7 mg/kg (95 % of the minimum incorporation rate),
12,2 mg/kg (70 % of the minimum incorporation rate).
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation between 17,7 mg/kg and 12,2 mg/kg.
8.3.Concentrated butter
8.3.1.The incorporation rate for concentrated butter, taking into account background absorbance, is 24 mg/kg
The results for the three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
19,2 mg/kg (95 % of the minimum incorporation rate),
13,2 mg/kg (70 % of the minimum incorporation rate).
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation between 19,2 mg/kg and 13,2 mg/kg.
ANNEX VIII(Article 5)
DETERMINING SITOSTEROL OR STIGMASTEROL IN BUTTER OR CONCENTRATED BUTTER BY CAPILLARY-COLUMN GAS CHROMATOGRAPHY
1.SCOPE AND FIELD OF APPLICATION
The method describes a procedure for the quantitative determination of sitosterol or stigmasterol in butter and concentrated butter. Sitosterol is taken to be the sum of β-sitosterol and 22 dihydro-β-sitosterol, other sitosterols are assumed to be insignificant.
2.PRINCIPLE
The butter or concentrated butter is saponified with potassium hydroxide in ethanolic solution and the unsaponifiables are extracted with diethyl ether.
The sterols are transformed into trimethyl-silyl ethers and are analysed by capillary-column gas chromatography with reference to an internal standard/betulin.
3.APPARATUS
3.1.150 ml saponification flask fitted with a reflux condenser having ground-glass joints
3.2.500 ml separating funnels
3.3.250 ml flasks
3.4.Pressure equalising funnels, 250 ml or similar, to collect waste diethyl ether
3.5.Glass column, 350 mm × 20 mm fitted with sintered glass plug
3.6.Waterbath or isomantle
3.7.Reaction vials, 2 ml
3.8.
Gas chromatograph suitable for use with a capillary column, provided with a splitting system consisting of:
3.8.1.a thermostatic chamber for columns capable of maintaining the desired temperature with an accuracy of ±1 °C
3.8.2.a temperature-adjustable vaporisation unit
3.8.3.a flame ionisation detector and converter-amplifier
3.8.4.an integrator-recorder suitable for use with the converter-amplifier (3.8.3)
3.9.A fused-silica capillary column entirely coated with BP1 or equivalent (or any other column of at least equal resolution) in a uniform thickness 0,25 μm; the column must be capable of resolving trimethyl-silyl derivatives of lanosterol and sitosterol. A column, length 12 m, internal diameter 0,2 mm, is suitable
3.10.A 1 μl gas chromatography microsyringe with hardened needle.
4.REAGENTS
All reagents must be of recognised analytical grade. The water used must be distilled water or water of at least equivalent purity.
4.1.Ethanol, at least 95 % pure
4.2.Potassium hydroxide, 60 % solution, dissolve 600 g potassium hydroxide (minimum 85 %) in water and make up to one litre with water
4.3.
Betulin of at least 99 % purity
4.3.1.
Solutions of betulin in diethyl ether (4.4)
4.3.1.1.The concentration of betulin solution used for sitosterol determination should be 1,0 mg/ml
4.3.1.2.The concentration of betulin solution used for stigmasterol determination should be 0,4 mg/ml
4.4.Diethyl ether, analytical purity (free from peroxides or residue)
4.5.Sodium sulphate, anhydrous, granular, previously dried at 102 °C for two hours
4.6.Silylating reagent, for example TRI-SIL (available from Pierce Chemical Co, Cat No 49001) or equivalent (Important: TRI-SIL is inflammable, toxic, corrosive and possibly carcinogenic. Laboratory personnel must be familiar with TRI-SIL safety data and take the appropriate precautions.)
4.7.Lanosterol
4.8.
Sitosterol, of known purity not less than 90 % pure (P)
Note 1: The purity of standard materials used for calibration must be determined using the method of normalisation. Assume that all sterols present in the sample are represented on the chromatogram, the total area of the peaks represents 100 % of the sterol constituents and that the sterols give the same detector response. Linearity of the system must be validated over the concentration ranges of interest.
4.8.1.Sitosterol standard solution — prepare a solution containing, to the nearest 0,001 mg/ml, approximately 0,5 mg/ml (W1) sitosterol (4.8) in diethyl ether (4.4)
4.9.
Stigmasterol, of known purity not less than 90 % pure (P)
4.9.1.Stigmasterol standard solution — prepare a solution containing, to the nearest 0,001 mg/m, approximately 0,2 mg/ml (W1) stigmasterol (4.9) in diethyl ether (4.4)
4.10.Resolution test mixture. Prepare a solution containing 0,05 mg/ml lanosterol (4.7) and 0,5 mg/ml sitosterol (4.8) in diethyl ether (4.4).
5.METHOD
5.1.Preparation of standard solutions for chromatography
The internal standard solution (4.3.1) must be added to the appropriate sterol standard solution at the same time as it is added to the saponified sample (see 5.2.2)
5.1.1.Sitosterol standard chromatographic solution: transfer 1 ml of sitosterol standard solution (4.8.1) to each of two reaction vials (3.7) and remove the diethyl ether with a stream of nitrogen. Add 1 ml of internal solution (4.3.1.1) and remove the diethyl ether with a stream of nitrogen
5.1.2.Stigmasterol standard chromatographic solution: transfer 1 ml of stigmasterol standard solution (4.9.1) to each of two reaction vials (3.7) and remove the diethyl ether with a stream of nitrogen. Add 1 ml of internal standard solution (4.3.1.2) and remove the diethyl ether with a stream of nitrogen
5.2.Preparation of the unsaponifiables
5.2.1.Melt the butter sample at a temperature not exceeding 35 °C, mix the sample thoroughly by stirring
Weigh, to the nearest 1 mg, approximately 1 g of butter (W2) or concentrated butter (W2) into a 150 ml flask (3.1). Add 50 ml ethanol (4.1) and 10 ml potassium hydroxide solution (4.2). Fit the reflux condenser and heat at approximately 75 °C for 30 minutes. Detach the condenser and cool the flask to approximately ambient temperature.
5.2.2.Add 1,0 ml of internal standard solution (4.3.1.1) to the flask if sitosterol is to be determined, or (4.3.1.2) if stigmasterol is to be determined. Mix thoroughly. Transfer the contents of the flask quantitatively into a 500 ml separating funnel (3.2), washing the flask in turn with 50 ml water and 250 ml diethyl ether (4.4). Shake the separating funnel vigorously for two minutes and allow the phases to separate. Run off the lower aqueous layer and wash the ether layer by shaking with four successive 100 ml aliquots of water
Note 2: To avoid formation of an emulsion, it is essential that the first two water washes are carried out gently (10 inversions). The third wash can be shaken vigorously for 30 seconds. If an emulsion is formed it can be destroyed by the addition of 5-10 ml of ethanol. If ethanol is added it is essential to carry out a further two vigorous water washes.
5.2.3.Pass the clear, soap-free ether layer through a glass column (3.5) containing 30 g anhydrous sodium sulphate (4.5). Collect the ether in a 250 ml flask (3.3). Add one anti-bumping granule and evaporate to near dryness in a waterbath or isomantle, taking care to collect the waste solvents.
Note 3: If sample extracts are taken to complete dryness at too high temperature sterol losses may occur.
5.3.Preparation of trimethyl silyl ethers
5.3.1.Transfer the ether solution remaining in the flask to a 2 ml reaction vial (3.7) with 2 ml diethyl ether and remove the ether using a stream of nitrogen. Wash the flask with two further 2 ml aliquots of diethyl ether, transferring to the vial and removing the ether with nitrogen each time
5.3.2.Silylate the sample by addition of 1 ml TRI-SIL (4.6). Close the vial and shake vigorously to dissolve. If dissolution is incomplete, warm to 65-70 °C. Allow to stand for at least five minutes before injecting into the gas-chromatograph. Silylate standards in the same way as samples. Silylate the resolution test mixture (4.10) in the same way as samples
Note 4: Silylation must be effected in a water-free environment. Incomplete silylation of betulin is indicated by a second peak close to that of betulin.
The presence of ethanol at the silylation stage will interfere with silylation. This may result from inadequate washing at the extraction stage. If this problem persists, a fifth wash may be introduced at the extraction stage, shaking vigorously for 30 seconds.
5.4.Gas-chromatographic analysis
5.4.1.Choice of operating conditions
Set up the gas-chromatograph according to the manufacturer’s instructions.
The guideline operating conditions are as follows:
column temperature: 265 °C
injector temperature: 265 °C
detector temperature: 300 °C
carrier gas flow rate: 0,6 ml/min.
hydrogen pressure: 84 kPa
air pressure: 155 kPa
sample split: 10:1 to 50:1; the split ratio must be optimised in accordance with the manufacturer’s instructions and linearity of detector response, then validated over the concentration range of interest.
Note 5: It is especially important that the injection liner is regularly cleared.
amount of substance injected: 1 μl of TMSE solution.
Allow the system to equilibrate and obtain a satisfactory stable response before commencing any analysis.
These conditions must be varied in the light of column and gas-chromatograph characteristics so as to obtain chromatograms which meet the following requirements:
the sitosterol peak must be adequately resolved from lanosterol. Figure 1 shows a typical chromatogram which should be obtained from a silylated resolution test mixture (4.10),
the relative retention times of the following sterols should be approximately:
cholesterol: 1,0
stigmasterol: 1,3
sitosterol: 1,5
betulin: 2,5
the retention time for betulin should be approximately 24 minutes.
5.4.2.Analytical procedure
Inject 1 μl of silylated standard solution (stigmasterol or sitosterol) and adjust the integrator calibration parameters.
Inject a further 1 μl of silylated standard solution to determine the response factors with reference to betulin.
Inject 1 μl of silylated sample solution and measure peak areas. Each chromatographic run must be bracketed by an injection of standards.
As a guide, six injections of sample should be included in each bracketed run.
Note 6: Integration of the stigmasterol peak should include any tailing as defined by points 1, 2 and 3 in Figure 2b.
Integration of the sitosterol peak should include the area of the 22 dihydro-β-sitosterol (stigmastanol) peak which elutes immediately after sitosterol (see Figure 3b) when evaluating total sitosterol.
6.CALCULATION OF RESULTS
6.1.Determine the area of the sterol peaks and betulin peaks in both standards bracketing a batch, and calculate R1:
R1 = (average area of the sterol peak in the standard)/(average area of the betulin peak in the standard)
Determine the area of the sterol peak (stigmasterol and sitosterol) and betulin peak in the sample and calculate R2:
R2 = (area of the sterol peak in the sample)/(area of the betulin peak in the sample)
=
sterol content of the standard (mg) contained in 1 ml of standard solution (4.8.1 or 4.9.1)
=
weight of sample (g) (5.2.1)
=
purity of standard sterol (4.8 or 4.9)
Sterol content of the sample (mg/kg) = ((R 2) / (R 1)) × ((W 1) / (W 2)) × P × 10.
7.ACCURACY OF THE METHOD
7.1.Butter
7.1.1.Repeatability
7.1.1.1.Stigmasterol
The difference between the results of two determinations carried out within the shortest feasible time interval, by one operator using the same apparatus on identical test material, may not exceed 19,3 mg/kg.
7.1.1.2.Sitosterol
The difference between the results of two determinations carried out within the shortest feasible time interval, by one operator using the same apparatus on identical test material, may not exceed 23,0 mg/kg.
7.1.2.Reproducibility
7.1.2.1.Stigmasterol
The difference between the results of two determinations carried out by operators in different laboratories, using different apparatus on identical test material, may not exceed 31,9 mg/kg.
7.1.2.2.Sitosterol
The difference between the results of two determinations carried out by operators in different laboratories, using different apparatus on identical test material, may not exceed 8,7 % relative to the mean of the determination.
7.1.3.Source of precision data
The precision data were determined from an experiment conducted in 1992 involving eight laboratories and six samples (three blind duplicates) for stigmasterol and six samples (three blind duplicates) for sitosterol.
7.2.Concentrated butter
7.2.1.Repeatability
7.2.1.1.Stigmasterol
The difference between the results of two determinations carried out within the shortest feasible time interval, by one operator using the same apparatus on identical test material, may not exceed 10,2 mg/kg.
7.2.1.2.Sitosterol
The difference between the results of two determinations carried out within the shortest feasible time interval, by one operator using the same apparatus on identical test material, may not exceed 3,6 % relative to the mean of the determinations.
7.2.2.Reproducibility
7.2.2.1.Stigmasterol
The difference between the results of two determinations carried out by operators in different laboratories, using different apparatus on identical test material, may not exceed 25,3 mg/kg.
7.2.2.2.Sitosterol
The difference between the results of two determinations carried out by operators in different laboratories, using different apparatus on identical test material, may not exceed 8,9 % relative to the mean of the determinations.
7.2.3.Source of precision data
The precision data were determined from an experiment conducted in 1991 involving nine laboratories and six samples (three blind duplicates) for stigmasterol and six samples (three blind duplicates) for sitosterol.
8.TOLERANCE LIMITS
8.1.Three samples must be taken from the traced product in order to check on the correct tracing of the product.
8.2.Butter
8.2.1.Stigmasterol
8.2.1.1.The incorporation rate for stigmasterol is 150 g of at least 95 % pure stigmasterol per tonne of butter, i.e. 142,5 mg/kg, or 170 g of at least 85 % pure stigmasterol per tonne of butter, i.e. 144,5 mg/kg.
8.2.1.2.The results for the three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
115,8 mg/kg (95 % of the minimum incorporation rate for 95 % pure stigmasterol),
117,7 mg/kg (95 % of the minimum incorporation rate for 85 % pure stigmasterol),
80,1 mg/kg (70 % of the minimum incorporation rate for 95 % pure stigmasterol),
81,5 mg/kg (70 % of the minimum incorporation rate for 85 % pure stigmasterol).
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation respectively between 115,8 mg/kg and 80,1 mg/kg or 117,7 mg/kg and 81,5 mg/kg.
8.2.2.Sitosterol
8.2.2.1.The incorporation rate for sitosterol is 600 g of at least 90 % pure sitosterol per tonne of butter, i.e. 540 mg/kg
8.2.2.2.The results for the three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
482,6 mg/kg (95 % of the minimum incorporation rate for 90 % pure sitosterol),
347,6 mg/kg (70 % of the minimum incorporation rate for 90 % pure sitosterol).
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation between 482,6 mg/kg and 347,6 mg/kg.
8.3.Concentrated butter
8.3.1.Stigmasterol
8.3.1.1.The incorporation rate for stigmasterol is 150 g of at least 95 % pure stigmasterol per tonne of concentrated butter, i.e. 142,5 mg/kg; or 170 g of at least 85 % pure stigmasterol per tonne of concentrated butter, i.e. 144,5 mg/kg
8.3.1.2.The results for the three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
118,5 mg/kg (95 % of the minimum incorporation rate for 95 % pure stigmasterol),
120,4 mg/kg (95 % of the minimum incorporation rate for 85 % pure stigmasterol),
82,9 mg/kg (70 % of the minimum incorporation rate for 95 % pure stigmasterol),
84,3 mg/kg (70 % of the minimum incorporation rate for 85 % pure stigmasterol).
The tracer concentration of the sample giving the lowest result is used in conjunction with interpolation respectively between 118,5 mg/kg and 82,9 mg/kg or 120,4 mg/kg and 84,3 mg/kg.
8.3.2.Sitosterol
8.3.2.1.The incorporation rate for sitosterol is 600 g of at least 90 % pure sitosterol per tonne of concentrated butter, i.e. 540 mg/kg
8.3.2.2.The results for the three samples obtained from the analysis of the product are used to check the rate and the homogeneity of tracer incorporation and the lowest of these results is compared with the following limits:
480,9 mg/kg (95 % of the minimum incorporation rate for 90 % pure sitosterol),
345,9 mg/kg (70 % of the minimum incorporation rate for 90 % pure sitosterol).
The tracer concentration in the sample giving the lowest result is used in conjunction with interpolation between 480,9 mg/kg and 345,9 mg/kg.
Figure 1
Chromatogram of resolution test mixture
Complete resolution is preferable, i.e. the peak trace for lanosterol should return to baseline before leaving for the sitosterol peak although incomplete resolution is tolerable.

| Note: Integration of the stigmasterol peak should include any tailing as defined by points 1, 2 and 3. | |
| Figure 2a | Figure 2b |
| Stigmasterol standard | Butter sample denatured with stigmasterol |
 |  |
| Note: β-sitosterol often contains an impurity (identified as stigmastanol) which elutes immediately after β-sitosterol. The areas of these two peaks should be summed when evaluating the total β-sitosterol present. | |
| Figure 3a | Figure 3b |
| Sitosterol standard | Butter sample denatured with β-Sitosterol |
 |  |
ANNEX IX(Article 6)
REFERENCE METHOD FOR THE DETECTION OF COWS’ MILK AND CASEINATE IN CHEESES FROM EWES’ MILK, GOATS’ MILK OR BUFFALOS’ MILK OR MIXTURES OF EWES’, GOATS’ AND BUFFALOS’ MILK
1.SCOPE
Detection of cows’ milk and caseinate in cheeses made from ewes’ milk, goats’ milk, buffalos’ milk or mixtures of ewes’, goats’ and buffalos’ milk by isoelectric focusing of γ-caseins after plasminolysis.
2.FIELD OF APPLICATION
The method is suitable for sensitive and specific detection of native and heat-treated cows’ milk and caseinate in fresh and ripened cheeses made from ewes’ milk, goats’ milk, buffalos’ milk or mixtures of ewes’, goats’ and buffalos’ milk. It is not suitable for the detection of milk and cheese adulteration by heat-treated bovine whey protein concentrates.
3.PRINCIPLE OF THE METHOD
3.1.Isolation of caseins from cheese and the reference standards
3.2.Dissolving of the isolated caseins and submitting to plasmin (EC.3.4.21.7) cleavage
3.3.Isoelectric focusing of plasmin-treated caseins in the presence of urea and staining of proteins
3.4.Evaluation of stained γ3 and γ2-casein patterns (evidence of cows’ milk) by comparison of the pattern obtained from the sample with those obtained in the same gel from the reference standards containing 0 % and 1 % cows’ milk.
4.REAGENTS
Unless otherwise indicated, analytical grade chemicals must be used. Water must be double-distilled or of equivalent purity.
Note: The following details apply to laboratory prepared polyacrylamide gels containing urea, of dimensions 265 × 125 × 0,25 mm. Where other sizes and types of gel are used, the separation conditions may have to be adjusted.
Isoelectric focusing
4.1.Reagents for production of the urea containing polyacrylamide gels
4.1.1.Stock gel solution
Dissolve:
4,85 g acrylamide
0,15 g N, N'-methylene-bis-acrylamide (BIS)
48,05 g urea
15,00 g glycerol (87 % w/w),
in water and make up to 100 ml and store in a brown glass bottle in the refrigerator.
Note: A commercially available preblended acrylamide/BIS solution can be used in preference to the quoted fixed weights of the neurotoxic acrylamides. Where such a solution contains 30 % w/v acrylamide and 0,8 % w/v BIS, a volume of 16,2 ml must be used for the formulation instead of the fixed weights. The shelf life of the stock solution is a maximum of 10 days; if its conductivity is more than 5 μS, de-ionize by stirring with 2 g Amberlite MB-3 for 30 minutes, then filter through a 0,45 μm membrane.
4.1.2.Gel solution
Prepare a gel solution by mixing additives and ampholytes with the stock gel solution (see 4.1.1).
9,0 ml stock solution
24 mg β-alanine
500 μl ampholyte pH 3,5-9,5(8)
250 μl ampholyte pH 5-7(8)
250 μl ampholyte pH 6-8(8).
Mix the gel solution and de-gas for two to three minutes in an ultrasonic bath or in vacuum.
Note: Prepare the gel solution immediately prior to pouring it (see 6.2).
4.1.3.Catalyst solutions
4.1.3.1.N, N, N' N' — tetramethylethylenediamine (Temed)
4.1.3.2.40 % w/v ammonium persulphate (PER):
Dissolve 800 mg PER in water and make up to 2 ml.
Note: Always use freshly prepared PER solution.
4.2.Contact fluid
Kerosene or liquid paraffin
4.3.Anode solution
Dissolve 5,77 g phosphoric acid (85 % w/w) in water and dilute to 100 ml.
4.4.Cathode solution
Dissolve 2,00 g sodium hydroxide in water and dilute to 100 ml with water.
Sample preparation
4.5.Reagents for protein isolation
4.5.1. Dilute acetic acid (25,0 ml of glacial acetic acid made up to 100 ml with water)
4.5.2. Dichloromethane
4.5.3. Acetone
4.6.Protein dissolving buffer
Dissolve
5,75 g glycerol (87 % w/w)
24,03 g urea
250 mg dithiothreitol,
in water and make up to 50 ml
Note: Store in a refrigerator, maximum shelf-life one week.
4.7.Reagents for plasmin cleavage of caseins
4.7.1.Ammonium carbonate buffer
Titrate a 0,2 mol/l ammonium hydrogencarbonate solution (1,58 g/100 ml water) containing 0,05 mol/l ethylenediaminetetraacetic acid (EDTA, 1,46 g/100 ml with a 0,2 mol/l ammonium carbonate solution (1,92 g/100 ml water) containing 0,05 mol/l EDTA to ph 8.
4.7.2. Bovine plasmin (EC. 3.4.21.7), activity at least 5 U/ml
4.7.3.ε-Aminocaproic acid solution for enzyme inhibition
Dissolve 2,624 g ε-aminocaproic acid (6 amino-n-hexanoic acid) in 100 ml of 40 % (v/v) ethanol.
4.8.Standards
4.8.1. Certified reference standards of a mixture of renneted ewes’ and goats’ skimmed milk containing 0 % and 1 % of cows’ milk are available from the Commission’s Institute for Reference Materials and Measurements, B-2440 Geel, Belgium
4.8.2.Preparation of laboratory interim-standards of buffalos’ renneted milk containing 0 % and 1 % of cows’ milk
Skimmed milk is prepared by centrifuging of either buffalo or bovine raw bulk milk at 37 °C at 2 500 g for 20 minutes. After cooling the tube and contents rapidly to 6 to 8 °C, the upper fat layer is removed completely. For the preparation of the 1 % standard add 5,00 ml of bovine skimmed milk to a 495 ml of buffalos’ skimmed milk in a 1 l beaker, adjust the pH to 6,4 by the addition of dilute lactic acid (10 % w/v). Adjust the temperature to 35 °C and add 100 μl of calf rennet (rennet activity 1: 10 000, c. 3 000 U/ml), stir for 1 minute and then leave the beaker covered with an aluminium foil at 35 °C for one hour to allow formation of the curd. After the curd has formed, the whole renneted milk is freeze-dried without prior homogenization or draining of the whey. After freeze-drying it is finely ground to produce a homogeneous powder. For the preparation of the 0 % standard, carry out the same procedure using genuine buffalo skimmed milk. The standards must be stored at -20 °C.
Note: It is advisable to check the purity of the buffalos’ milk by isoelectric focusing of the plasmin-treated caseins before preparation of the standards.
Reagents for protein staining
4.9.Fixative
Dissolve 150 g trichloroacetic acid in water and make up to 1 000 ml.
4.10.Destaining solution
Dilute 500 ml methanol and 200 ml glacial acetic acid to 2 000 ml with distilled water.
Note: Prepare the destaining solution fresh every day; it can be prepared by mixing equal volumes of stock solutions of 50 % (v/v) methanol and 20 % (v/v) glacial acetic acid.
4.11.Staining solutions
4.11.1.Staining solution (stock solution 1)
Dissolve 3,0 g Coomassie Brilliant Blue G-250 (C.I. 42655) in 1 000 ml 90 % (v/v) methanol using a magnetic stirrer (approximately 45 minutes), filter through two medium-speed folded filters.
4.11.2.Staining solution (stock solution 2)
Dissolve 5,0 g copper sulphate pentahydrate in 1 000 ml 20 % (v/v) acetic acid.
4.11.3.Staining solution (working solution)
Mix together 125 ml of each of the stock solutions (4.11.1, 4.11.2) immediately prior to staining.
Note: The staining solution should be prepared on the day that it is used.
5.EQUIPMENT
5.1.Glass plates (265 × 125 × 4 mm); rubber roller (width 15 cm); levelling table
5.2.Gel carrier sheet (265 × 125 mm)
5.3.Covering sheet (280 × 125 mm). Stick on strip of adhesive tape (280 × 6 × 0,25 mm) to each long edge (see Figure 1)
5.4.Electrofocusing chamber with cooling plate (e.g. 265 × 125 mm) and suitable power supply (≥ 2,5 kV) or automatic electrophoresis device
5.5.Circulation cryostat, thermostatically controlled at 12 ± 0,5 °C
5.6.Centrifuge, adjustable to 3 000 g
5.7.Electrode strips (≥ 265 mm long)
5.8.Plastic dropping bottles for the anode and cathode solutions
5.9.Sample applicators (10 × 5 mm, viscose or low protein-adsorption filter paper)
5.10.Stainless steel scissors, scalpels and tweezers
5.11.Stainless steel or glass staining and destaining dishes (e.g. 280 × 150 mm instrument trays)
5.12.Adjustable rod homogenizer (10 mm shaft diameter), rpm range 8 000 to 20 000
5.13.Magnetic stirrer
5.14.Ultrasonic bath
5.15.Film welder
5.16.25 μl micropipettes
5.17.Vacuum concentrator or freeze-dryer
5.18.Thermostatically controlled water bath adjustable to 35 and 40 ± 1 °C with shaker
5.19.Densitometer equipment reading at λ = 634 nm
6.PROCEDURE
6.1.Sample preparation
6.1.1.Isolation of caseins
Weigh the amount equivalent to 5 g dry mass of cheese or the reference standards into a 100 ml centrifuge tube, add 60 ml distilled water and homogenize with a rod homogenizer (8 000 to 10 000 rpm). Adjust to pH 4,6 with dil. acetic acid (4.5.1) and centrifuge (5 minutes, 3 000 g). Decant the fat and whey, homogenize the residue at 20 000 rpm in 40 ml distilled water adjusted to pH 4,5 with dil. acetic acid (4.5.1), add 20 ml dichloromethane (4.5.2), homogenize again and centrifuge (5 minutes, 3 000 g). Remove the casein layer that lies between the aqueous and organic phases (see Figure 2) with a spatula and decant off both phases. Rehomogenize the casein in 40 ml distilled water (see above) and 20 ml dichloromethane (4.5.2) and centrifuge. Repeat this procedure until both extraction phases are colourless (two to three times). Homogenize the protein residue with 50 ml acetone (4.5.3) and filter through a medium-speed folded filter paper. Wash the residue on the filter with two separate 25 ml portions of acetone each time and allow to dry in the air or a stream of nitrogen, then pulverize finely in a mortar.
Note: Dry casein isolates should be kept at -20 °C.
6.1.2.Plasmin cleavage of β-caseins to intensify γ-caseins
Disperse 25 mg of isolated caseins (6.1.1) in 0,5 ml ammonium carbonate buffer (4.7.1) and homogenize for 20 minutes by e.g. using ultrasonic treatment. Heat to 40 °C and add 10 μl plasmin (4.7.2), mix and incubate for one hour at 40 °C with continuous shaking. To inhibit the enzyme add 20 μl ε-aminoproic acid solution (4.7.3), then add 200 mg of solid urea and 2 mg of dithiothreitol.
Note: To obtain more symmetry in the focused casein bands it is advisable to freeze-dry the solution after adding the ε-aminocaproic acid and then dissolving the residues in 0,5 ml protein dissolving buffer (4.6).
6.2.Preparation of the urea containing polyacrylamide gels
With the aid of a few drops of water roll the gel carrier sheet (5.2) onto a glass plate (5.1), removing any extraneous water with paper towel or tissue. Roll the cover sheet (5.3) with spacers (0,25 mm) onto another glass plate in the same way. Lay the plate horizontally on a levelling table.
Add 10 μl Temed (4.1.3.1) to the prepared and de-aerated gel solution (4.1.2), stir and add 10 μl PER-solution (4.1.3.2), mix thoroughly and immediately pour out evenly onto the centre of the cover sheet. Place one edge of the gel carrier plate (sheet side down) on the cover sheet plate and lower it slowly so that a gel film forms between the sheets and spreads out regularly and free of bubbles (Figure 3). Carefully lower the gel carrier plate completely using a thin spatula and place three more glass plates on top of it to act as weights. After polymerization is complete (about 60 minutes) remove the gel polymerized onto the gel carrier sheet along with the cover sheet by tipping the glass plates. Clean the reverse of the carrier sheet carefully to remove gel residues and urea. Weld the gel sandwich into a film tube and store in a refrigerator (maximum six weeks).
Note: The cover sheet with the spacers can be re-used. The polyacrylamide gel can be cut to smaller sizes, recommended when there are few samples or if an automatic electrophoresis device is used (two gels, size 4,5 × 5 cm).
6.3.lsoelectric focusing
Set the cooling thermostat to 12 °C. Wipe off the reverse of the gel carrier sheet with kerosene, then drip a few drops of kerosene (4.2) onto the centre of the cooling block. Then roll the gel sandwich, carrier side down, onto it, taking care to avoid bubbles. Wipe off any excess kerosene and remove the cover sheet. Soak the electrode strips with the electrode solutions (4.3, 4.4), cut to gel length and place in the positions provided (distance of electrodes 9,5 cm).
Conditions for isoelectric focusing:
6.3.1.Gel size 265 × 125 × 0,25 mm
| a Sample application: After pre-focusing (step 1), pipette 18 μl of the sample and standard solutions onto the sample applicators (10 × 5 mm), place them on the gel at 1 mm intervals from each other and 5 mm longitudinally from the anode and press lightly. Carry out focusing using the above conditions, carefully removing the sample applicators after the 60 minutes of sample focusing. | |||||
| Step | Time(min.) | Voltage(V) | Current(mA) | Power(W) | Volt-hours(Vh) |
|---|---|---|---|---|---|
1.Pre-focusing | 30 | maximum 2 500 | maximum 15 | constant 4 | c. 300 |
2.Sample focusinga | 60 | maximum 2 500 | maximum 15 | constant 4 | c. 1 000 |
3.Final focusing | 60 | maximum 2 500 | maximum 5 | maximum 20 | c. 3 000 |
| 40 | maximum 2 500 | maximum 6 | maximum 20 | c. 3 000 | |
| 30 | maximum 2 500 | maximum 7 | maximum 25 | c. 3 000 | |
Note: If thickness or width of the gels are changed, the values for current and power have to be suitably adjusted (e.g. double the values for electric current and power if a 265 × 125 × 0,5 mm gel is used).
6.3.2.Example of a voltage programme for an automatic electrophoresis device (2 gels of 5,0 × 4,5 cm), electrodes without strips applied directly to the gel
Place sample applicator in step 2 at 0 Vh.
Remove sample applicator in step 2 at 30 Vh.
6.4.Protein staining
6.4.1.Protein fixation
Remove the electrode strips immediately after turning off the power and put the gel immediately into a staining/destaining dish filled with 200 ml fixative (4.9); leave for 15 minutes, shaking continuously.
6.4.2.Washing and staining the gel plate
Thoroughly drain off the fixative and wash the gel plate twice for 30 seconds each time with 100 ml destaining solution (4.10). Pour off the destaining solution and fill the dish with 250 ml staining solution (4.11.3); allow to stain for 45 minutes with gentle shaking.
6.4.3.Destaining the gel plate
Pour off the staining solution, wash the gel plate twice using a 100 ml destaining solution (4.10) each time, then shake with 200 ml destaining solution for 15 minutes and repeat the destaining step at least two or three times until the background is clear and uncoloured. Then rinse the gel plate with distilled water (2 × 2 minutes) and dry in the air (2 to 3 hours) or with a hairdryer (10 to 15 minutes).
Note 1: Carry out fixing, washing, staining and destaining at 20 °C. Do not use elevated temperatures.
Note 2: If more sensitive silver staining (e.g. Silver Staining Kit, Protein, Pharmacia Biotech, Code No 17-1150-01) is preferred, plasmin-treated casein samples have to be diluted to 5 mg/ml.
7.EVALUATION
Evaluation is performed by comparing the protein patterns of the unknown sample with reference standards on the same gel. Detection of cows’ milk in cheeses from ewes’ milk, goats’ milk and buffalos’ milk and mixtures of ewes’, goats’ and buffalos’ milk is done via the γ3- and γ2-caseins, whose isoelectric points range between pH 6,5 and pH 7,5 (Figures 4 a, b, Figure 5). The detection limit is less than 0,5 %.
7.1.Visual estimation
For visual evaluation of the amount of bovine milk it is advisable to adjust the concentrations of samples and standards to obtain the same level of intensity of the ovine, caprine and/or buffalo γ2- and γ3-caseins (see ‘γ2 E,G,B’ and ‘γ3 E,G,B’ in Figures 4 a, b and Figure 5). After which the amount of bovine milk (less than, equal to or greater than 1 %) in the unknown sample can be judged directly by comparing the intensity of the bovine γ3- and γ2-caseins (see ‘γ3 C’ and ‘γ2 C’ in Figures 4a, b and Figure 5) to those of the 0 % and 1 % reference standards (ewe, goat) or, laboratory interim-standards (buffalo).
7.2.Densitometric estimation
If available, apply densitometry (5.19) for the determination of the peak area ratio of bovine to ovine, caprine and/or buffalo γ2- and γ3-caseins (see Figure 5). Compare this value to γ2- and γ3-casein peak area ratio of the 1 % reference standard (ewe, goat) or laboratory interim-standard (buffalo) analysed on the same gel.
Note: The method is operating satisfactorily, if there is a clear positive signal for both bovine γ2- and γ3-caseins in the 1 % reference standard but not in the 0 % reference standard. If not, optimize the procedure following the details of the method precisely.
A sample is judged as being positive, if both bovine γ2- and γ3-caseins or the corresponding peak area ratios are equal to or greater than the level of the 1 % reference standard.
8.REFERENCES
Addeo F., Moio L., Chianese L., Stingo C., Resmini P., Berner I, Krause I., Di Luccia A., Bocca A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft 45, 708-711 (1990).
Addeo F., Nicolai M.A., Chianese L., Moio L., Spagna Musso S., Bocca A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft 50, 83-85 (1995).
Krause I., Berner I, Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte — and carrier ampholyte/immobilized pH gradient — isoelectric focusing of γ-caseins using plasmin as signal amplifier. in: Electrophoresis-Forum 89 (B. J. Radola, ed.) pp 389-393, Bode-Verlag, München (1989).
Krause Ι., Belitz H.-D., Kaiser K.-P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. –käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen. Z. Lebensm. Unters. Forsch. 174, 195-199 (1982).
Radola B.J.: Ultrathin-layer isoelectric focusing in 50-100 μm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis 1, 43-56 (1980).



a = spacer tape (0,25 mm); b = covering sheet (5.3); c, e = glass plates (5.1); d = gel solution (4.1.2); f = gel carrier sheet (5.2)
Figure 4a
Isoelectric focusing of plasmin-treated caseins from ewes’ and goats’ milk cheese containing different amounts of cows’ milk.

% CM = percentage of cows’ milk, C = cow, E = ewe, G = goat
Upper half of the IEF gel is shown.
Figure 4b
Isoelectric focusing of plasmin treated caseins from cheese made from mixtures of ewes’, goats’ and buffalos’ milk containing different amounts of cows’ milk.

% CM = percentage of cows’ milk; 1 + = sample containing 1 % of cows’ milk and spiked with pure bovine casein at the middle of the track. C = cow, E = ewe, G = goat, B = buffalo.
Total separation distance of the IEF gel is shown.
Figure 5
Superposition of densitograms of standards (STD) and cheese samples made from a mixture of ewes’ and goats’ milk after isoelectric focusing

a,b = standards containing 0 and 1 % of cows’ milk; c-g = cheese samples containing 0, 1, 2, 3 and 7 % of cows’ milk. C = cow, E = ewe, G = goat.
Upper half of the IEF gel was scanned at λ = 634 nm.
ANNEX X(Article 7)
REFERENCE METHOD FOR DETECTING COLIFORMS IN BUTTER, SKIMMED-MILK POWDER, CASEIN AND CASEINATES
1.PREPARATION OF SAMPLES
ISO standard 8261
2.PROCEDURE
ISO standard 4831
Samples corresponding to 1 g of butter or 0,1 g of skimmed-milk powder or casein/caseinates are inoculated in the culture medium.
Three tubes are inoculated per sample.
3.RESULTS
If the 3 tubes produce 3 negative results, the result is ‘compliant’
If the 3 tubes produce 2 or 3 positive results, the result is ‘non-compliant’
If the 3 tubes produce 2 negative results, the analysis is redone twice (with two tubes)
If the 2 results are negative, the result is ‘compliant’
If at least 1 result is positive, the result is ‘non-compliant’.
ANNEX XI(Article 8)
DETERMINATION OF LACTOSE IN COMPOUND FEEDINGSTUFFS
1.SCOPE AND FIELD OF APPLICATION
Determination of lactose in compound feedingstuffs.
2.REFERENCE
The content of lactose is defined as the percentage by mass as determined by the procedure described.
3.DEFINITION
The anhydrous lactose content is expressed as g per 100 g.
4.PRINCIPLE
The compound feedingstuff is reconstituted with water. A ‘Biggs’ solution is added to a dilute weighed aliquot to precipitate out the fat and the protein component fractions of the compound feedingstuff. The sample is filtered (or centrifuged) and the filtrate (or supernatant) is injected on a cation exchange HPLC column in the lead form using HPLC grade water as the mobile phase. The eluted lactose is detected by a differential refractometer(9).
5.REAGENTS
5.1.General
Use only reagents of recognized analytical grade, unless otherwise specified, and degassed HPLC grade water.
5.2.Lactose
D-Lactose monohydrate ((C12H22)O11H2O) can take up additional moisture. Before use measure the real amount of water by Karl-Fisher or remove the additional moisture by placing lactose in an oven at 105 °C for 8 hours (the lactose does not lose its crystal water by this treatment).
5.3.Concentrated Biggs/Szijarto solution(10)
Dissolve 9,10 g zinc acetate ehydrate (Zn(CH3COO)2.2H2O) and 5,46 g phosphotungstic acid monohydrate (H3[P(W3O10)4.xH2O]) in about 70 ml with HPLC grade water (6.8) in a 100 ml volumetric flask.
Add 5,81 ml glacial acetic acid (CH3COOH). Dilute to the 100 ml mark with HPLC grade water (6.8) water and mix. The solution can be stored at room temperature for 1 year.
5.4.Diluted Biggs/Szijarto solution
Dilute 25 ml concentrated Biggs/Szijarto solution (5.3) with water to 500 ml using a volumetric flask. The solution can be stored at room temperature for 1 month.
5.5.Preparation of HPLC grade water
Filter the ultra pure water (6.8) by using the vacuum system filtration (6.9). To improve the pump performance and to obtain a stable baseline, degas the mobile phase daily by selecting one of the available techniques such as sparging by helium, sonication, vacuum or in-line degassing system.
Note: In order to prolong column life it is essential that the carbon dioxide content of the eluent is as low as possible and that re-uptake is prevented.
6.APPARATUS
Usual laboratory equipment and, in particular, the following:
6.1.HPLC ion exchange resin column
Column packing: 8 % cross-linked polystyrene-divinylbenzene copolymer functionalised with cation-exchange groups in the lead form.
Column dimensions: Length 300 mm, internal diameter ca. 8 mm.
Use of other diameters is possible provided that the flow-rate is adjusted accordingly.
6.2.Guard column
The guard column is a combination of a separate cation exchanger (H+) and an anion exchanger (CO3-), each packed in columns of ca. 30 mm × 4,6 mm (L × ID) (e.g. micro-guard columns in a micro-guard holder) and connected in series or in the form of a mixed bed consisting of AG 50W-X4, -400 mesh (H+) and AG3-X4A, 200–400 mesh (OH-) in the ratio of 35:65 (m/m) manually packed in a column of ca. 20 × 9 mm (L × ID).
6.3.Column oven
Oven capable of maintaining a constant temperature of 85 °C ± 1 °C.
6.4.HPLC pump
Pump capable of generating a constant flow-rate (< 0,5 % fluctuations) at 0,2-1,0 m/min.
6.5.HPLC injection device
Auto sampler capable of injecting 25 µL and having a repeatability < 0,5 %.
Alternatively a manual device may be used (same requirements as the auto sampler).
6.6.HPLC detector
Highly sensitive refractive index detector having a noise < 5,10-9 RI units.
6.7.Integrator
Software or a dedicated integrator to perform data acquisition, processing and generating peak areas and peak heights, which can be converted to lactose concentrations.
6.8.Water purification unit
System capable to provide ultra pure water (type 1) having a resistivity >14 MΩ.cm.
6.9.Solvent filtration unit
System that enables filtration of the water using 0,45 µm pore size membrane filter.
Note: Many water purification units (6.8) have a built in 0,45 or 0,2 µm filtration. Additional filtration can be omitted if this water is directly used.
6.10.Analytical balance
Balance having a read-out of 0,1 mg.
6.11.Water bath
Water bath capable of maintaining a temperature at 40 °C (±0,5).
6.12.Centrifuge
Having a capacity of generating at least 3 000 g for Eppendorf vials or equivalent or larger type of vials.
6.13.Volumetric flask 50 mL
Capacity of 50 mL, class A.
Note: Flasks of other capacity can be used by taking into account the volume factor.
6.14.Volumetric flask 100 mL
Capacity of 100 mL, class A.
6.15.Graduated pipette
Graduated pipette of 10 mL
Note: Alternatively, a handpipettor having a capacity of 5 mL can be used by adding twice a volume of 5 mL of reagent (5.3).
7.SAMPLING
It is important that the laboratory receives a sample taken according to ISO 707/IDF 50(11), which is truly representative and has not been damaged during transport or storage.
8.PREPARATION OF LACTOSE STANDARD SOLUTION
8.1.Standard 1
Dissolve an accurately (read-out at 0,1 mg) weighed amount of ca. 50 mg lactose monohydrate (5.2) in a volumetric flask of 100 mL (6.14) and make up to the mark with water.
8.2.Standard 2
Dissolve an accurately (read-out at 0,1 mg) weighed amount of ca. 100 mg lactose monohydrate (5.2) in a volumetric flask of 100 mL (6.14) and make up to the mark with water.
Note: The standard solutions can be stored for maximal 1 week at ca. 5 °C.
9.PREPARATION OF THE TEST SAMPLE
9.1.Reconstitution of the sample
Weigh ca. 5 g of the powder into a flask of 50 mL (6.13) and note the weight at 1 mg accuracy (W1, (11)). Add 50 mL of water and note the increase of the weight (W2 (11)) at 0,01 g accuracy. Place the closed flask in the water bath (6.11) for 30 min and invert it a few times during this period. Let it subsequently cool to room temperature.
9.2.Sample treatment
Take ca. 1 g of this solution and deposit it in a 50 mL volumetric flask (6.13), note the weight at 1 mg accuracy (W3 (11)), add 20 mL of water followed by the addition of 10 mL diluted Biggs/Szijarto reagent (5.4), make up to the mark with water. Gently invert the flask 5 times during the first 30 min.
After 1 hour take an aliquot and centrifuge (6.12) at 3 000 g for 10 min (higher g can be used at correspondingly shorter time). Use an aliquot of the supernatant for HPLC analysis.
10.HPLC DETERMINATION
10.1.Preliminary preparation of HPLC
10.1.1.Installation of the column and pre-column
Install the pre-column (6.2) outside the column oven (6.3) and the column (6.1) in the oven.
Note: If the oven does not contain tubing to preheat the eluent, it is necessary that the eluent passes ca. 15 cm stainless steel tubing in the oven before entering the column (it is absolutely necessary that the eluent has heated up before entering the column, otherwise peak broadening shall occur).
10.1.2.Detector and initial flow
In order to get a stable baseline, turn on the detector (6.6) at least 24 h before starting the analysis. Set the internal temperature of the detector at 35 °C. Set the flow rate at 0,2 ml/min (6.4) for at least 20 min while the column oven (6.3) is set to room temperature.
10.1.3.Column oven and final flow-rate
Set the column oven (6.3) at 85 °C. When that temperature is reached, increase after 30 minutes gradually the flow rate from 0,2 ml/min to 0,6 ml/min (6.4). Allow the system to equilibrate with that flow rate and at 85 °C for 2 h or until a stable baseline is obtained.
10.1.4.Integration
Carefully chose the acquisition and integration parameters (6.7) such as data rate, sensitivity, time constant, peak width and threshold.
The retention time of lactose is ca. 11 min.
Note: Many data acquisition software programs (6.7) afford easy measurement of the theoretical plate count. Measure the theoretical plate count of standard 1 (8.1) regularly and replace the column (6.1) when the plate count is 25 % lower than that of the initial value of a new column.
10.1.5.Guard column test
Check regularly (at least once in every sequence) the ability of the guard column (6.2) to eliminate salts from the sample by injecting 25 µL of a 0,05 % sodium chloride solution. Whenever peaks are appearing, the guard column should be replaced.
10.2.Running standards
Inject at the beginning of each series of analyses 25 µL (6.5) of standard 1 (8.1) and subsequently of standard 2 (8.2). Repeat this every 10 to 20 samples and apply this also at the end of the sequence.
10.3.Running samples
Inject 25 µL of the supernatant (9.2) of the sample.
11.CALCULATION AND EXPRESSION OF THE RESULTS
11.1.Calibration
Normally peak heights are used to calculate the results, however, if the signal contains to much noise peak area can be used (quantitation by peak height is less influenced by peaks of components in low concentration and which are partly, but insufficiently separated from the lactose peak).
The software (6.7) should calculate a linear calibration curve forced through the origin. Check the curve for possible non linearity (apparent non linearity is most likely caused by a mistake in preparing the standards 1 (8.1) or 2 (8.2), bad integration and, less likely, by a mall functioning injector).
Use as input the calculated lactose concentrations in mg/mL of the standards 1 (8.1) and 2 (8.2) as water free lactose.
The slope (RF) of the calibration line is defined by area/concentration in mg/mL.
11.2.Samples
The result of the analysis is obtained as g/100 g and calculated using the software (6.7) or using the following formula:
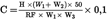
Where:
:
concentration of lactose in g/100 g powder
:
peak height of lactose of sample
:
Response factor (or slope) of calibration plot in mV/mg/mL
:
sample weight of powder sample in g (9.1)
:
weight of added water in g to powder sample (9.1)
:
sample weight of reconstituted solution of powder in g (9.2)
:
Volume of volumetric flask used in (9.2)
:
conversion of the result in g/100 g
12.PRECISION
The values derived from this inter laboratory test may not be applicable to concentration ranges and matrices other than those given. The values for the repeatability and reproducibility will be derived from the result of an inter laboratory test carried out in accordance with ISO 5725(12)
12.1.Repeatability
The absolute difference between two single test results, obtained using the same method on identical test material in the same laboratory with the same operator using the same equipment within a short time of interval, will in not more than 5 % of cases be greater than xxx (to be determined by a collaborative trial)(13).
12.2.Reproducibility
The absolute difference between two single test results, obtained using the same method on identical test material in different laboratories with different operators using different equipment, will in not more than 5 % of cases be greater than 0,5 g/100g (to be determined by a collaborative trial)
ANNEX XII(Article 9)
DETECTION OF RENNET WHEY IN SKIMMED-MILK POWDER FOR PUBLIC STORAGE BY DETERMINATION OF CASEINOMACROPEPTIDES HIGH-PERFORMANCE LIQUID CHROMATOGRAPHY (HPLC)
1.SCOPE AND FIELD OF APPLICATION
This method allows detection of rennet whey in skimmed-milk powder intended for public storage by determination of the caseinomacropeptides.
2.REFERENCE
International Standard ISO 707 — Milk and Milk Products — Methods of sampling, conforming to the guidelines contained in Annex I(2)I last paragraph.
3.DEFINITION
The content of rennet whey solids is defined as the percentage by mass as determined by the caseinomacropeptide content by the procedure described.
4.PRINCIPLE
Reconstitution of the skimmed-milk powder, removal of fat and proteins with trichloroacetic acid, followed by centrifugation or filtration.
Determination of the quantity of caseinomacropeptides (CMP) in the supernatant by high-performance liquid chromatography (HPLC).
Evaluation of the result obtained for the samples by reference to standard samples consisting of skimmed-milk powder with or without the addition of a known percentage of whey powder.
5.REAGENTS
All reagents must be of recognised analytical grade. The water used must be distilled water or water of at least equivalent purity.
5.1.Trichloroacetic acid solution
Dissolve 240 g of trichloroacetic acid (CCl3COOH) in water and make up to 1 000 ml. The solution should be clear and colourless.
5.2.Eluent solution, pH 6,0
Dissolve 1,74 g of dipotassium hydrogen phosphate (K2HPO4), 12,37 g of potassium dihydrogen phosphate (KH2PO4) and 21,41 g of sodium sulphate (Na2SO4) in about 700 ml of water. Adjust, if necessary, to pH 6,0, using a solution of phosphoric acid or potassium hydroxide.
Make up to 1 000 ml with water and homogenise.
Note: The composition of the eluent can be updated to comply with the certificate of the standards or the recommendations of the manufacturer of the column packing material.
Filter the eluent solution, prior to use, through a membrane filter with a 0,45 μm pore diameter.
5.3.Flushing solvent
Mix one volume acetonitrile (CH3CN) with nine volumes water. Filter the mixture prior to use through a membrane filter with a 0,45 μm pore diameter.
Note: Any other flushing solvent with a bactericidal effect which does not impair the columns’ resolution efficiency may be used.
5.4.Standard samples
5.4.1. Skimmed-milk powder meeting the requirements of this Resolution (i.e. [0])
5.4.2. The same skimmed-milk powder adulterated with 5 % (m/m) rennet-type whey powder of standard composition (i.e. [5]).
6.APPARATUS
6.1.Analytical balance
6.2.Optional centrifuge capable of attaining a centrifugal force of 2 200 g, fitted with stoppered or capped centrifuge tubes of about 50 ml capacity
6.3.Mechanical shaker
6.4.Magnetic stirrer
6.5.Glass funnels, diameter about 7 cm
6.6.Filter papers, medium filtration, diameter about 12,5 cm
6.7.Glass filtration equipment with 0,45 μm pore diameter membrane filter
6.8.Graduated pipettes allowing delivery of 10 ml (ISO 648, Class A, or ISO/R 835) or a dispensing system capable of delivering 10,0 ml in two minutes
6.9.Dispensing system capable of delivering 20,0 ml water at ca. 50 °C
6.10.Thermostatic water bath, set at 25 ± 0,5 °C
6.11.
HPLC equipment, consisting of:
6.11.1.Pump
6.11.2.Injector, hand or automatic, with a 15 to 30 μl capacity
6.11.3.Two TSK 2 000-SW columns in series (length 30 cm, internal diameter 0,75 cm) or equivalent columns (e.g. single TSK 2 000-SWxl, single Agilent Technologies Zorbax GF 250) and a precolumn (3 cm × 0,3 cm) packed with I 125 or material of equivalent effectiveness
6.11.4.Thermostatic column oven, set at 35 ± 1 °C
6.11.5.Variable wavelength UV detector, permitting measurements at 205 nm with a sensitivity of 0,008 A
6.11.6.Integrator capable of valley-to-valley integration
Note: Working with columns kept at room temperature is possible, but their power of resolution is slightly lower. In that case, the temperature should vary by less than ± 5 °C in any one range of analyses.
7.SAMPLING
7.1.Samples must be taken in accordance with the procedure laid down in International Standard ISO 707. However, Member States may use another method of sampling provided that it complies with the principles of the abovementioned standard.
7.2.Store the sample in conditions which preclude any deterioration or change in composition.
8.PROCEDURE
8.1.Preparation of the test sample
Transfer the milk powder into a container with a capacity of about twice the volume of the powder, fitted with an airtight lid. Close the container immediately. Mix the milk powder well by means of repeated inversion of the container.
8.2.Test portion
Weight 2,000 ± 0,001 g of test sample into a centrifuge tube (6.2) or a suitable stoppered flask (50 ml).
8.3.Removal of fat and proteins
8.3.1.Add 20,0 ml of warm water (50 °C) to the test portion. Dissolve the powder by shaking for five minutes using a mechanical shaker (6.3). Place the tube into the water bath (6.10) and allow to equilibrate to 25 °C
8.3.2.Add 10,0 ml of the trichloroacetic acid solution (5.1) of ca. 25 °C in two minutes, while stirring vigorously with the aid of the magnetic stirrer (6.4). Place the tube in a water bath (6.10) and leave for 60 minutes
8.3.3.Centrifuge (6.2) for 10 minutes at 2 200 g, or filter through paper (6.6), discarding the first 5 ml of filtrate
8.4.Chromatographic determination
8.4.1.Inject 15 to 30 μl of accurately measured supernatant or filtrate (8.3.3) into the HPLC apparatus (6.11) operating at a flow rate of 1,0 ml of eluent solution (5.2) per minute
Note 1. Another flow rate may be used, dependent of the internal diameter of the columns used or the instructions of the manufacturer of the column.
Note 2. Keep the eluent solution (5.2) at 85 °C throughout the chromatographic analysis in order to keep the eluent degassed and to prevent bacterial growth. Any precaution with a similar effect may be used.
Note 3. Rinse the columns with water during each interruption. Never leave the eluent solution in them (5.2).
Prior to any interruption of more than 24 hours, rinse the columns with water then wash them with solution (5.3) for at least three hours at a flow rate of 0,2 ml per minute.
8.4.2.The results of chromatographic analysis of the test sample [E] are obtained in the form of chromatogram in which each peak is identified by its retention time RT as follows:
| Peak II: | The second peak of the chromatogram having an RT of about 12,5 minutes. |
| Peak III: | The third peak of the chromatogram, corresponding to the CMP, having an RT of 15,5 minutes. |
The choice of the column(s) can effect the retention times of the individual peaks considerably.
The integrator (6.11.6) automatically calculates the area A of each peak:
| AII: | area of peak II, |
| AIII: | area of peak III, |
It is essential to examine the appearance of each chromatogram prior to quantitative interpretation, in order to detect any abnormalities due either to malfunctioning of the apparatus or the columns, or to the origin and nature of the sample analysed.
If in doubt, repeat the analysis.
8.5.Calibration
8.5.1.Apply exactly the procedure described from point 8.2 to point 8.4.2 to the standard samples (5.4)
Use freshly prepared solutions, because CMP degrade in an 8 % trichloroacetic environment. The loss is estimated at 0,2 % per hour at 30 °C.
8.5.2.Prior to chromatographic determination of the samples, condition the columns by repeatedly injecting the standard sample (5.4.2) in solution (8.5.1) until the area and retention time of the peak corresponding to the CMP are constant
8.5.3.Determine the response factors R by injecting the same volume of filtrates (8.5.1) as used for the samples
9.EXPRESSION OF RESULTS
9.1.Method of calculation and formulae
9.1.1.Calculation of the response factors R:
| Peak II: | RII = 100/(AII[0]) |
where:
=
the response factors of peaks II,
=
the areas of peaks II of the standard sample [0] obtained in 8.5.3.
| Peak III: | RIII = W/(AIII[5] – AIII[0]) |
where:
=
the response factor of peak III,
=
the areas of peak III in standard samples [0] and [5] respectively obtained in 8.5.3,
=
the quantity of whey in standard sample [5], i.e. 5.
9.1.2.Calculation of the relative area of the peaks in the sample [E]
SII[E] = RII × AII[E]
SIII[E] = RIII × AIII[E]
SIV[E] = RIV × AIV[E]
where:
=
the relative areas of peaks II, III and IV respectively in the sample [E],
=
the areas of peaks II and III respectively in the sample [E] obtained in 8.4.2,
=
the response factors calculated in 9.1.1.
9.1.3.Calculation of the relative retention time of peak III in sample [E]: RRTIII[E] = (RTIII[E])/(RTIII[5])
where:
=
the relative retention time of peak III in sample [E],
=
the retention time of peak III in sample [E] obtained in 8.4.2,
=
the retention time of peak III in control sample [5] obtained in 8.5.3.
9.1.4.Experiments have shown that there is a linear relation between the relative retention time of peak III, i.e. RRTIII [E] and the percentage of whey powder added up to 10 %
The RRTIII [E] is < 1,000 when the whey content is > 5 %;
the RRTIII [E] is ≥ 1,000 when the whey content is ≤ 5 %.
The uncertainty allowed for the values of RRTIII is ±0,002.
Normally the value of RRTIII [0] deviates little from 1,034. Depending on the condition of the columns, the value may approach 1,000, but it must always be greater.
9.2.Calculation of the percentage of rennet whey powder in the sample:
W = SIII[E] – [1, 3 + (SIII[0] – 0, 9)]
where:
=
the percentage m/m of rennet whey in the sample [E];
=
the relative area of peak III of test sample [E] obtained as in 9.1.2;
=
represents the relative average area of peak III expressed in grams of rennet whey per 100 g determined in non-adulterated skimmed-milk powder of various origins. This figure was obtained experimentally;
=
represents the relative area of peak III which is equal to RIII × AIII [0]. These values are obtained in 9.1.1 and 8.5.3 respectively;
=
represents the correction to be made to the relative average area 1,3 when SIII [0] is not equal to 0,9. Experimentally the relative average area of peak III of the control sample [0] is 0,9.
9.3.Accuracy of the procedure
9.3.1.Repeatability
The difference between the results of two determinations carried out simultaneously or in rapid succession by the same analyst using the same apparatus on identical test material shall not exceed 0,2 % m/m.
9.3.2.Reproducibility
The difference between two single and independent results, obtained in two different laboratories on identical test material shall not exceed 0,4 % m/m.
9.4.Interpretation
9.4.1.Assume the absence of whey if the relative area of peak III, SIII [E] expressed in grams of rennet whey per 100 g of the product is ≤ 2,0 + (SIII[0] – 0,9) where
=
is the maximum value allowed for the relative area of peak III taking into account the relative area of peak III, i.e. 1,3, the uncertainty due to variations in the composition of skimmed-milk powder and the reproducibility of the method (9.3.2),
=
is the correction to be made when the area SIII [0] is different from 0,9 (see point 9.2)
9.4.2.If the relative area of peak III, SIII [E] is > 2,0 + (SIII[0] – 0,9) and the relative area of peak II, SII [E] ≤ 160, determine the rennet whey content as indicated in point 9.2.
9.4.3.
If the relative area of peak III, SIII [E] is > 2,0 + (SIII[0] – 0,9) and the relative area of peak II, SII [E] ≤ 160, determine the total protein content (P %); then examine graphs 1 and 2.
9.4.3.1.The data obtained after analysis of samples of unadulterated skimmed-milk powders with a high total protein content have been assembled in graphs 1 and 2.
The continuous line represents the linear regression, the coefficients of which are calculated by the least squares method.
The dashed straight line fixes the upper limit of the relative area of peak III with a probability of not being exceeded in 90 % of cases.
The equations for the dashed straight lines of graphs 1 and 2 are:
| SIII = 0,376 P % – 10,7 | (graph 1), |
| SIII = 0,0123 SII [E] + 0,93 | (graph 2), |
respectively where:
=
the relative area of peak III calculated either according to total protein content or according to the relative area of peak SII [E],
=
the total protein content expressed as a percentage, by weight,
=
the relative area of sample calculated in point 9.1.2.
These equations are equivalent to the figure of 1,3 mentioned in point 9.2.
The discrepancy (T1 and T2) between the relative area SIII [E] found and the relative area SIII is given by means of the following: T1 = SIII[E] – [(0,376 P% – 10,7) + (SIII[0] – 0,9)]T2 = SIII[E] – [(0,0123 SII[E] + 0,93) + (SIII[0] – 0,9)]
9.4.3.2.
are zero or less, the presence of rennet whey cannot be determined.
exceed zero, rennet whey is present.
The rennet whey content is calculated according to the following formula: W = T20,91
where:
0,91 is the distance on the vertical axis between the continuous and dotted straight lines.
Skimmed-milk powder

Skimmed-milk powder

ANNEX XIII(Article 9)
DETERMINING RENNET WHEY SOLIDS IN SKIMMED-MILK POWDER AND THE MIXTURES REFERRED TO IN REGULATION (EC) No 2799/1999
1.PURPOSE: DETECTING THE ADDITION OF RENNET WHEY SOLIDS TO:
skimmed-milk powder as defined in Article 2 of Regulation (EC) No 2799/1999; and
mixtures as defined in Article 4 of Regulation (EC) No 2799/1999.
3.DEFINITION
The content of rennet whey solids is defined as the percentage by mass as determined by caseinomacropeptide content by the procedure described.
4.PRINCIPLE
Caseinomacropeptides content is determined in accordance with Annex XII. Samples giving positive results are analysed for caseinomacropeptide A by a reversed-phase high-performance liquid chromatography procedure (HPLC procedure). Alternatively samples are directly analysed by the reversed-phase HPLC procedure. Evaluation of the result is obtained by reference to standard samples consisting of skimmed-milk powder with and without a known percentage of whey powder. Results higher than 1 % (m/m) show that rennet whey solids are present.
5.REAGENTS
All reagents must be of recognised analytical grade. The water used must be distilled water or water of at least equivalent purity. Acetonitrile should be of spectroscopic or HPLC quality.
Reagents needed for the procedure are described in Annex XII to this Regulation.
Reagents for reversed phase HPLC.
5.1.Trichloroacetic acid solution
Dissolve 240 g of trichloroacetic acid (CCl3COOH) in water and make up to 1 000 ml. The solution should be clear and colourless.
5.2.Eluents A and B
Eluent A: 150 ml of acetonitrile (CH3CN), 20 ml of isopropanol (CH3CHOHCH3), and 1,00 ml of trifluoroacetic acid (TFA, CF3COOH) are placed in a 1 000 ml volumetric flask. Make up to 1 000 ml with water.
Eluent B: 550 ml of acetonitrile, 20 ml of isopropanol and 1,00 ml of TFA are placed in a 1 000 ml volumetric flask. Make up to 1 000 ml with water. Filter the eluent solution, prior to use, through a membrane filter with a 0,45 μm pore diameter.
5.3.Conservation of the column
After the analyses the column is flushed with eluent B (via a gradient) and subsequently flushed with acetonitrile (via a gradient for 30 minutes). The column is stored in acetonitrile.
5.4.Standard samples
5.4.1.Skimmed-milk powder meeting the requirements for public storage (i e. [0]).
5.4.2.The same skimmed-milk powder adulterated with 5 % (m/m) rennet-type whey powder of standard composition (i.e. [5]).
5.4.3.The same skimmed-milk powder adulterated with 50 % (m/m) rennet-type whey powder of standard composition (i.e. [50])(14).
6.APPARATUS
Apparatus needed for the procedure described is described in Annex XII to this Regulation.
6.1.Analytical balance
6.2.Optional centrifuge capable of attaining a centrifugal force of 2 200 g, fitted with stoppered or capped centrifuge tubes of about 50 ml capacity
6.3.Mechanical shaker
6.4.Magnetic stirrer
6.5.Glass funnels, diameter about 7 cm
6.6.Filter papers, medium filtration, diameter about 12,5 cm
6.7.Glass filtration equipment with 0,45 μm pore diameter membrane filter
6.8.Graduated pipettes, allowing delivery of 10 ml (ISO 648, Class A, or ISO/R 835), or a dispensing system capable of delivering 10,0 ml in two minutes
6.9.Dispensing system capable of delivering 20,0 ml water at ca. 50 °C
6.10.Thermostatic water bath, set at 25 ± 0,5 °C
6.11.
HPLC equipment, consisting of:
6.11.1.Binary gradient pumping system
6.11.2.Injector, hand or automatic, with a 100 μl capacity
6.11.3.Agilent Technologies Zorbax 300 SB-C3 column (length 25 cm, 0,46 cm internal diameter) or an equivalent wide-pore silica based reversed-phase column
6.11.4.Thermostatic column oven, set at 35 ± 1 °C
6.11.5.Variable wavelength UV detector, permitting measurements at 210 nm (if necessary, a higher wavelength up to 220 nm may be used) with a sensitivity of 0,02 Å
6.11.6.Integrator capable of setting the integration to common baseline or valley-to-valley
Note: Operation of the column at room temperature is possible, provided that the room temperature does not fluctuate more than 1 °C, otherwise too much variation in the retention time of CMPA takes place.
7.SAMPLING
7.1.Samples must be taken in accordance with the procedure laid down in International Standard ISO 707. However, Member States may use another method of sampling provided that it complies with the principles of the abovementioned standard
7.2.Store the sample in conditions which preclude any deterioration or change in composition.
8.PROCEDURE
8.1.Preparation of the test sample
Transfer the milk powder into a container with a capacity of about twice the volume of the powder, fitted with an airtight lid. Close the container immediately. Mix the milk powder well by means of repeated inversion of the container
8.2.Test portion
Weigh 2,00 ± 0,001 g of test sample into a centrifuge tube (6.2) or suitable stoppered flask (50 ml).
Note: In the case of mixtures, weigh such an amount of the test sample that the defatted sample portion corresponds to 2,00 g.
8.3.Removal of fat and proteins
8.3.1.Add 20,0 ml of warm water (50 °C) to the test portion. Dissolve the powder by shaking for five minutes or 30 minutes in the case of acid buttermilk using a mechanical shaker (6.3). Place the tube into the water bath (6.10) and allow to equilibrate to 25 °C
8.3.2.Add 10,0 ml of the trichloroacetic acid solution of ca. 25 °C (5.1) constantly over two minutes, while stirring vigorously with the aid of the magnetic stirrer (6.4). Place the tube in a water bath (6.10) and leave for 60 minutes
8.3.3.Centrifuge (6.2) 2 200 g for 10 minutes, or filter through paper (6.6), discarding the first 5 ml of filtrate
8.4.Chromatographic determination
8.4.1.Perform HPLC-analysis as described in Annex XII. If a negative result is obtained, the sample analysed does not contain rennet-whey solids in detectable amounts. If the result is positive, the reversed-phase HPLC procedure described below must be applied. Alternatively the reversed-phase HPLC method can directly be applied. The presence of acid buttermilk powder may give rise to false-positive results using the method described in Annex XII. The reversed-phase HPLC method excludes this possibility
8.4.2.Before the reversed phase HPLC-analysis is carried out, the gradient conditions should be optimised. A retention time of 26 ± 2 minutes for CMPA is optimal for gradient systems having a dead volume of about 6 ml (volume from the point where the solvents come together to the volume of the injector loop, inclusive). Gradient systems having a lower dead volume (e.g. 2 ml) should use 22 minutes as an optimal retention time
Take solutions of the standard samples (5.4) without and with 50 % rennet whey.
Inject 100 μl of supernatant or filtrate (8.3.3) into the HPLC apparatus operating at the scouting gradient conditions given in Table 1.
Table 1
Scouting gradient conditions for optimisation of the chromatography
| Time(min) | Flow(ml/min) | % A | % B | Curve |
|---|---|---|---|---|
| Initial | 1,0 | 90 | 10 | * |
| 27 | 1,0 | 60 | 40 | linear |
| 32 | 1,0 | 10 | 90 | linear |
| 37 | 1,0 | 10 | 90 | linear |
| 42 | 1,0 | 90 | 10 | linear |
Comparison of the two chromatograms should reveal the location of the peak of CMPΑ.
Using the formula given below, the initial solvent composition to be used for the normal gradient (see 8.4.3) can be calculated % B = 10 – 2,5 + (13,5 + (RTcmpA – 26) / 6) × 30 / 27 % B = 7,5 + (13,5 + (RTcmpA – 26) / 6) × 1,11
Where:
:
retention time of CMPΑ in the scouting gradient
:
the initial % B of the scouting gradient
:
% B at midpoint minus % B at initial in the normal gradient
:
midpoint time of the scouting gradient
:
required retention time of CMPΑ
:
ratio of slopes of the scouting and normal gradient
:
% B at initial minus % B at 27 minutes in the scouting gradient
:
run-time of the scouting gradient.
8.4.3.Take solutions of the test samples
Inject 100 μl of accurately measured supernatant or filtrate (8.3.3) into the HPLC apparatus operating at a flow rate of 1,0 ml of eluent solution (5.2) per minute.
The composition of the eluent of the start of the analysis is obtained from 8.4.2. It is normally close to A:B = 76:24 (5.2). Immediately after the injection a linear gradient is started, which results in a 5 % higher percentage of B after 27 minutes. Subsequently a linear gradient is started, which brings the eluent composition to 90 % B in five minutes. This composition is maintained for five minutes, after which the composition is changed, via a linear gradient in five minutes to the initial composition. Depending on the internal volume of the pumping system, the next injection can be made 15 minutes after reaching the initial conditions.
Note 1. The retention time of the CMPA should be 26 ± 2 minutes. This can be achieved by varying the initial and end conditions of the first gradient. However, the difference in the % B for the initial and end conditions of the first gradient must remain 5 % B.
Note 2. The eluents should be degassed sufficiently and should also remain degassed. This is essential for proper functioning of the gradient pumping system. The standard deviation for the retention time of the CMPA peak should be smaller than 0,1 minutes (n = 10).
Note 3. Every five samples the reference sample (5) should be injected and used to calculate a new response factor R. (9.1.1).
8.4.4.The results of the chromatographic analysis of the test sample (E) are obtained in the form of a chromatogram in which the CMPA peak is identified by its retention time of about 26 minutes
The integrator (6.11.6) automatically calculates the peak height H of the CMPA peak. The baseline location should be checked in every chromatogram. The analysis or the integration should be repeated if the baseline was incorrectly located.
Note: If the CMPA peak is sufficiently separated from other peaks valley-to-valley baseline allocation should be used, otherwise use dropping perpendiculars to a common baseline, which should have starting point close to the CMPA peak (thus not at t = 0 min!). Use for the standard and the samples the same type integration type and check in case of common baseline its consistency for the samples and the standard.
It is essential to examine the appearance of each chromatogram prior to quantitative interpretation, in order to detect any abnormalities due either to malfunctioning of the apparatus or the column, or to the origin and nature of the sample analysed. If in doubt, repeat the analysis.
8.5.Calibration
8.5.1.Apply exactly the procedure described from point 8.2 to point 8.4.4 to the standard samples (5.4.1 to 5.4.2). Use freshly prepared solutions, because CMP degrades in an 8 % trichloroacetic acid environment at room temperature. At 4 °C the solution remains stable for 24 hours. In the case of long series of analyses the use of a cooled sample tray in the automatic injector is desirable
Note: 8.4.2. may be omitted if the % B at initial conditions is known from previous analyses.
The chromatogram of the reference sample [5] should be analogous to Figure. 1. In this figure the CMPA peak is preceded by two small peaks. It is essential to obtain a similar separation.
8.5.2.Prior to chromatographic determination of the samples inject 100 μl of the standard sample without rennet whey [0] (5.4.1)
The chromatogram should not show a peak at the retention time of the CMPA peak.
8.5.3.Determine the response factors R by injecting the same volume of filtrate (8.5.1) as used for the samples.
9.EXPRESSION OF RESULTS
9.1.Method of calculation and formulae
9.1.1.Calculation of the response factor R:
CMPA peak: R = W/H
Where:
=
the response factor of the CMPA peak
=
the height of the CMPA peak
=
the quantity of whey in the standard sample [5].
9.2.Calculation of the percentage of rennet whey powder in the sample
W(E) = R × H(E)
Where:
=
the percentage (m/m) of rennet whey in the sample (E).
=
the response factor of the CMPA peak (9.1.1)
=
the height of the CMPA peak of the sample (E)
If W(E) is greater than 1 % and the difference between the retention time and that of the standard sample [5] is smaller than 0,2 minutes then rennet whey solids are present.
9.3.Accuracy of the procedure
9.3.1.Repeatability
The difference between the results of two determinations carried out simultaneously or in rapid succession by the same analyst using the same apparatus on identical test material shall not exceed 0,2 % m/m.
9.3.2.Reproducibility
Not determined.
9.3.3.Linearity
From 0 to 16 % of rennet whey a linear relationship should be obtained with a coefficient of correlation > 0,99.
9.4.Interpretation
The 1 % limit is fixed in agreement with the provisions of points 9.2 and 9.4.1 of Annex XIX to Regulation (EEC) No 214/2001, which includes the uncertainty due to reproducibility.
Table 1Ni –4,6 standard

ANNEX XIV(Article 10)
SKIMMED-MILK POWDER: QUANTITATIVE DETERMINATION OF PHOSPHATIDYLSERINE AND PHOSPHATIDYLETHANOLAMINE
Method: reversed-phase HPLC
1.PURPOSE AND FIELD OF APPLICATION
The method describes a procedure for the quantitative determination of phosphatidylserine (PS) and phosphatidylethanolamine (PE) in skimmed-milk powder (SMP) and is suitable for detecting buttermilk solids in SMP.
2.DEFINITION
PS + PE content: the mass fraction of substance determined using the procedure here specified. The result is expressed as milligrams of phosphatidylethanolamine dipalmitoyl (PEDP) per 100 g powder.
3.PRINCIPLE OF THE METHOD
Extraction of aminophospholipids by methanol from reconstituted milk powder. Determination of PS and PE as o-phthaldialdehyde (OPA) derivatives by reversed-phase (RP) HPLC and fluorescence detection. Quantification of PS and PE content in the test sample by reference to a standard sample containing a known amount of PEDP.
4.REAGENTS
All reagents must be of recognised analytical grade. Water must be distilled or water or water of at least equivalent purity, unless otherwise specified.
4.1.Standard material: PEDP, at least 99 % pure
Note: Standard material must be stored at -18 °C.
4.2.Reagents for standard sample and test sample preparation
4.2.1.HPLC-grade methanol
4.2.2.HPLC-grade chloroform
4.2.3.Tryptamine-monohydrochloride
4.3.Reagents for o-phthaldialdehyde derivatisation
4.3.1.Sodium hydroxide, 12 M water solution
4.3.2.Boric acid, 0,4 M water solution adjusted to pH 10,0 with sodium hydroxide (4.3.1)
4.3.3.2-mercaptoethanol
4.3.4.o-phthaldialdehyde (OPA)
4.4.HPLC elution solvents
4.4.1.Elution solvents must be prepared using HPLC-grade reagents.
4.4.2.HPLC-grade water
4.4.3.Methanol of fluorimetric tested purity
4.4.4.Tetrahydrofuran
4.4.5.Sodium dihydrogen phosphate
4.4.6.Sodium acetate
4.4.7.Acetic acid.
5.APPARATUS
5.1.Analytical balance, capable of weighing to the nearest 1 mg, with a readability of 0,1 mg
5.2.Beakers, 25 and 100 ml capacity
5.3.Pipettes, capable of delivering 1 and 10 ml
5.4.Magnetic stirrer
5.5.Graduated pipettes, capable of delivering 0,2, 0,5 and 5 ml
5.6.Volumetric flasks, 10, 50 and 100 ml capacity
5.7.Syringes, 20 and 100 μl capacity
5.8.Ultrasonic bath
5.9.Centrifuge, capable of operating at 27 000 × g
5.10.Glass vials, about 5 ml capacity
5.11.Graduated cylinder, 25 ml capacity
5.12.pH-meter, accurate to 0,1 pH units
5.13.
HPLC equipment
5.13.1.Gradient pumping system, capable of operating at 1,0 ml/min at 200 bar
5.13.2.Autosampler with derivatisation capability
5.13.3.Column heater, capable of maintaining the column at 30 °C ± 1 °C
5.13.4.Fluorescence detector, capable of operating at 330 nm excitation wavelength and 440 nm emission wavelength
5.13.5.Integrator or data processing software capable of peak area measurement
5.13.6.A Lichrosphere — 100 column (250 × 4,6 mm) or an equivalent column packed with octadecylsilane (C 18), 5 μm particle size.
6.SAMPLING
Sampling must be carried out in accordance with ISO Standard 707.
7.PROCEDURE
7.1.Preparation of the internal standard solution
7.1.1.Weigh 30,0 ± 0,1 mg of tryptamine-monohydrochloride (4.2.3) into a 100 ml volumetric flask (5.6) and make up to the mark with methanol (4.2.1)
7.1.2.Pipette 1 ml (5.3) of this solution into a 10 ml volumetric flask (5.6) and make up to the mark with methanol (4.2.1) in order to obtain a 0,15 mM tryptamine concentration
7.2.Preparation of the test sample solution
7.2.1.Weigh 1,000 ± 0,001 g of the SMP sample into a 25 ml beaker (5.2). Add 10 ml of distilled water at 40 °C ± 1 °C by a pipette (5.3) and stir with a magnetic stirrer (5.4) for 30 minutes in order to dissolve any lumps
7.2.2.Pipette 0,2 ml (5.5) of the reconstituted milk into a 10 ml volumetric flask (5.6), add 100 μl of the 0,15 mM tryptamine solution (7.1) using a syringe (5.7) and make up to the volume with methanol (4.2.1). Mix carefully by inversion and sonicate (5.8) for 15 min
7.2.3.Centrifuge (5.9) at 27 000 g × g for 10 minutes and collect the supernatant in a glass vial (5.10)
Note: Test sample solution should be stored at 4 °C until the HPLC analysis is performed.
7.3.Preparation of the external standard solution
7.3.1.Weigh 55,4 mg PEDP (4.1) into a 50 ml volumetric flask (5.6) and add about 25 ml of chloroform (4.2.2) using a graduated cylinder (5.11). Heat the stoppered flask to 50 °C ± 1 °C and mix carefully till the PEDP dissolves. Cool the flask to 20 °C, make up to the volume with methanol (4.2.1) and mix by inversion
7.3.2.Pipette 1 ml (5.3) of this solution into a 100 ml volumetric flask (5.6) and make up to the volume with methanol (4.2.1). Pipette 1 ml (5.3) of this solution into a 10 ml volumetric flask (5.6), add 100 μl (5.7) of 0,15 mM tryptamine solution (7.1) and make up to the volume with methanol (4.2.1). Mix by inversion
Note: Reference sample solution should be stored at 4 °C until the HPLC analysis is performed.
7.4.Preparation of the derivatising reagent
Weigh 25,0 ± 0,1 mg of OPA (4.3.4) into a 10 ml volumetric flask (5.6), add 0,5 ml (5.5) of methanol (4.2.1) and mix carefully to dissolve the OPA. Make up to the mark with boric acid solution (4.3.2) and add 20 μl of 2-mercaptoethanol (4.3.3) by syringe (5.7).
Note: The derivatising reagent should be stored at 4 °C in a brown glass vial and is stable for one week.
7.5.Determination by HPLC
7.5.1.Elution solvents (4.4)
Solvent A: Solution of 0,3 mM sodium dihydrogen phosphate and 3 mM sodium acetate solution (adjusted to pH 6,5 ± 0,1 with acetic acid): methanol: tetrahydrofuran = 558:440:2 (v/v/v)
Solvent B: methanol
7.5.2.Suggested eluting gradient:
| Time(min) | Solvent A(%) | Solvent B(%) | Flow rate(ml/min) |
|---|---|---|---|
| Initial | 40 | 60 | 0 |
| 0,1 | 40 | 60 | 0,1 |
| 5,0 | 40 | 60 | 0,1 |
| 6,0 | 40 | 60 | 1,0 |
| 6,5 | 40 | 60 | 1,0 |
| 9,0 | 36 | 64 | 1,0 |
| 10,0 | 20 | 80 | 1,0 |
| 11,5 | 16 | 84 | 1,0 |
| 12,0 | 16 | 84 | 1,0 |
| 16,0 | 10 | 90 | 1,0 |
| 19,0 | 0 | 100 | 1,0 |
| 20,0 | 0 | 100 | 1,0 |
| 21,0 | 40 | 60 | 1,0 |
| 29,0 | 40 | 60 | 1,0 |
| 30,0 | 40 | 60 | 0 |
Note: The eluting gradient may require slight modification in order to achieve the resolution shown in figure 1.
Column temperature: 30 °C.
7.5.3. Injection volume: 50 μl derivatising reagent and 50 μl sample solution
7.5.4.Column equilibration
Starting up the system on a daily basis, flush the column with 100 % solvent B for 15 minutes, then set at A:B = 40:60 and equilibrate at 1 ml/min for 15 minutes. Perform a blank run by injecting methanol (4.2.1).
Note: Before long-term storage flush the column with methanol: chloroform = 80:20 (v/v) for 30 minutes.
7.5.5. Determine the PS + PE content in the test sample
7.5.6.Perform the sequence of the chromatographic analyses keeping constant the run-to-run time in order to obtain constant retention times. Inject the external standard solution (7.3) every 5-10 test sample solutions in order to calculate the response factor 0
Note: The column must be cleaned by flushing with 100 % solvent B (7.5.1) for at least 30 minutes every 20-25 runs.
7.6.Integration mode
7.6.1.PEDP peak
PEDP is eluted as a single peak. Determine the peak area by valley-to-valley integration.
7.6.2.Tryptamine peak
Tryptamine is eluted as a single peak (Figure 1). Determine the peak area by valley-to-valley integration.
7.6.3.PS and PE peaks groups
Under the described conditions (Figure 1), PS elutes as two main partially unresolved peaks preceded by a minor peak. PE elutes as three main partially unresolved peaks. Determine the whole area of each peak cluster setting the baseline as reported in Figure 1.
8.CALCULATION AND EXPRESSION OF RESULTS
PS and PE content in the test sample shall be calculated as follows: C = 55,36 × ((A2)/(A1)) × ((T1)/(T2))
where:
=
PS or PE content (mg/100 g powder) in the test sample
=
PEDP peak area of the standard sample solution (7.3)
=
PS or PE peak area of the test sample solution (7.2)
=
Tryptamine peak area of the standard sample solution (7.3)
=
Tryptamine peak area of the test sample solution (7.2).
9.ACCURACY OF THE METHOD
Note: The values for repeatability were calculated according to the IDF International Standard(15). The provisional reproducibility limit was calculated according to the procedure defined in Annex III(b) hereto.
9.1.Repeatability
The relative standard derivation of the repeatability, which expresses the variability of independent analytical results obtained by the same operator using the same apparatus under the same conditions on the same test sample and in a short interval of time, should not exceed 2 % relative. If two determinations are obtained under these conditions, the relative difference between the two results should not be greater than 6 % of the arithmetic mean of the results.
9.2.Reproducibility
If two determinations are obtained by operators in different laboratories using different apparatus under different conditions for the analysis on the same test sample, the relative difference between the two results should not be greater than 11 % of the arithmetic mean of the results.
10.REFERENCES
10.1. Resmini P., Pellegrino L., Hogenboom J.A., Sadini V., Rampilli M., ‘Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids’. Sci. Tecn. Latt.-Cas., 39,395 (1988).
Figure 1
HPLC pattern of OPA-derivatives of phosphatidylserine (PS) and phosphatidylethanolamine (PE) in methanol extract of reconstituted skim-milk powder. Integration mode for the peaks of PS, PE and tryptamine (internal standard) is reported

ANNEX XV(Article 11)
DETECTION OF ANTIMICROBIAL RESIDUES IN SKIMMED MILK POWDER
A microbial inhibitor screening test using Geobacillus stearothermophilus var. calidolactis ATCC 10149 (identical to strain C953) as test micro-organism and being sufficiently sensitive to detect 4 μg benzylpenicillin per kg milk and 100 μg sulfadimidine per kg milk shall be used. Commercial tests kits are available and can be used if they have the required sensitivity for benzylpencillin and sulfadimidine.
For the test, reconstituted skimmed milk powder (1 g powder 9 ml aqua dest) is used. The test is carried out as described in ISO/TS 26844:2006 Milk and milk products — Determination of antimicrobial residues — Tube diffusion test IDF — Bulletin No 258/1991, section 1, Chapter 2, or according to the instructions of the test kit manufacturer(16).
Positive results are to be interpreted as follows:
The presence of β-lactams can be confirmed by repeating the test adding penicillinase to the test system(17):
Negative result: Inhibiting substance is a β-lactam antibiotic.
Positive result remains: Inhibiting substance cannot be identified by this procedure, continue with 2.
The presence of sulfonamides can be confirmed by repeating the test adding p-amino benzoic acid to the test system:
Negative result: Inhibiting substance is a sulfonamide.
Positive result remains: Inhibiting substance cannot be identified by this procedure, continue with 3.
The presence of a combination of a β-lactam and sulfonamide can be confirmed by repeating the test adding pencillinase + p-amino benzoic acid to the test system:
Negative result: Inhibiting substances are a β-lactam antibiotic and a sulfonamide.
Positive result: Inhibiting substance cannot be identified by this procedure.
ANNEX XVI(Article 12)
QUANTITATIVE DETERMINATION OF SKIMMED-MILK POWDER IN COMPOUND FEEDINGSTUFFS BY ENZYMATIC COAGULATION OF PARA-CASEIN
1.PURPOSE
Quantitative determination of skimmed-milk powder in compound feedingstuffs by enzymatic coagulation of para-casein.
2.SCOPE
This method applies to compound feedingstuffs containing at least 10 % skimmed-milk powder; large quantities of buttermilk and/or of certain non-milk proteins may lead to interferences.
3.PRINCIPLE OF THE METHOD
3.1.Dissolving of casein contained in the compound feedingstuff by extraction with sodium citrate solution
3.2.Adjustment of the calcium ion concentration to the required level to precipitate para-casein by addition of rennet
3.3.The nitrogen content of the para-casein precipitate is determined by the Kjeldahl method as described by standard ISO 8968-2:2001/IDF 20-2:2001; the quantity of skimmed-milk powder is calculated on the basis of a minimum casein content of 27,5 % (see 8.1).
4.REAGENTS
The reagents used must be of analytical grade. The water used must be distilled water or water of equivalent purity. With the exception of the rennet (4.5), all the reagents and solutions must be free of nitrogenous substances.
4.1.Trisodium citrate, dihydrate (1 % w/v solution)
4.2.Calcium chloride (about 5 M solution)
Dissolve 75g of CaCl2 · 2 H2O in 100 ml of distilled water by shaking (draw attention to exothermic reaction). Leave overnight and then filter the solution. Store the solution in a refrigerator.
4.3.0,1 N sodium hydroxide
4.4.0,1 N hydrochloric acid
4.5.Liquid calf rennet (strength about 100 IMCU/ml according to standard ISO 11815/IDF 157). Store in a refrigerator at 4 to 6 °C
4.6.Reagents for the quantitative determination of nitrogen according to the Kjeldahl method as described by ISO 8968-2:2001/IDF 20-2:2001.
5.APPARATUS
Common laboratory apparatus, including:
5.1.Mortar or homogeniser
5.2.Analytical balance capable of weighing to the nearest 1 mg, with a readability of 0,1 mg.
5.3.Bench-top centrifuge (500 g or 2 000 to 3 000 rpm) with 50 ml tubes and 2 000 g
5.4.Magnetic stirrer with (10 to 15 mm) followers
5.5.150 to 200 ml beakers
5.6.250 and 500 ml flasks
5.7.Glass funnels of 60 to 80 mm diameter
5.8.Fast-filtering ashless filters of diameter 150 mm (Whatman No 41 or equivalent)
5.9.Pipettes of various nominal volume
5.10.Thermostatically controlled water bath at 37 °C ± 1 °C
5.11.pH meter, accurate to 0,1 units
5.12.Thermometers, accurate to 1 °C.
6.PROCEDURE
6.1.Preparation of the sample
Grind in the mortar or homogenise in the mill 10 to 20 g of the sample to obtain a homogeneous mixture.
6.2.Dissolving of milk powder and separation of the insoluble residue
6.2.1.Weigh 1,000 ± 0,002 g of well-homogenised compound feedingstuff (6.1) directly into a 50 ml centrifuge tube. Add 30 ml of trisodium citrate solution (4.1) previously heated to 45 °C ± 2 °C. Mix with the aid of the magnetic stirrer for at least five minutes or by vigorous manual shaking.
6.2.2.Centrifuge at 500 g (2 000 to 3 000 rpm) for 10 minutes and decant the clear aqueous supernatant into a 150 to 200 ml beaker, taking care that no loose material on the bottom goes over.
6.2.3.Carry out two further extractions on the residue, according to the same procedure, adding the extracts to the first one.
6.2.4.If a layer of oil forms at the surface, cool in the refrigerator until the fat solidifies and remove the solid layer with a spatula.
6.3.Coagulation of casein with the enzymes of rennet
6.3.1.While stirring continuously, add dropwise 2 ml of calcium chloride (4.2) to the total aqueous extract (about 100 ml). Adjust the pH to 6,4-6,5 with solutions of NaOH (4.3) or HCl (4.4). Place in the thermostatically-controlled water bath at 37 °C ± 1 °C for 15 to 20 minutes to obtain saline balance. It becomes more evident by the formation of a light turbidity.
6.3.2.Transfer the liquid into one centrifuge tubes and centrifuge at 2 000 g for 10 minutes in order to remove the precipitated material. Transfer the supernatant, without washing the sediment, into one another centrifuge tubes.
6.3.3.Bring the temperature of the supernatant back to 37 °C ± 1 °C. While stirring the extract, add, dropwise, 0,5 ml of the liquid rennet (4.5). Coagulation occurs in two minutes.
6.3.4.Return the sample to the water bath and leave at a temperature of 37 °C ± 1 °C for 15 minutes. Remove the sample from the bath and break the coagulum by stirring. Centrifuge at 2 000 g for 10 minutes. Filter the supernatant through a suitable filter paper (5.8) and retain the filter paper. Wash the precipitate in the centrifuge tube with 50 ml of water at approximately 35 °C by stirring the precipitate.
Centrifuge again at 2 000 g for 10 minutes. Filter the supernatant through the filter paper retained previously.
6.4.Determination of casein nitrogen
6.4.1.After washing, transfer quantitatively the precipitate to the filter paper retained from 6.3.4 using distilled water. Transfer the dried filter paper to the Kjeldahl flask. Determine the nitrogen by the Kjeldahl method as described by standard ISO 8968-2:2001/IDF 20-2:2001.
7.BLANK TEST
7.1.A blank test shall be made regularly by submitting to mineralisation by the Kjeldahl method as described at standard ISO 8968-2:2001/IDF 20-2:2001. An ashless filter paper (5.8) moistened with a mixture of 90 ml (4.1) sodium citrate solution, 2 ml solution of calcium chloride (4.2), 0,5 ml of liquid rennet (4.5), and washed with 3 × 15 ml of distilled water
7.2.The volume of acid used for the blank test must be subtracted from the volume of acid (4.4) used for titration of the sample.
8.EXPRESSION OF RESULTS
8.1.The percentage of skimmed-milk powder in the compound feedingstuff is calculated by the following formula:

where:
N is the percentage of para-casein nitrogen;
27,5 is the factor for converting determined casein into the percentage of skimmed-milk powder;
2,81 and 0,908 are correction factors obtained from regression analysis.
9.ACCURACY OF THE METHOD
9.1.Repeatability
In at least 95 % of the cases studied, duplicate analysis of the same sample by the same operator in the same laboratory must give differences in the results equivalent to no greater than 2,3 g of skimmed-milk powder in 100 g of compound feedingstuff.
9.2.Reproducibility
In at least 95 % of the cases studied, the same sample analysed by two laboratories, must give differences in the results no greater than 6,5 g of skimmed-milk powder in 100 g of compound feedingstuff.
10.OBSERVATIONS
10.1.The addition of large percentages of certain non-milk proteins and especially of soya proteins, when heated together with skimmed-milk powder, may lead to too high results due to co-precipitation with the para-casein of milk.
10.2.Addition of buttermilk may lead to somewhat low figures, due to the fact that only the non-fat portion is determined. Addition of certain acid buttermilk may give considerably low figures, due to incomplete dissolving in the citrate solution.
10.3.Lecithin additions of 0,5 % or more may also lead to low results.
10.4.Incorporation of high-heat skimmed-milk powder may lead to too high figures due to the co-precipitation of certain whey proteins with the para-casein of milk.
ANNEX XVII(Article 13)
DETECTION OF STARCH IN SKIMMED-MILK POWDER, DENATURED MILK POWDER AND COMPOUND FEEDINGSTUFFS
1.SCOPE
This method is for the detection of starch which is issued as a tracer in denatured milk powders.
Limit of detection of the method is approximately 0,05 g of starch per 100 g of sample.
2.PRINCIPLE
The reaction is based on the one used in iodometry:
fixation by the colloids of the free iodine in aqueous solution,
absorption by the starch micelles and by colour formation.
3.REAGENTS
3.1.Iodine solution
Iodine: 1,0 g,
Potassium iodine: 2,0 g,
Distilled water: 100 ml,
Dissolve 1,0 g of iodine and 2,0 g of potassium iodine in water in a 100 ml one-mark volumetric flask. Dilute to the 100 ml mark with water and mix.
4.APPARATUS
4.1.Analytical balance
4.2.Boiling water bath
4.3.Test tubes, 25 mm × 200 mm.
5.PROCEDURE
Weigh 1,0 g of the sample to the nearest 0,1g and transfer it into the test tube (4.3).
Add 20 ml of distilled water and shake in order to disperse the sample.
Place in the boiling water bath (4.2) and leave for 5 minutes.
Remove from the bath and cool to room temperature.
Add 0,5 ml of the iodine solution (3.1), shake and observe the resulting colour.
6.EXPRESSION OF RESULTS
A blue colouration indicates the presence of native starch in the sample.
When the sample contains modified starch the colour may not be blue.
7.REMARKS
The colour, the intensity of the colour and the microscopic appearance of the starch, will vary depending on the origin of native starch (e.g. maize or potato) and the type of modified starch present in the sample.
In the presence of modified starches the colour produced turns violet, red or brown, according to the degree of modification of the crystalline structure of native starch.
ANNEX XVIII(Article 14)
DETERMINATION OF MOISTURE CONTENT IN DRIED CREAM
1.SCOPE
This Annex specifies a method for the determination of the moisture content of dried cream.
2.TERMS AND DEFINITIONS
For the purpose of this Annex, the following definition applies.
Moisture content: the loss of mass determined by the procedure specified in this International Standard.
It is expressed as a percentage by mass.
3.PRINCIPLE
Drying of a test portion at 102 ± 2 °C to constant mass and weighing to determine the loss of mass.
4.APPARATUS
Usual laboratory equipment and, in particular, the following:
4.1.Analytical balance, capable of weighing to the nearest 1 mg, with a readability of 0,1 mg.
4.2.Drying oven, well ventilated, capable of being maintained thermostatically at 102 ± 2 °C throughout the working space.
4.3.Desiccator, provided with freshly dried silica gel with hygrometric indicator or another effective desiccant.
4.4.Flat-bottomed dishes, depth of approximately 25 mm, diameter of approximately 50 mm, and made of appropriate material (for example glass, stainless steel, nickel or aluminium), provided with well-fitting, readily removable lids.
4.5.Bottles, with tight-fitting stoppers, for mixing the laboratory samples.
5.SAMPLING
It is important that the laboratory receive a test sample which is truly representative and has not been damaged or changed during transport or storage.
Sampling is not part of the method specified in this International Standard. A recommended sampling method is given in ISO 707|IDF 50.
Store the sample in such a way that deterioration and change in composition are prevented.
6.PREPARATION OF TEST SAMPLE
Thoroughly mix the test sample by repeatedly shaking and inverting the container (if necessary, after having transferred all test samples to an air-tight container of sufficient capacity to allow this operation to be carried out).
In case complete homogeneity is not attained by this procedure, take the test portions (for two single determinations) from the prepared test sample at two points as far apart as possible.
7.PROCEDURE
7.1.Preparation of the dish
7.1.1.Heat an uncovered dish and its lid (4.4) in the oven (4.2), set at 102 ± 2 °C, for at least 1 h.
7.1.2.Place the lid on the dish, transfer the covered dish to the desiccator (4.3), allow to cool to the temperature of the balance room and weigh to the nearest 1 mg recording the weight to 0,1 mg.
7.2.Test portion
Transfer approximately 1 to 3 g of the prepared test sample (6) into the dish, cover with the lid and weigh to the nearest 1 mg recording the weight to 0,1 mg.
7.3.Determination
7.3.1.Uncover the dish and place it with its lid in the oven (4.2), set at 102 ± 2 °C, for 2 h.
7.3.2.Replace the lid, transfer the covered dish to the desiccator, allow to cool to the temperature of the balance room and weigh to the nearest 1 mg recording the weight to 0,1 mg.
7.3.3.Uncover the dish and heat it again, with its lid, in the oven for 1 h. Then repeat operation 7.3.2.
7.3.4.Repeat the heating and the weighing procedure until the mass decreases by 1 mg or less, or increases between two successive weightings.
Take for the calculation the lowest mass recorded.
8.CALCULATION AND EXPRESSION OF RESULTS
8.1.Calculation
The moisture content, expressed in g/100g, is equal to:

where:
m0 is the mass, in grams, of the dish and the lid (7.1.2);
m1 is the mass, in grams, of the dish, the lid and the test portion before drying (7.2);
m2 is the mass, in grams, of the dish, the lid and the test portion after drying (7.3.4).
Report the result to two decimal places.
9.PRECISION
Note: The values for repeatability and reproducibility were derived from the results of an interlaboratory test (see Steiger, G. Bulletin of IDF No 285/1993, p. 21-28) carried out in accordance with IDF Standard 135B:1991. Milk and milk products — Precision characteristics of analytical methods — Outline of collaborative study procedure.
9.1.Repeatability
The absolute difference between two independent single test results, obtained using the same method on identical test material in the same laboratory by the same operator using the same equipment within a short interval of time, will in not more than 5 % of cases be greater than 0,20 g of moisture per 100 g of product.
9.2.Reproducibility
The absolute difference between two independent single test results, obtained using the same method on identical test material in different laboratories by different operators using different equipment will in not more than 5 % of cases be greater than 0,40 g of moisture per 100 g of product.
10.TEST REPORT
The test report shall specify
all information necessary for the complete identification of the sample,
the sampling method used, if known,
the test method used with reference to this International Standard,
all operating details not specified in this International Standard, or regarded as optional, together with details of any incidents which may have influenced the test result(s).
The test result(s) obtained, and if the repeatability has been checked, the final quoted result obtained.
ANNEX XIX(Article 15)
DETERMINATION OF MOISTURE IN ACID BUTTERMILK POWDER
1.SCOPE
To determine the moisture content of acid buttermilk powder originally intended for animal feedingstuffs.
2.PRINCIPLE
The sample is dried under vacuum. The loss of mass is determined by weighing.
3.APPARATUS
3.1.Analytical balance, capable of weighing to the nearest 1 mg, with a readability of 0,1 mg
3.2.Dishes of non-corrodible metal or of glass with lids ensuring airtight closure; working surface allowing the test sample to be spread at about 0,3 g/cm2
3.3.Adjustable electrically heated vacuum oven fitted with an oil pump and a mechanism for introducing hot air dried through a tower containing for example calcium oxide or calcium sulphate (containing moisture indicator)
3.4.Desiccator with an efficient drying agent
3.5.Drying oven ventilated, thermostatically controlled, at 102 ± 2 °C.
4.PROCEDURE
Heat a dish (3.2) with its lid in the oven (3.5) for at least one hour. Place the lid on the container, immediately transfer to a desiccator (3.4) allow to cool to room temperature and weigh to the nearest 1 mg, recording the mass to 0,1 mg.
Uncover the dish and transfer about 5g of sample into the dish and weigh to the nearest 1 mg, recording the mass to 0,1 mg. Place the dish with its lid in the vacuum oven (3.3) preheated to 83 °C. To prevent the oven temperature from falling unduly, introduce the dish as rapidly as possible.
Bring the pressure up to 100 Torr (13,3 kPa) and leave to dry to constant weight (approximately 4 hours) at this pressure in a current of hot dry air.
Reckon drying time from the moment when the oven returns to 83 °C. Carefully bring the oven back to atmospheric pressure. Open the oven, place the lid on the dish immediately, remove the dish from the oven, leave to cool for 30 to 45 minutes in a desiccator (3.4) and weigh to the nearest 1 mg recording the mass to 0,1 mg. Dry for an additional 30 minutes in the vacuum oven (3.3) at 83 °C and reweigh. Repeat the heating and weighing procedure until the mass of the dish with its lid decreases by 1 mg or less, or increases between two successive weighings. Take for the calculation the lowest mass recorded.
5.CALCULATION
% Moisture = (m1 – m2) / (m1 – m0) × 100 %
Where:
is the mass of the dish and the lid;
is the mass of the dish, the lid and the test portion before drying;
is the mass of the dish, the lid and the test portion after drying.
Record the result to the nearest 0,1 g / 100 g.
6.PRECISION
6.1.Repeatability limit
The absolute difference between two independent single test results, obtained using the same method on identical test material in the same laboratory by the same operator using the same equipment within a short interval of time, will in not more than 5 % of the cases be greater that 0,4 g water/ 100 g buttermilk powder.
6.2.Reproducibility limit
The absolute difference between two independent single test results, obtained using the same method on identical test material in different laboratories by different operators using different equipment will in not more than 5 % of cases be greater than 0,6 g water/100 g acid buttermilk powder.
6.3.Source of precision data
The precision data were determined from an experiment conducted in 1995 involving eight laboratories and 12 samples (6 blind duplicates).
ANNEX XX(Article 16)
REFERENCE METHOD FOR THE DETERMINATION OF MILK FAT PURITY BY GAS CHROMATOGRAPHIC ANALYSIS OF TRIGLYCERDIES — REVISION 2
1.SCOPE AND FIELD OF APPLICATION
This standard specifies a reference method for the determination of milk fat purity using gas chromatographic analysis of triglycerides. Both vegetable fats and animal fats such as beef tallow and lard can be detected.
Using defined triglyceride equations, the integrity of milk fat is determined. Basically, the method applies to bulk bovine milk, or products made thereof, irrespective of feeding, breed or lactation conditions. Only exceptionally high feeding of pure vegetable oils, such as rapeseed oil, can result in a false positive result. Milk products obtained from individual cows can also cause a false positive result.
In particular, the method is applicable to fat extracted from milk products purporting to contain pure milk fat with unchanged composition, such as butter, cream, milk, and milk powder. Technological treatment of milk fat such as removal of cholesterol or fractionation can cause a false positive result. This is also true for milk fat obtained from skim milk or buttermilk. The method is not always applicable to fat extracted from cheese, because the ripening process can affect the fat composition so strongly that a false positive result is obtained.
Note 1: Butyric (n-butanoic) acid (C4) occurs exclusively in milk fat and enables quantitative estimations of low to moderate amounts of milk fat in vegetable and animal fats to be made. Due to the large variation in C4 in the approximate percentage mass fraction range 3,1 % and 3,8 %, however, it is difficult to provide qualitative and quantitative information for foreign fat to pure milk fat mass fractions of up to 20 % [1].
Note 2: Practically, quantitative results cannot be derived from the sterol content of vegetable fats, because they depend on the production and processing conditions. Even more, the qualitative determination of foreign fat using sterols is ambiguous.
2.DEFINITION
Milk fat purity: absence of vegetable and animal fats determined by the procedure specified in this standard.
Note: The purity is determined using S-values, which are calculated from the triglyceride composition. The triglyceride mass fractions are expressed as a percentage.
3.PRINCIPLE OF THE METHOD
The fat extracted from milk or milk products is analysed by gas chromatography using a packed or a short capillary column to determine the triglycerides (TGs), separated by total carbon numbers. By inserting the mass fraction, expressed as a percentage, of fat molecules of different sizes (C24 to C54, using even C numbers only) into suitable TG equations, S-values are calculated. If the S-values exceed the limits established with pure milk fat, the presence of foreign fat is detected.
Note 1: The suitability and equivalence of both packed and capillary columns have been demonstrated previously [2-4].
Note 2: The S-value is a sum of TG mass fractions being multiplied by defined factors respectively.
4.REAGENTS
All reagents shall be of recognized analytical grade.
4.1.Carrier gas: nitrogen or, alternatively, helium or hydrogen, all with a purity of at least 99,995 %.
4.2.
Fat standards, for standardizing a milk fat standard according to Clause 7.3.3.
4.2.1.Triglyceride standards, saturated, suited products are available commercially.
4.2.2.Cholesterol standard.
4.3.Methanol (CH3OH), free of water.
4.4.n-Hexane (CH3(CH2)4CH3).
4.5.n-Heptane (CH3(CH2)5CH3).
4.6.Other gases: hydrogen, purity at least 99,995 %, free from organic impurities (CnHm < 1 µl/l); synthetic air, free from organic impurities (CnHm < 1 µl/l).
4.7.Anhydrous sodium sulfate (Na2SO4).
5.APPARATUS
Usual laboratory equipment and, in particular, the following:
5.1.High-temperature gas chromatograph
The high-temperature gas chromatograph shall be suited for temperatures of at least 400 °C and be equipped with a flame ionization detector (FID). Septa used in the injector shall withstand high temperatures and exhibit a very low degree of ‘bleeding’. For capillary GC, use an on-column injector. Always use graphite seals to connect the column as well as injector and/or detector inserts (where applicable).
5.2.Chromatography column
5.2.1.Packed column
Use a glass column of internal diameter 2 mm and length 500 mm, packed with a stationary phase of 3 % OV-1 on 125 µm to 150 µm (100 to 120 mesh) Gas ChromQ(18). The preparation, silanization, packing and conditioning of the packed column is described in Annex A.
Alternatively a capillary column may be used (5.2.2).
5.2.2.Capillary column
Use a short capillary column, e.g. of length 5 m with a non-polar stationary phase that can withstand temperatures up to 400 °C or more(19)). Condition the column by performing 20 analyses of a milk fat solution (7.2) within 2 to 3 days by using the settings given in 7.3.4.2. After that the response factors (7.3.3) shall be close to 1 and less than 1,20.
Note: Columns with different dimensions and a different non-polar, highly temperature-resistant phase can be used, as long as their performance is consistent with this standard. See also 7.3.4.2.
5.3.Extrelut column, of capacity 1 ml to 3 ml, filled with silica gel, needed for the extraction of milk fat according to 7.1.3 only.
5.4.Graphite seals, capable of withstanding temperatures of at least 400 °C; to be used for the connection of the GC column as well as for the injector and/or detector inserts.
5.5.Water bath, capable of maintaining a temperature of 50 °C ± 2 °C.
5.6.Oven, capable of operating at 50 °C ± 2 °C and 100 °C ± 2 °C.
5.7.Microlitre pipette.
5.8.Graduated pipette, of capacity 5 ml.
5.9.Round-bottomed flask, of capacity 50 ml.
5.10.Erlenmeyer flask, of nominal capacity 250 ml.
5.11.Funnel.
5.12.Fine-pored filter paper.
5.13.Rotary evaporator.
5.14.Ampoules, of nominal capacity 1 ml, fitted with a polytetrafluoroethylene-lined aluminium crimp cap or screw cap.
5.15.Injection syringe, with syringe plunger not reaching into the tip of the needle (packed column GC).
Note: With these syringes better repeatability of the results is obtained.
5.16.Analytical balance, capable of weighing to the nearest 1 mg, with a readability of 0,1 mg.
6.SAMPLING
A representative sample should have been sent to the laboratory. It should not have been damaged or changed during transport or storage.
Sampling is not part of the method specified in this International Standard. A recommended sampling method is given in ISO 707 | IDF 50 [5].
7.PROCEDURE
7.1.Preparation of test samples
Use for test sample preparation one of the three following methods of milk fat extraction.
7.1.1.Isolation from butter or butteroil
Melt 50 g to 100 g of test sample at 50 °C using a water bath (5.5) or an oven (5.6). Place 0,5 to 1,0 g of sodium sulfate (4.7) in a folded filter paper (5.12). Preheat a 250 ml Erlenmeyer flask (5.10) and a funnel (5.11) with inserted filter paper in the oven (5.6) set at 50 °C. Filter, while maintaining the preheated flask, funnel and inserted filter device in the oven, the fat layer of the molten sample. Take care that no serum is transferred.
Only in cases where a limited amount of test sample is available, a smaller test sample may be used and the procedure should be adapted accordingly. However, the handling of a smaller test portion involves a higher risk of obtaining a non-representative sample.
Note 1: Butter can be obtained from cream by churning and thorough washing of the resulting butter grain.
Note 2: The milk fat obtained using the procedure in 7.1.1 will be almost free of phospholipids.
7.1.2.Extraction according to the Röse–Gottlieb gravimetric method
Extract the fat fraction from the test sample by using the gravimetric method described in one of the Standards ISO 1211 | IDF 001D, ISO 2450 | IDF 016C or ISO 7328 | IDF 116A.
Note: If phospholipids are present in the milk fat obtained, a cholesterol peak will be obtained which is increased by approximately 0,1 %. The TG composition standardized to 100 % including cholesterol is thereby influenced only to a negligible extent.
7.1.3.Extraction from milk using silica gel columns
Add, using a microlitre pipette (5.7), 0,7 ml of test sample tempered to 20 °C to a 1 ml to 3 ml Extrelut column (5.3). Allow to distribute uniformly on the silica gel for approximately 5 min.
To denature the protein–lipid complexes, add, using the graduated pipette (5.8), 1,5 ml of methanol (4.3) to the Extrelut column. Subsequently, extract the fat fraction from the test sample with 20 ml of n-hexane (4.4). Add the n-hexane slowly in small amounts. Collect the solvent draining off in a 50 ml round-bottomed flask (5.9) previously dried to a constant, known mass weighed to the nearest 1 mg, recording the mass to 0,1 mg.
Allow the column to drain until empty after the extraction. Distil off the solvents from the eluate on a rotary evaporator (5.13) with its water bath set at between 40 °C and 50 °C. After the solvents have been distilled off, dry and subsequently weigh the round-bottomed flask and its contents to the nearest 1 mg, recording the mass to 0,1 mg. Determine the fat mass yield by subtracting the mass of the dried empty round-bottomed flask from the mass obtained.
Note: Fat extractions by the Gerber, Weibull–Berntrop or Schmid–Bondzynski–Ratzlaff methods or isolation of milk fat using detergents (BDI method) are not suitable for TG analysis, because substantial quantities of partial glycerides or phosholipids can pass into the fat phase. Consequently, the application of this International Standard is limited regarding certain products, particularly cheese.
7.2.Preparation of sample solution
For gas chromatography with a packed column, prepare a 5 % (volume fraction) solution of the fat (obtained according to 7.1) in n-hexane (4.4) or n-heptane (4.5). Depending on the column dimensions, use a concentration of 1 % (0,53 mm, ID wide-bore) or lower for on-column injection with a capillary column.
Based on the column used and the mass of fat obtained in 7.1.3, determine the amount of solvent (4.4 or 4.5) to be added to the test sample material in the flask on the basis of weighing to the nearest 1 mg, and recording the mass to 0,1 mg. Completely dissolve the remainder.
Transfer approximately 1 ml of the sample solution into an ampoule (5.14).
7.3.Chromatographic triglyceride determination
7.3.1.Baseline drift
To minimize baseline rising, the column shall be conditioned as specified in 5.2.2 (capillary column) or in Annex A.4 (packed column).
Note: Because of the high column temperature, the analysis of TGs is particularly susceptible to a rise of the baseline in the high carbon-number range.
7.3.2.Injection technique
7.3.2.1.Packed column
To avoid discrimination effects, apply the hot-needle technique for improving the quantification of the high-boiling TG components. Fill the needle with air by drawing up the fat solution in the syringe. Insert the needle into the injector. Heat the needle up prior to injection for about 3 s. Then, rapidly inject the syringe content.
7.3.2.2.Capillary column
When using cold on-column injection (7.3.4.2), insert the needle of the syringe and inject immediately. The dwell time of the needle in the injection port should be such that broad tailing of the solvent peak is avoided.
Note: The optimum dwell time typically is about 3 s.
7.3.3.Calibration
7.3.3.1.General
For the calibration of test samples, perform two to three analyses of standardized milk fat at the beginning of every day. Use the last analysis of the standardized milk fat to determine the response factors, RFsi (mass fraction/area fraction) of the TGs and of cholesterol and apply these to the subsequent test samples (see 9.1):

where:
is the mass fraction, expressed as a percentage, of each TG or cholesterol in the standardized milk fat;
is the numerical value of the peak area of each TG or cholesterol in the standardized milk fat.
Use either 7.3.3.2 or 7.3.3.3 to obtain a standardized milk fat with a known TG composition.
7.3.3.2.Commercial milk fat standard
The best way to determine the response factor of each constituent of the test sample is to use a standardized milk fat with a certified TG composition.
Note: A suitable standard is CRM 519 (anhydrous milk fat) obtainable from the Institute for Reference Materials and Measurements (IRMM), Geel, Belgium(20)).
7.3.3.3.Laboratory milk fat standard
Prepare about 1 g of a mixture of the fat standards (see 4.2, containing at least the saturated TGs, C24, C30, C36, C42, C48 and C54, as well as cholesterol; plus, preferably, C50 and C52) by weighing to the nearest 1 mg, recording the mass to 0,1 mg, to obtain a relative TG composition similar to milk fat.
Analyse repeatedly a solution of the fat standards mixture in n-hexane (4.4) or n-heptane (4.5) according to 7.3.4. In the same sequence, analyse repeatedly averagely composed milk fat.
Determine the TG response factors from the fat standards mixture. Intermediate response factors of TGs not present in the mixture can be calculated by mathematical interpolation. Apply the response factors obtained to the milk fat, in order to obtain a standardized composition. The standardized milk fat thus obtained has a stock life of several years, if stored under nitrogen at a maximum temperature of -18 °C.
7.3.4.Chromatographic conditions
Note: Use of either packed or capillary columns generally results in a resolution similar to Figure 1. Splitting of the even-numbered TGs is not normally observed and shall be avoided.
7.3.4.1.Packed column
Temperature programme: Set the initial oven temperature at 210 °C. Maintain it at that temperature for 1 min. Then increase the temperature at a rate of 6 °C/min to 350 °C. Maintain it at that (final) temperature for 5 min.
Detector and injector temperatures: Set both at 370 °C.
Carrier gas: Use nitrogen at a constant flow rate of about 40 ml/min. Adjust the exact carrier gas flow in such a manner that C54 is eluted at 341 °C.
Duration of analysis: 29,3 min.
Injection volume: Inject 0,5 µl of a 5 % (volume fraction) sample solution.
If no TG analyses are carried out, maintain the initial oven temperature as given in a), the detector and injector temperatures as in b) and the carrier gas flow rate as in c) at constant level, also overnight and during weekends and holidays. This ensures best performance of the column.
7.3.4.2.Capillary column
Temperature programme: Set the initial oven temperature at 80 °C. Maintain it at that temperature for 0,5 min. Then increase the temperature at a rate of 50 °C/min to 190 °C and subsequently at a rate of 6 °C/min to 350 °C. Maintain it at that (final) temperature for 5 min.
Detector temperature: Set at 370 °C.
Carrier gas: Use nitrogen at a constant flow rate of about 3 ml/min.
Duration of analysis: 34,4 min.
Injection volume: Inject 0,5 µl of a 1 % (volume fraction) sample solution.
Maintain these settings during standby to ensure best performance (see 7.3.4.1).
The analytical settings given in 7.3.4.2 are suitable for use with a wide-bore column (0,53 mm ID) as specified in 5.2.2. Different conditions may be applied if another column dimension or phase is used.
8.INTEGRATION, EVALUATION AND CONTROL OF THE ANALYTICAL PERFORMANCE
Evaluate the chromatogram peaks with an integration system capable of baseline drawing and reintegration. Figure 1 shows a correctly integrated chromatogram, whereas Figure 2 demonstrates a sporadic error in the baseline ending after C54 that influences the percentages of all TGs. Nevertheless, exclude peaks eluting after C54 from the evaluation.
Combine TGs with an odd acyl-C number (2n + 1) with the preceding even-numbered TG (2n). Do not take into account the low C56 content. Multiply the area percentages of the remaining TGs including cholesterol by the corresponding response factors of the standardized milk fat (latest calibration) and normalize altogether to 100 % according to 9.1.


To control measuring conditions, compare with the coefficients of variation, CVs, expressed as percentages, of the various TGs given in Table 1 which are based on 19 consecutive analyses of the same milk fat sample.
If the CVs are considerably higher than the values given in Table 1, the chromatographic conditions are not appropriate.
Note: The values given in Table 1 are not mandatory, but are indicative for quality control purposes.
In case, however, that higher CV-values are accepted, the repeatability and reproducibility limits given in Clause 10 shall nonetheless be complied with.
Table 1
Coefficients of variation of triglyceride contents (19 consecutive analyses)
| Triglyceride | CV % |
|---|---|
| C24 | 10,00 |
| C26 | 2,69 |
| C28 | 3,03 |
| C30 | 1,76 |
| C32 | 1,03 |
| C34 | 0,79 |
| C36 | 0,25 |
| C38 | 0,42 |
| C40 | 0,20 |
| C42 | 0,26 |
| C44 | 0,34 |
| C46 | 0,37 |
| C48 | 0,53 |
| C50 | 0,38 |
| C52 | 0,54 |
| C54 | 0,60 |
9.CALCULATION AND EXPRESSION OF RESULTS
9.1.Triglyceride composition
9.1.1.Calculation
Calculate the mass fraction of each TG (for i = C24, C26, C28, C30, C32, C34, C36, C38, C40, C42, C44, C46, C48, C50, C52, and C54) plus cholesterol, wi , expressed as a percentage, of the total TG content of the test sample by using the following equation:

where
is the numerical value of the peak area of each TG in the test sample;
is the response factor of each TG determined by calibration (7.3.3).
9.1.2.Expression of test results
Express the results to two decimal places.
9.2. S-values
9.2.1.Calculation
9.2.1.1.Calculate the S-values, expressed as a percentage, by inserting the calculated wi (9.1.1) of the appropriate TG percentages into Equations (3) to (7). Use all equations irrespective of the kind of foreign fat suspected.
9.2.1.2.Soy bean, sunflower, olive, rapeseed, linseed, wheat germ, maize germ, cotton seed and fish oil.
S = 2,098 3 · w C30 + 0,728 8 · w C34 + 0,692 7 · w C36 + 0,635 3 · w C38 + 3,745 2 · w C40 – 1,292 9 · w C42 + 1,354 4 · w C44 + 1,701 3 · w C46 + 2,528 3 · w C50 (3)
9.2.1.3.Coconut and palm kernel fat.
S = 3,745 3 · w C32 + 1,113 4 · w C36 + 1,364 8 · w C38 + 2,154 4 · w C42 + 0,427 3 · w C44 + 0,580 9 · w C46 + 1,292 6 · w C48 + 1,030 6 · w C50 + 0,995 3 · w C52 + 1,239 6 · w C54 (4)
9.2.1.4.Palm oil and beef tallow.
S = 3,664 4 · w C28 + 5,229 7 · w C30 – 12,507 3 · w C32 + 4,428 5 · w C34 – 0,201 0 · w C36 + 1,279 1 · w C38 + 6,743 3 · w C40 – 4,271 4 · w C42 +6,373 9 · w C46 (5)
9.2.1.5.Lard.
S = 6,512 5 · w C26 + 1,205 2 · w C32 + 1,733 6 · w C34 + 1,755 7 · w C36 + 2,232 5 · w C42 + 2,800 6 · w C46 + 2,543 2 · w C52 + 0,989 2 · w C54 (6)
9.2.1.6.Total.
S = – 2,757 5 · w C26 + 6,407 7 · w C28 + 5,543 7 · w C30 – 15,324 7 · w C32 + 6,260 0 · w C34 + 8,010 8 · w C40 – 5,033 6 · w C42 + 0,635 6 · w C44 + 6,017 1 · w C46 (7)
9.2.2.Expression of test results
Express the results to two decimal places.
9.3.Detection of foreign fat
Compare the five S-values obtained in 9.2.1 with the corresponding S-limits given in Table 2.
Consider the test sample as a pure milk fat, when all five S-values fall inside the limits mentioned in Table 2. However, if any S-value falls outside the corresponding limits, the sample is considered to contain a foreign fat.
Though individual Equations (3) to (6) are more sensitive for certain foreign fats than total Equation (7) (see Table B.1), a positive result obtained with only one of Equations (3) to (6) does not allow conclusions to be drawn on the kind of foreign fat.
Annex B describes a procedure for the calculation of the content of vegetable or animal fat in the adulterated milk fat. This procedure is not validated and is informative only.
Table 2
S-limits for pure milk fats
| a Calculated on a 99 % confidence level, so that foreign fat addition is only indicated if the detection limits of the relevant equation are exceeded (see Table B.1). | ||
| Foreign fat | Equation | S-limitsa |
|---|---|---|
| Soy bean, sunflower, olive, rapeseed, linseed, wheat germ, maize germ, cotton seed, fish oil | (3) | 98,05 to 101,95 |
| Coconut and palm kernel fat | (4) | 99,42 to 100,58 |
| Palm oil and beef tallow | (5) | 95,90 to 104,10 |
| Lard | (6) | 97,96 to 102,04 |
| Total | (7) | 95,68 to 104,32 |
10.PRECISION
10.1.Interlaboratory test
The repeatability and reproducibility values were determined on the basis of Equations (3) to (7) using pure milk fat and may not be applicable to matrices other than those given.
10.2.Repeatability
The absolute difference between two single test results, obtained using the same method on identical test material in the same laboratory by the same operator using the same equipment within a short interval of time, will not exceed the limits listed in Table 3 in more than 5 % of cases.
Table 3
Repeatability limits, r, for Equations (3) to (7)
| Foreign fat | Equation | r % |
|---|---|---|
| Soy bean, sunflower, olive, rapeseed, linseed, wheat germ, maize germ, cotton seed, fish oil | (3) | 0,67 |
| Coconut and palm kernel fat | (4) | 0,12 |
| Palm oil and beef tallow | (5) | 1,20 |
| Lard | (6) | 0,58 |
| Total | (7) | 1,49 |
10.3.Reproducibility
The absolute difference between two single test results, obtained using the same method on identical test material in different laboratories with different operators using different equipment, will not exceed the limits listed in Table 4 in more than 5 % of cases.
Table 4
Reproducibility limits, R, for Equations (3) to (7)
| Foreign fat | Equation | R % |
|---|---|---|
| Soy bean, sunflower, olive, rapeseed, linseed, wheat germ, maize germ, cotton seed, fish oil | (3) | 1,08 |
| Coconut and palm kernel fat | (4) | 0,40 |
| Palm oil and beef tallow | (5) | 1,81 |
| Lard | (6) | 0,60 |
| Total | (7) | 2,07 |
11.UNCERTAINTY OF MEASUREMENT
With the repeatability, r, and the reproducibility, R, the expanded uncertainty for an S-value can be calculated.
Inclusion of the expanded uncertainty (based on duplicate analyses) into the S-limits of Table 2 results in extended S-limits which are given in Table 5.
Table 5
Extended S-limits for pure milk fats including the expanded uncertainty
| Foreign fat | Equation | Extended S-limits |
|---|---|---|
| Soy bean, sunflower, olive, rapeseed, linseed, wheat germ, maize germ, cotton seed, fish oil | (3) | 97,36 to 102,64 |
| Coconut and palm kernel fat | (4) | 99,14 to 100,86 |
| Palm oil and beef tallow | (5) | 94,77 to 105,23 |
| Lard | (6) | 97,65 to 102,35 |
| Total | (7) | 94,42 to 105,58 |
12.TEST REPORT
The test report shall specify:
all information necessary for the complete identification of the sample,
the sampling method used, if known,
the test method used, with reference to this International Standard,
all operational details not specified in this International Standard, or regarded as optional, together with details of any incidents which may have influenced the test result(s),
the test result(s) obtained, and, if the repeatability has been checked, the final quoted result obtained.
ANNEX A(normative)
PREPARATION OF THE PACKED COLUMN
A.1REAGENTS AND APPARATUS
A.1.1 Toluene (C6H5CH3).
A.1.2 Dimethyldichlorosilane [Si(CH3)2Cl2] solution.
Dissolve 50 ml dimethyldichlorosilane in 283 ml toluene (A.1.1).
A.1.3 Cocoa butter solution, with a mass fraction of 5 % cocoa butter in n-hexane (4.4) or n-heptane (4.5).
A.1.4 Stationary phase, 3 % OV-1 on 125 µm to 150 µm (100 to 120 mesh) Gas ChromQ(21)).
Note: The indication of grain was converted to micrometres in accordance with BS 410 (all parts) [6].
A.1.5 Glass column, of internal diameter 2 mm and length 500 mm, U-shaped.
A.1.6
Apparatus, for filling the packed column.
A.1.6.1 Filling column, with screwed-on end caps, provided with a mark up to which the required quantity of stationary phase can be filled.
A.1.6.2 Fine sieve, with mesh size of about 100 µm, with screw cap suited for sealing the glass column according to Figure A.3.
A.1.6.3 Silanized glass wool, deactivated.
A.1.6.4 Vibrator, for uniform distribution of the stationary phase during filling.
A.1.6.5 Silanizing devices, for silanizing the glass surface of the column.
A.1.6.6 Woulff bottle.
A.1.6.7 Water suction pump.
A.2SILANIZATION (DEACTIVATION OF THE GLASS SURFACE)
After connecting the Woulff bottle (A.1.6.6) to the water suction pump (A.1.6.7), dip tube 2 (see Figure A.1) into the dimethyldichlorosilane solution (A.1.2). Fill the column (A.1.5) with that solution by closing the stopcock. Open the stopcock again and subsequently remove the two tubes. Fix the column on a stand. Completely fill it using a pipette with dimethyldichlorosilane solution (A.1.2).

Key
tube 1
tube 2
water suction pump
stopcock
glass column
dimethyldichlorosilane and toluene
Let the column stand for 20 min to 30 min. Then replace the Woulff bottle by a filter flask. Empty the column by connecting it to the water suction pump (A.1.6.7) (see Figure A.2). Rinse the emptied column successively using 75 ml of toluene (A.1.1) and 50 ml of methanol (4.3) by dipping tube 2 into the solvent. Dry the rinsed column in the oven (5.6) set at 100 °C for approximately 30 min.

Key
tube 1
tube 2
water suction pump
filter flask
glass column
rinsing agent
A.3FILLING
Fill the column by using the apparatus represented in Figure A.3. Fill the stationary phase (A.1.4) in the filling column (A.1.6.1) up to the mark. Seal the lower end of the glass column to be filled with an approximately 1 cm long plug of silanized, compressed glass wool (A.1.6.3). Close the end of the column with the fine sieve (A.1.6.2).

Key
nitrogen inlet
filling column, to be filled up to the mark with OV-1
glass column to be filled
screw cap with filter, against which the glass fibre and stationary phase are pressed
Fill the column under pressure (300 kPa and a flow of nitrogen) with the stationary phase. To obtain a uniform, continuous, and firm packing, move a vibrator up and down the glass column during filling. After filling, press a solid plug of silanized glass wool (A.1.6.3) into the other end of the packed column. Cut off the protruding ends. Press the plug into the column for a few millimetres with a spatula.
A.4CONDITIONING
During steps a) to c), do not connect the back end of the column to the detector to avoid contamination. Condition the filled column (A.3) as follows:
flush the column with nitrogen for 15 min, with the flow speed set at 40 ml/min and the GC oven set at 50 °C;
heat the column at a rate of 1 °C/min up to 355 °C, with the nitrogen flow rate set at 10 ml/min;
hold the column at 355 °C for 12 h to 15 h;
inject two times 1 µl of cocoa butter solution (A.1.3) using the temperature program for the packed column given in 7.3.4.1;
Note: Cocoa butter consists almost exclusively of high-boiling C50 to C54 TGs and, thus, reduces the effort of column conditioning with regard to the respective response factors.
inject 20 times 0,5 µl of a milk fat solution according to 7.2 within 2 to 3 days using the settings for the packed column given in 7.3.4.1.
Use only columns with response factors close to 1 for the analysis of test samples. Response factors should not be higher than 1,20.
ANNEX B(informative)
QUANTIFICATION OF THE FOREIGN FAT CONTENT
B.1GENERAL
Table B.1 indicates the detection limits for various foreign fats calculated on a 99 % confidence level. The middle column shows the detection limits of the best individual Equation of (3) to (6).
The detection limits of the total Equation (7), shown in the rightmost column, are somewhat higher. In principle, Equation (7) is only needed for the quantification of foreign fat.
With all equations, combinations of various foreign fats also can be detected. The variation of the TG composition between individual samples of one kind of foreign fat has no significant influence on detection limits.
When using both the individual equations and the total equation, the detection limits of the individual equations apply. However, the S-value of the total equation is needed for quantification in certain cases (B.2).
Table B.1
99 % limits of detection of foreign fat added to milk fat as percentages
| Foreign fat | Individual equation% | Total equation% |
|---|---|---|
| Soy bean oil | 2,1 | 4,4 |
| Sunflower oil | 2,3 | 4,8 |
| Olive oil | 2,4 | 4,7 |
| Coconut oil | 3,5 | 4,3 |
| Palm oil | 4,4 | 4,7 |
| Palm kernel fat | 4,6 | 5,9 |
| Rapeseed oil | 2,0 | 4,4 |
| Linseed oil | 2,0 | 4,0 |
| Wheat germ oil | 2,7 | 6,4 |
| Maize germ oil | 2,2 | 4,5 |
| Cotton seed oil | 3,3 | 4,4 |
| Lard | 2,7 | 4,7 |
| Beef tallow | 5,2 | 5,4 |
| Hydrogenated fish oil | 5,4 | 6,1 |
B.2CALCULATION
Perform a quantitative foreign fat determination only if at least one of the S-limits (Table 2 or Table 5) is exceeded. In order to obtain quantitative information, calculate the foreign fat mass fraction or foreign fat mixture mass fraction, w f, expressed as a percentage, in the test sample using the following equation:

where
is the result obtained by inserting TG data from milk fat to which a foreign fat or foreign fat mixture has been added into one of Equations (3) to (7);
is a constant, depending on the kind of foreign fat added.
If the kind of foreign fat added to milk fat is not known, use a general S f-value of 7,46 (Table B.2). Always use the S-value obtained from Equation (7), even if its S-limits are not exceeded but those of another equation are.
With known foreign fats, insert their individual S f-values (Table B.2) into Equation (B.1). Choose the relevant foreign fat equation from Equations (3) to (6) to calculate S.
Table B.2
S f-values of various foreign fats
| Foreign fat | S f |
|---|---|
| Unknown | 7,46 |
| Soy bean oil | 8,18 |
| Sunflower oil | 9,43 |
| Olive oil | 12,75 |
| Coconut oil | 118,13 |
| Palm oil | 7,55 |
| Palm kernel oil | 112,32 |
| Rapeseed oil | 3,30 |
| Linseed oil | 4,44 |
| Wheat germ oil | 27,45 |
| Maize germ oil | 9,29 |
| Cotton seed oil | 41,18 |
| Lard | 177,55 |
| Beef tallow | 17,56 |
| Fish oil | 64,12 |
B.3EXPRESSION OF TEST RESULTS
Express the test results to two decimal places.
Bibliography
Molkentin, J., Precht, D. Representative determination of the butyric acid content in European milk fats. Milchwissenschaft, 52, 1987, pp. 82-85
Precht, D., Molkentin, J. Quantitative triglyceride analysis using short capillary columns. Chrompack News, 4, 1993, pp. 16-17
Molkentin, J., Precht, D. Comparison of packed and capillary columns for quantitative gas chromatography of triglycerides in milk fat. Chromatographia, 39, 1994, pp. 265-270
Molkentin, J., Precht, D. Equivalence of packed and capillary GC columns with respect to suitability for foreign fat detection in butter using the triglyceride formula method. Chromatographia, 52, 2000, pp. 791-797
ISO 707|IDF 50, Milk and milk products — Guidance on sampling
BS 410:1988, Test sieves — Technical requirements and testing
Precht, D. Control of milk fat purity by gas chromatographic triglyceride analysis. Kieler Milchwirtsch. Forschungsber., 43, 1991, pp. 219-242
Precht, D. Detection of adulterated milk fat by fatty acid and triglyceride analyses. Fat Sci. Technol., 93, 1991, pp. 538-544
DIN 10336:1994, Nachweis und Bestimmung von Fremdfetten in Milchfett anhand einer gaschromatographischen Triglyceridanalyse [Detection and determination of foreign fats in milk fat using a gas chromatographic triglyceride analysis]
Commission of the European Communities: Consideration of results from the first, second, third, fourth, fifth and sixth EEC collaborative trial: Determination of triglycerides in milk fat; Doc. No VI/2644/91, VI/8.11.91, VI/1919/92, VI 3842/92. VI/5317/92, VI/4604/93
Molkentin, J. Detection of foreign fat in milk fat from different continents by triacylglycerol analysis. Eur. J. Lipid Sci. Technol., 109, 2007, pp. 505-510.
ANNEX XXI(Article 18)
PROCEDURE APPLICABLE WHEN THE RESULTS OF AN ANALYSIS ARE DISPUTED (CHEMICAL ANALYSIS)
1.A further analysis is carried out in another laboratory approved by the competent authority using the relevant method at the request of the manufacturer, provided that sealed duplicate samples of the product are available and have been stored appropriately with the competent authority. The request is expressed within seven working days after notification of the results of the first analysis. The analysis is carried out within 21 working days after receipt of the request. The competent authority will send these samples to a second laboratory at the request and the expense of the manufacturer. The laboratory must be authorized to carry out official analyses and must have proven competence for the relevant analysis under question.
2.The expanded uncertainties (k = 2) of the mean  of the
of the  repeated measurements in laboratory 1 and of the mean
repeated measurements in laboratory 1 and of the mean  of the
of the  repeated measurements in laboratory 2 are
repeated measurements in laboratory 2 are
3.  and
and  resp., where
resp., where  is the repeatability standard deviation and
is the repeatability standard deviation and  is the reproducibility standard deviation of the relevant method. If the final result y of measurement in the laboratories is calculated using a formula of the form
is the reproducibility standard deviation of the relevant method. If the final result y of measurement in the laboratories is calculated using a formula of the form  ,
,  ,
,  or
or  the usual procedures for combining standard deviations in such cases must be followed in order to obtain the uncertainty.
the usual procedures for combining standard deviations in such cases must be followed in order to obtain the uncertainty.
4.In order to test whether the results of the two laboratories are in compliance with the reproducibility standard deviation  of the method, the expanded uncertainty of the difference
of the method, the expanded uncertainty of the difference  is calculated:
is calculated:
5.  If the absolute value of the difference of the laboratory means,
If the absolute value of the difference of the laboratory means,  , is not larger than its uncertainty
, is not larger than its uncertainty  ,
,

the results of the two laboratories are in compliance with the reproducibility standard deviation 

is reported as the final result. Its expanded uncertainty is

The consignment is rejected as being not in compliance with an upper legal limit UL if

otherwise it is accepted as being in compliance with UL.
The consignment is rejected as being not in compliance with a lower legal limit LL if

otherwise it is accepted as being in compliance with LL.
If the absolute value of the difference of the laboratory means, 


The results of the two laboratories are not in compliance with the reproducibility standard deviation.
In this case the consignment is rejected as non-compliant if the second analysis confirms the first. Otherwise, the consignment is accepted as compliant.
The final result must be notified by the competent authority to the manufacturer as soon as possible. The costs of the second analysis are to be borne by the manufacturer, if the consignment is rejected.
ANNEX XXIICORRELATION TABLE
| Regulation (EC) No 213/2001 | This Regulation |
|---|---|
| Article 1 | Article 1 |
| Article 2 | Article 1 |
| Article 3 | Article 2 |
| — | Article 3 |
| Article 4 | — |
| Article 5 | — |
| Article 6 | Article 4 |
| Article 7 | Article 18 |
| Article 8 | — |
| Article 9 | Article 5 |
| Article 10 | Article 6 |
| Article 11 | Article 7 |
| Article 12 | Article 8 |
| Article 13 | Article 9 |
| Article 14 | Article 10 |
| Article 15 | Article 11 |
| Article 16 | Article 12 |
| Article 17 | Article 13 |
| — | Article 14 |
| Article 18 | Article 15 |
| Article 19 | Article 16 |
| Article 17 | |
| Article 19 | |
| Article 20 | — |
| Article 21 | — |
| Article 22 | Article 20 |
| Article 23 | Article 21 |
OJ L 160, 26.6.1999, p. 48. Regulation as last amended by Regulation (EC) No 1152/2007 (OJ L 258, 4.10.2007, p. 3). Regulation (EC) No 1255/1999 will be replaced by Regulation (EC) No 1234/2007 (OJ L 299, 16.11.2007, p. 1) as from 1 July 2008.
OJ L 308, 25.11.2005, p. 1. Regulation as last amended by Regulation (EC) No 1546/2007 (OJ L 337, 21.12.2007, p. 68).
OJ L 279, 11.10.1990, p. 22. Regulation as last amended by Regulation (EC) No 1487/2006 (OJ L 278, 10.10.2006, p. 8).
OJ L 256, 7.9.1987, p. 1. Regulation as last amended by Commission Regulation (EC) No 1352/2007 (OJ L 303, 21.11.2007, p. 3).
= FAME method.
The produce Ampholine® pH 3,5-9,5 (Pharmacia) and Resolyte® pH 5-7 and pH 6-8 (BDH, Merck) have proved particularly suitable for obtaining the required separation of γ-caseins.
J. Koops en C. Olieman, Netherlands Milk and Dairy Journal, 39 (1985) 89-106.
D.A. Biggs en L. Szijarto, Journal of Dairy Science, 46 (1963) 1196.
ISO 707 (IDF 50), Milk and milk products — Methods of sampling.
ISO 5725-1, Accuracy (trueness and precision) of measurement methods and results. Part 1: General principles and definitions.
ISO 5725-2, Accuracy (trueness and precision) of measurement methods and results. Part 2: A basic method for the determination of repeatability and reproducibility of a standard measurement method.
Rennet-type whey powder of standard composition and also the adulterated skimmed-milk powder are available from NIZO, Kernhemseweg 2, PO Box 20 — NL-6710 BA Ede. However, powders giving equivalent results to the NIZO powders may also be used.
International IDF Standard 135B/1991. Milk and milk products. Precision characteristics of analytical methods. Outline of collaborative study procedure.
Important notice: False-positive results may be obtained, when skimmed-milk powder is analysed. It is important, therefore, to verify that the test system used does not yield false-positive results.
Some β-lactams are less sensitive to β-lactamase. In such cases an additional pre-treatment of the sample, (1 ml of test sample with 0,3 ml of penase concentrate at 37 °C for 2 h) is recommended.
Example of a suitable product available commercially. This information is given for the convenience of users of this International Standard and does not constitute an endorsement of this product.
CP-Ultimetal SimDist (5 m × 0,53 mm × 0,17 µm) is an example of a suitable product available commercially. This information is given for the convenience of users of this International Standard and does not constitute an endorsement of this product.
Example of a suitable product available commercially. This information is given for the convenience of users of this International Standard and does not constitute an endorsement of this product.
Example of a suitable product available commercially. This information is given for the convenience of users of this International Standard and does not constitute an endorsement by ISO or by IDF of this product.