ANNEX XI(Article 8)
DETERMINATION OF LACTOSE IN COMPOUND FEEDINGSTUFFS
1.SCOPE AND FIELD OF APPLICATION
Determination of lactose in compound feedingstuffs.
2.REFERENCE
The content of lactose is defined as the percentage by mass as determined by the procedure described.
3.DEFINITION
The anhydrous lactose content is expressed as g per 100 g.
4.PRINCIPLE
The compound feedingstuff is reconstituted with water. A ‘Biggs’ solution is added to a dilute weighed aliquot to precipitate out the fat and the protein component fractions of the compound feedingstuff. The sample is filtered (or centrifuged) and the filtrate (or supernatant) is injected on a cation exchange HPLC column in the lead form using HPLC grade water as the mobile phase. The eluted lactose is detected by a differential refractometer(1).
5.REAGENTS
5.1.General
Use only reagents of recognized analytical grade, unless otherwise specified, and degassed HPLC grade water.
5.2.Lactose
D-Lactose monohydrate ((C12H22)O11H2O) can take up additional moisture. Before use measure the real amount of water by Karl-Fisher or remove the additional moisture by placing lactose in an oven at 105 °C for 8 hours (the lactose does not lose its crystal water by this treatment).
5.3.Concentrated Biggs/Szijarto solution(2)
Dissolve 9,10 g zinc acetate ehydrate (Zn(CH3COO)2.2H2O) and 5,46 g phosphotungstic acid monohydrate (H3[P(W3O10)4.xH2O]) in about 70 ml with HPLC grade water (6.8) in a 100 ml volumetric flask.
Add 5,81 ml glacial acetic acid (CH3COOH). Dilute to the 100 ml mark with HPLC grade water (6.8) water and mix. The solution can be stored at room temperature for 1 year.
5.4.Diluted Biggs/Szijarto solution
Dilute 25 ml concentrated Biggs/Szijarto solution (5.3) with water to 500 ml using a volumetric flask. The solution can be stored at room temperature for 1 month.
5.5.Preparation of HPLC grade water
Filter the ultra pure water (6.8) by using the vacuum system filtration (6.9). To improve the pump performance and to obtain a stable baseline, degas the mobile phase daily by selecting one of the available techniques such as sparging by helium, sonication, vacuum or in-line degassing system.
Note: In order to prolong column life it is essential that the carbon dioxide content of the eluent is as low as possible and that re-uptake is prevented.
6.APPARATUS
Usual laboratory equipment and, in particular, the following:
6.1.HPLC ion exchange resin column
Column packing: 8 % cross-linked polystyrene-divinylbenzene copolymer functionalised with cation-exchange groups in the lead form.
Column dimensions: Length 300 mm, internal diameter ca. 8 mm.
Use of other diameters is possible provided that the flow-rate is adjusted accordingly.
6.2.Guard column
The guard column is a combination of a separate cation exchanger (H+) and an anion exchanger (CO3-), each packed in columns of ca. 30 mm × 4,6 mm (L × ID) (e.g. micro-guard columns in a micro-guard holder) and connected in series or in the form of a mixed bed consisting of AG 50W-X4, -400 mesh (H+) and AG3-X4A, 200–400 mesh (OH-) in the ratio of 35:65 (m/m) manually packed in a column of ca. 20 × 9 mm (L × ID).
6.3.Column oven
Oven capable of maintaining a constant temperature of 85 °C ± 1 °C.
6.4.HPLC pump
Pump capable of generating a constant flow-rate (< 0,5 % fluctuations) at 0,2-1,0 m/min.
6.5.HPLC injection device
Auto sampler capable of injecting 25 µL and having a repeatability < 0,5 %.
Alternatively a manual device may be used (same requirements as the auto sampler).
6.6.HPLC detector
Highly sensitive refractive index detector having a noise < 5,10-9 RI units.
6.7.Integrator
Software or a dedicated integrator to perform data acquisition, processing and generating peak areas and peak heights, which can be converted to lactose concentrations.
6.8.Water purification unit
System capable to provide ultra pure water (type 1) having a resistivity >14 MΩ.cm.
6.9.Solvent filtration unit
System that enables filtration of the water using 0,45 µm pore size membrane filter.
Note: Many water purification units (6.8) have a built in 0,45 or 0,2 µm filtration. Additional filtration can be omitted if this water is directly used.
6.10.Analytical balance
Balance having a read-out of 0,1 mg.
6.11.Water bath
Water bath capable of maintaining a temperature at 40 °C (±0,5).
6.12.Centrifuge
Having a capacity of generating at least 3 000 g for Eppendorf vials or equivalent or larger type of vials.
6.13.Volumetric flask 50 mL
Capacity of 50 mL, class A.
Note: Flasks of other capacity can be used by taking into account the volume factor.
6.14.Volumetric flask 100 mL
Capacity of 100 mL, class A.
6.15.Graduated pipette
Graduated pipette of 10 mL
Note: Alternatively, a handpipettor having a capacity of 5 mL can be used by adding twice a volume of 5 mL of reagent (5.3).
7.SAMPLING
It is important that the laboratory receives a sample taken according to ISO 707/IDF 50(3), which is truly representative and has not been damaged during transport or storage.
8.PREPARATION OF LACTOSE STANDARD SOLUTION
8.1.Standard 1
Dissolve an accurately (read-out at 0,1 mg) weighed amount of ca. 50 mg lactose monohydrate (5.2) in a volumetric flask of 100 mL (6.14) and make up to the mark with water.
8.2.Standard 2
Dissolve an accurately (read-out at 0,1 mg) weighed amount of ca. 100 mg lactose monohydrate (5.2) in a volumetric flask of 100 mL (6.14) and make up to the mark with water.
Note: The standard solutions can be stored for maximal 1 week at ca. 5 °C.
9.PREPARATION OF THE TEST SAMPLE
9.1.Reconstitution of the sample
Weigh ca. 5 g of the powder into a flask of 50 mL (6.13) and note the weight at 1 mg accuracy (W1, (11)). Add 50 mL of water and note the increase of the weight (W2 (11)) at 0,01 g accuracy. Place the closed flask in the water bath (6.11) for 30 min and invert it a few times during this period. Let it subsequently cool to room temperature.
9.2.Sample treatment
Take ca. 1 g of this solution and deposit it in a 50 mL volumetric flask (6.13), note the weight at 1 mg accuracy (W3 (11)), add 20 mL of water followed by the addition of 10 mL diluted Biggs/Szijarto reagent (5.4), make up to the mark with water. Gently invert the flask 5 times during the first 30 min.
After 1 hour take an aliquot and centrifuge (6.12) at 3 000 g for 10 min (higher g can be used at correspondingly shorter time). Use an aliquot of the supernatant for HPLC analysis.
10.HPLC DETERMINATION
10.1.Preliminary preparation of HPLC
10.1.1.Installation of the column and pre-column
Install the pre-column (6.2) outside the column oven (6.3) and the column (6.1) in the oven.
Note: If the oven does not contain tubing to preheat the eluent, it is necessary that the eluent passes ca. 15 cm stainless steel tubing in the oven before entering the column (it is absolutely necessary that the eluent has heated up before entering the column, otherwise peak broadening shall occur).
10.1.2.Detector and initial flow
In order to get a stable baseline, turn on the detector (6.6) at least 24 h before starting the analysis. Set the internal temperature of the detector at 35 °C. Set the flow rate at 0,2 ml/min (6.4) for at least 20 min while the column oven (6.3) is set to room temperature.
10.1.3.Column oven and final flow-rate
Set the column oven (6.3) at 85 °C. When that temperature is reached, increase after 30 minutes gradually the flow rate from 0,2 ml/min to 0,6 ml/min (6.4). Allow the system to equilibrate with that flow rate and at 85 °C for 2 h or until a stable baseline is obtained.
10.1.4.Integration
Carefully chose the acquisition and integration parameters (6.7) such as data rate, sensitivity, time constant, peak width and threshold.
The retention time of lactose is ca. 11 min.
Note: Many data acquisition software programs (6.7) afford easy measurement of the theoretical plate count. Measure the theoretical plate count of standard 1 (8.1) regularly and replace the column (6.1) when the plate count is 25 % lower than that of the initial value of a new column.
10.1.5.Guard column test
Check regularly (at least once in every sequence) the ability of the guard column (6.2) to eliminate salts from the sample by injecting 25 µL of a 0,05 % sodium chloride solution. Whenever peaks are appearing, the guard column should be replaced.
10.2.Running standards
Inject at the beginning of each series of analyses 25 µL (6.5) of standard 1 (8.1) and subsequently of standard 2 (8.2). Repeat this every 10 to 20 samples and apply this also at the end of the sequence.
10.3.Running samples
Inject 25 µL of the supernatant (9.2) of the sample.
11.CALCULATION AND EXPRESSION OF THE RESULTS
11.1.Calibration
Normally peak heights are used to calculate the results, however, if the signal contains to much noise peak area can be used (quantitation by peak height is less influenced by peaks of components in low concentration and which are partly, but insufficiently separated from the lactose peak).
The software (6.7) should calculate a linear calibration curve forced through the origin. Check the curve for possible non linearity (apparent non linearity is most likely caused by a mistake in preparing the standards 1 (8.1) or 2 (8.2), bad integration and, less likely, by a mall functioning injector).
Use as input the calculated lactose concentrations in mg/mL of the standards 1 (8.1) and 2 (8.2) as water free lactose.
The slope (RF) of the calibration line is defined by area/concentration in mg/mL.
11.2.Samples
The result of the analysis is obtained as g/100 g and calculated using the software (6.7) or using the following formula:
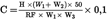
Where:
:
concentration of lactose in g/100 g powder
:
peak height of lactose of sample
:
Response factor (or slope) of calibration plot in mV/mg/mL
:
sample weight of powder sample in g (9.1)
:
weight of added water in g to powder sample (9.1)
:
sample weight of reconstituted solution of powder in g (9.2)
:
Volume of volumetric flask used in (9.2)
:
conversion of the result in g/100 g
12.PRECISION
The values derived from this inter laboratory test may not be applicable to concentration ranges and matrices other than those given. The values for the repeatability and reproducibility will be derived from the result of an inter laboratory test carried out in accordance with ISO 5725(4)
12.1.Repeatability
The absolute difference between two single test results, obtained using the same method on identical test material in the same laboratory with the same operator using the same equipment within a short time of interval, will in not more than 5 % of cases be greater than xxx (to be determined by a collaborative trial)(5).
12.2.Reproducibility
The absolute difference between two single test results, obtained using the same method on identical test material in different laboratories with different operators using different equipment, will in not more than 5 % of cases be greater than 0,5 g/100g (to be determined by a collaborative trial)
13.REFERENCES
J. Koops en C. Olieman, Netherlands Milk and Dairy Journal, 39 (1985) 89-106.
D.A. Biggs en L. Szijarto, Journal of Dairy Science, 46 (1963) 1196.
ISO 707 (IDF 50), Milk and milk products — Methods of sampling.
ISO 5725-1, Accuracy (trueness and precision) of measurement methods and results. Part 1: General principles and definitions.
ISO 5725-2, Accuracy (trueness and precision) of measurement methods and results. Part 2: A basic method for the determination of repeatability and reproducibility of a standard measurement method.