- Latest available (Revised)
- Original (As adopted by EU)
Commission Regulation (EC) No 429/2008Show full title
Commission Regulation (EC) No 429/2008 of 25 April 2008 on detailed rules for the implementation of Regulation (EC) No 1831/2003 of the European Parliament and of the Council as regards the preparation and the presentation of applications and the assessment and the authorisation of feed additives (Text with EEA relevance)
You are here:

What Version
More Resources
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


This item of legislation originated from the EU
Legislation.gov.uk publishes the UK version. EUR-Lex publishes the EU version. The EU Exit Web Archive holds a snapshot of EUR-Lex’s version from IP completion day (31 December 2020 11.00 p.m.).
Status:
This is the original version as it was originally adopted in the EU.
This legislation may since have been updated - see the latest available (revised) version
3.SECTION III: STUDIES CONCERNING SAFETY OF THE ADDITIVE
The studies included in this section and in the specific Annexes are intended to permit assessment of:
the safety of use of the additive in the target species;
any risk associated with the selection and/or transfer of resistance to antimicrobials and increased persistence and shedding of enteropathogens;
the risks to the consumer of food derived from animals given feedingstuffs containing or treated with the additive or which could result from the consumption of food containing residues of the additive or its metabolites;
the risks from respiratory, other mucosal tissue, eye or cutaneous contact for persons likely to handle the additive as such or as incorporated into premixtures or feedingstuffs; and
the risks of adverse effects on the environment, from the additive itself, or from products derived from the additive, either directly and/or excreted by animals.
3.1.Studies concerning the safety of use of the additive for the target animals
The studies included in this section are intended to assess:
the safety of use of the additive in the target species per se; and
any risk associated with the selection and/or transfer of resistance to antimicrobials and increased persistence and shedding of enteropathogens.
3.1.1.Tolerance studies for the target species
The aim of the tolerance test is to provide a limited evaluation of short-term toxicity of the additive to the target animals. It is also used to establish a margin of safety, if the additive is consumed at higher doses than recommended. Such tolerance tests must be conducted to provide evidence for safety for each of the target species/animal categories for which a claim is made. In some cases it is acceptable to include some elements of the tolerance test in one of the efficacy trials provided that the requirements given below for these tests are met. All studies reported in this section must be based on the additive described in Section II.
3.1.1.1.The design of a tolerance test includes a minimum of three groups:
an unsupplemented group;
a group with the highest recommended dose; and
an experimental group with the multi-fold level of the highest recommended dose.
In the experimental group the additive shall generally be given at ten times the highest recommended dose. Test animals shall be routinely monitored for visual evidence of clinical effects, performance characteristics, product quality where relevant, haematology and routine blood chemistry and for other parameters likely to be related to the biological properties of the additive. Critical end-points known from the toxicological studies in laboratory animals shall be considered. Any adverse effect detected during efficacy trials shall also be reported in this section. Unexplained deaths in the tolerance test shall be investigated by necropsy and, if appropriate, histology.
If a 100 times the maximum recommended dose can be shown to be tolerated, no haematology or routine blood chemistry would be required. If the product is tolerated only at lower level than ten times of the highest recommended dose, the study shall be designed in such a way that a margin of safety for the additive can be calculated and additional end-points (by necropsy, histology if relevant, and other appropriate criteria) shall be provided.
For some additives depending on their toxicology and metabolism or use, it may not be necessary to carry out tolerance tests.
The experimental design used must include consideration of adequate statistical power.
3.1.1.2.Duration of tolerance trials
Table 1
Duration of tolerance trials: Pigs
| Target animals | Duration of the studies | Characteristic of the target animals |
|---|---|---|
| Suckling piglets | 14 days | Preferably from 14 days to weaning |
| Weaned piglets | 42 days | For 42 days after weaning |
| Pigs for fattening | 42 days | Body weight at start of the study ≤ 35 kg |
| Sows for reproduction | 1 cycle | From insemination to the end of the weaning period |
If suckling and weaned piglets are applied for, a combined study (14 days suckling piglets and 28 days weaned piglets) would be considered sufficient. If the tolerance for weaned piglets has been shown, no separate study for pigs for fattening is required.
Table 2
Duration of tolerance trials: Poultry
| Target animals | Duration of the studies | Characteristic of the target animals |
|---|---|---|
| Chickens for fattening/reared for laying | 35 days | From hatching |
| Laying hens | 56 days | Preferably during the first third of the laying period |
| Turkeys for fattening | 42 days | From hatching |
Tolerance data from chickens for fattening or turkeys for fattening can be used to demonstrate tolerance for chickens or turkeys reared for laying/breeding respectively.
Table 3
Duration of tolerance trials: Bovines
| Target animals | Duration of the studies | Characteristic of the target animals |
|---|---|---|
| Calves for fattening | 28 days | Initial bodyweight ≤ 70kg |
| Calves for rearing; cattle for fattening or reproduction | 42 days | |
| Dairy cows | 56 days |
If calves for rearing and cattle for fattening were applied for, a combined study (28 days for each period) would be considered sufficient.
Table 4
Duration of tolerance trials: Sheep
| Target animals | Duration of the studies | Characteristic of the target animals |
|---|---|---|
| Lambs for rearing and for fattening | 28 days |
Table 5
Duration of tolerance trials: Salmonidae and other fish
| Target animals | Duration of the studies | Characteristic of the target animals |
|---|---|---|
| Salmon and trout | 90 days |
As an alternative to a 90-day duration, a study could be performed where the fish increase their initial body weight at the start of the trial by least a factor of two.
If the additive is intended to be used for brood stock only, the tolerance tests shall be carried out as close to the spawning period as possible. The tolerance tests shall last for 90 days and attention shall be paid to the egg quality and survival of the eggs.
Table 6
Duration of tolerance trials: Pets and other non food-producing animals
| Target animals | Duration of the studies | Characteristic of the target animals |
|---|---|---|
| Dogs and cats | 28 days |
Table 7
Duration of tolerance trials: Rabbits
| Target animals | Duration of the studies | Characteristic of the target animals |
|---|---|---|
| Rabbits for fattening | 28 days | |
| Breeding does | 1 cycle | From insemination to the end of the weaning period |
If rabbits suckling and weaned are applied for, a period of 49 days (beginning one week after birth) would be considered sufficient and must include the does until weaning.
If an additive is applied for a specific and shorter period than given by the animal category definition, it shall be administered according to the proposed conditions of use. However, the observation period shall not be shorter than 28 days and shall involve the relevant end-point (e.g. for sows for reproduction the number of piglets born alive when considering the gestation period, or the number and weight of weaned piglets when considering the lactation period).
3.1.1.3.Experimental conditions
The studies shall be reported individually, giving details of all experimental groups. The trial protocol shall be carefully drawn up with regard to general descriptive data. In particular, the following shall be recorded:
(1)
herd or flock: location and size; feeding and rearing conditions, method of feeding; for aquatic species, size and number of tanks or pens at the farm, light conditions and water quality including water temperature and salinity;
(2)
animals: species (for aquatic species intended for human consumption identification shall be made by their colloquial name followed in parenthesis by the Latin binomial), breed, age (size for aquatic species), sex, identification procedure, physiological stage and general health;
(3)
date and exact duration of testing: date and nature of the examinations performed;
(4)
diets: description of manufacture and quantitative composition of the diet(s) in terms of ingredients used, relevant nutrients (analysed values) and energy. Feed intake records;
(5)
concentration of the active substance(s) or agent(s) (and, where that is the case, substances used for comparative purposes) in the feedingstuffs shall be established by a control analysis, using the appropriate recognised methods: reference number(s) of the batches;
(6)
number of test and control groups, number of animals in each group: the number of animals involved in the trials must permit statistical analysis. The methods of statistical evaluation used should be stated. The report shall include all animals and/or experimental units involved in the trials. Cases which cannot be assessed due to a lack or loss of data shall be reported, and their distribution within the groups of animals classified;
(7)
the timing and prevalence of any undesirable consequences of treatment in individuals or groups must be reported (give details of the observation programme used in the study); and
(8)
therapeutic/preventive treatments, if necessary, shall not interact with the proposed mode of action of the additive and shall be recorded individually.
3.1.2.Microbial studies
Studies shall be provided to determine the ability of the additive to induce cross-resistance to antibiotics used in human or veterinary medicine, to select resistant bacterial strains under field conditions in target species, to give rise to effects on opportunistic pathogens present in the digestive tract, to cause shedding or to excrete zoonotic micro-organisms.
If the active substance(s) possesses antimicrobial activity at the feed concentration level, the minimum inhibitory concentration (MIC) for relevant bacterial species shall be determined, according to standardised procedures. Where relevant antimicrobial activity is demonstrated, the ability of the additive to select resistant bacterial strains in vitro and in the target species, and to induce cross-resistance to relevant antibiotics shall be established(1).
Tests at the recommended use level shall be provided for all microbial additives, and for those other additives in which an effect on the gut micro-flora can be anticipated. These studies shall demonstrate that use of the additive does not create conditions conducive to an overgrowth and shedding of potentially pathogenic micro-organisms.
The choice of micro-organisms to be monitored will depend on the target species, but shall include relevant zoonotic species, regardless of whether or not they produce symptoms in target animals.
3.2.Studies concerning the safety of use of the additive for consumers
The aim is to evaluate the safety of the additive for the consumer and to establish potential residues of the additive or its metabolites in food derived from animals given feed or water containing or treated with the additive.
3.2.1.Metabolic and residue studies
The establishment of the metabolic fate of the additive in the target species is a determinant step in the identification and quantification of the residues in the edible tissues or products derived from the animals given the feed or water containing the additive. Studies must be submitted concerning the absorption, distribution, metabolism and excretion of the substance (and its metabolites).
Studies must be carried out using internationally validated test methods and shall be performed in accordance with European legislation in force or OECD Guidelines for methodological details and according to the principles of GLP. The study shall respect the rules on animal welfare laid down by European Community legislation, and they shall not be repeated if not necessary.
Metabolic and residue studies on the target animal(s) shall be performed with the active substance incorporated in the feed (not given by gavage unless it is properly justified).
Structural identification of metabolites representing more than 10 % of the total residues in the edible tissues and products and more than 20 % of the total residues in the excreta shall be established. If the metabolic pathway of the active substance raises any toxicological concerns, metabolites below the above limits shall be identified.
Kinetic studies of the residues will form the basis for the calculation of consumer exposure and the establishment of a withdrawal period and MRLs, if necessary. A proposal for a marker residue shall be provided.
For some additives, depending on their nature or use, it may not always be necessary to carry out metabolic and residues studies.
3.2.1.1.Metabolic studies
The purpose of metabolic studies is to evaluate the absorption, distribution, biotransformation and excretion of the additive in the target species.
The studies required are:
(1)
metabolic balance following a single dose administration of the active substance at the doses proposed for use (total amount corresponding to the daily intake) and possibly a multiple dose (if justified) to assess an approximate rate and extent of the absorption, distribution (plasma/blood) and excretion (urine, bile, faeces, milk or eggs, expired air, excretion via gills) in male and female animals, where appropriate; and
(2)
metabolic profiling, identification of the metabolite(s) in excreta and tissues and distribution in tissues and products shall be established following repeated dose administration of the labelled compound to animals to the steady state (metabolic equilibrium) identified by plasma levels. The dose applied shall correspond to the highest dose proposed for use, and shall be incorporated into the feed.
3.2.1.2.Residue studies
Consideration shall be given to the amount and the nature of non-extractable residues in edible tissues or products.
Residue studies are required for all substances for which metabolic studies are needed.
If the substance is a natural constituent of body fluids or tissues or is naturally present in significant amounts in food or feedingstuffs, the requirement for residue studies is limited to the comparison of the tissue/product levels in an untreated group and in the group supplemented with the highest dose claimed.
For major species, studies shall simultaneously evaluate the total residues of toxicological significance and identify the marker residue of the active substance in edible tissue (liver, kidney, muscle, skin, skin + fat) and products (milk, eggs and honey). The marker residue is the residue selected for assay whose concentration has a known relationship to the total residue of toxicological concern in the tissues. Studies shall also show the permanence of the residues in the tissues or products to establish an appropriate withdrawal period.
For the determination of a withdrawal period, the suggested minimum number of animals sampled and/or products at each time point are the following:
edible tissues:
bovines, sheep, pigs and minor species 4;
poultry 6;
salmonids and other fish 10.
products:
milk 8 samples per time point;
eggs 10 eggs per time point;
honey 8 samples per time point.
Appropriate sex distribution shall be considered.
The residues shall be measured at zero withdrawal time (steady state) and at least three other time sampling points.
A proposal for a marker residue shall be provided.
Studies on the absorption, distribution and excretion, including the identification of main metabolites must be performed in the laboratory animal species in which the lowest NOAEL was obtained, or by default in the rat (both sexes). Additional studies on particular metabolites may be necessary if these metabolites are produced by target species and are not formed to a significant extent in the laboratory species.
3.2.1.3.Metabolic and disposition studies
A metabolism study including the metabolic balance, metabolic profile and identification of the main metabolites in the urine and faeces shall be performed. If another laboratory species shows a marked difference in the sensitivity from the rat, additional information will be required.
3.2.1.4.Bioavailability of residues
The assessment of the risks for the consumers related to bound residues in animal products may take into account an additional safety factor on the determination of their bioavailability using appropriate laboratory animals and recognised methods.
3.2.2.Toxicological studies
The safety of the additive is assessed on the basis of the toxicological studies performed in vitro and in vivo on laboratory animals. They generally include measurements of:
(1)
acute toxicity;
(2)
genotoxicity (mutagenicity, clastogenicity);
(3)
sub-chronic oral toxicity;
(4)
chronic oral toxicity/carcinogenicity;
(5)
reproduction toxicity including teratogenicity; and
(6)
other studies.
Further studies providing additional information necessary for the assessment of the safety of the active substance and its residues shall be conducted if there is any reason for concern.
On the basis of the results of these studies a toxicological NOAEL must be established.
Additional studies on particular metabolites may be necessary if these metabolites are produced by target species and are not formed to a significant extent in the laboratory test species. If metabolic studies are available in humans, data shall be taken into consideration in deciding the nature of eventual additional studies.
Toxicological studies must be carried out with the active substance. If the active substance is present in a fermentation product, the fermentation product shall be tested. The fermentation product tested must be identical to that to be used in the commercial product.
Studies must be carried out using internationally validated test methods and shall be performed in accordance with European legislation in force or OECD Guidelines for methodological details and according to the principles of GLP. The studies involving laboratory animals shall respect the rules on animal welfare laid down by European legislation and they shall not be repeated if not necessary.
3.2.2.1.Acute toxicity
Acute toxicity studies are required to classify and to provide limited characterisation of the toxicity of the compound.
Acute toxicity studies shall be carried out in at least two mammalian species. One laboratory species may be replaced by a target species, if appropriate.
It will be not necessary to determine a precise LD50; an approximate determination of the minimum lethal dose is considered sufficient. The maximum dosage shall not exceed 2 000 mg/kg body weight.
In order to reduce the number and the suffering of the animals involved, new protocols for acute dose toxicity testing are continually being developed. Studies carried out by these new procedures will be accepted, when properly validated.
OECD Guidelines 402 (acute dermal toxicity), 420 (Fixed Dose Method), 423 (Acute Toxic Class Method) and 425 (Up-and-Down Procedure) should be followed.
3.2.2.2.Genotoxicity studies including mutagenicity
To identify active substances and, if appropriate, their metabolites and degradation products with mutagenic and genotoxic properties, a selected combination of different genotoxicity tests must be carried out. If appropriate the tests shall be performed without and with mammalian metabolic activation and the compatibility of the test material with the test system shall be taken into account.
The core set comprises the following tests:
(1)
induction of gene mutations in bacteria and/or in mammalian cells (preferably the mouse lymphoma tk assay);
(2)
induction of chromosomal aberrations in mammalian cells; and
(3)
in vivo test in mammalian species.
Additional tests may be needed depending on the outcome of the above mentioned tests and taking into consideration the whole toxicity profile of the substance, as well as its intended use.
Protocols should be in line with OECD Guideline 471 (Salmonella typhimurium Reverse Mutation Test), 472 (Escherichia coli Reverse Mutation Test), 473 (in vitro Mammalian Chromosomal Aberration Test), 474 (Mammalian Erythrocyte Micronucleus Test), 475 (Mammalian Bone Marrow Chromosomal Aberration Test), 476 (in vitro Mammalian Cell Gene Mutation Test) or 482 (Unscheduled DNA Synthesis in Mammalian Cells in vitro), as well as other relevant OECD Guidelines for in vitro and in vivo assays.
3.2.2.3.Sub-chronic repeated dose oral toxicity studies
To investigate the sub-chronic toxic potential of the active substance, at least one study on a rodent species must be submitted with duration of at least 90 days. If deemed necessary, a second study must be performed with a non-rodent species. The test item must be administered orally with at least three levels in addition to a control group to obtain a dose response. The maximum dose used should normally be expected to reveal evidence of adverse effects. The lowest dose level should not be expected to produce any evidence of toxicity.
Protocols for these studies should be in line with the OECD Guidelines 408 (rodents) or 409 (non-rodents).
3.2.2.4.Chronic oral toxicity studies (including carcinogenicity studies)
To investigate the chronic toxic potential and carcinogenic potential, a chronic oral toxicity study must be carried out in at least one species, and shall be of at least 12 months' duration. The species chosen shall be the most appropriate on the basis of all available scientific data, including the results of the 90-day studies. The default species is the rat. If a second study is requested, a rodent or a non-rodent mammalian species shall be used. The test item must be administered orally with at least three levels in addition to a control group to obtain a dose response.
If the chronic toxicity study is combined with an examination of carcinogenicity, then the duration shall be extended to 18 months for mice and hamsters, and to 24 months for rats.
Carcinogenicity studies may not be necessary if the active substance and its metabolites:
(1)
give consistently negative results in the genotoxicity tests;
(2)
are not structurally related to known carcinogens; and
(3)
give no effects indicative of potential (pre)neoplasia in chronic toxicity assays.
Protocols should be in line with OECD Guideline 452 (chronic toxicity study) or 453 (combined chronic toxicity/carcinogenicity study).
3.2.2.5.Reproduction toxicity studies (including prenatal developmental toxicity)
To identify possible impairment of male or female reproductive function or harmful effects on progeny resulting from the administration of the active substance, studies of reproductive function must be carried out by:
(1)
two generation reproduction toxicity study; and
(2)
prenatal developmental toxicity study (teratogenicity study).
For new trials validated alternative methods reducing the use of animals can be used.
3.2.2.5.1.Two generation reproduction toxicity study
Studies of reproductive function must be carried out and extend over at least two filial generations (F1, F2) in at least one species, usually a rodent, and may be combined with a teratogenicity study. The substance under investigation shall be administered orally to males and females at an appropriate time prior to mating. Administration shall continue until the weaning of the F2 generation.
All relevant fertility, gestation, parturition, maternal behaviour, suckling, growth and development of the F1 offspring from fertilisation to maturity and the development of the F2 offspring to weaning must be carefully observed and reported. Protocols for the reproduction toxicity study should be in line with OECD Guideline 416.
3.2.2.5.2.Prenatal developmental toxicity study (teratogenicity study)
The objective is to detect any adverse effects on the pregnant female and the development of the embryo and foetus as a result of exposure from implantation through the entire gestation period. Such effects include enhanced toxicity in the pregnant females, embryo-foetal death, altered foetal growth and structural abnormalities and anomalies in the foetus.
The rat is usually the species of choice for the first study. If a negative or an equivocal result for teratogenicity is observed, another developmental toxicity study shall be conducted in a second species, preferably the rabbit. If the rat study is positive for teratogenicity, a study in a second species is not necessary except where a review of all the core studies indicates that the ADI would be based on the rat teratogenicity. In this case a study in a second species would be required to determine the most sensitive species for this endpoint. Protocols should be in line with OECD Guideline 414.
3.2.2.6.Other specific toxicological and pharmacological studies
Further studies providing additional information useful for the assessment of the safety of the active substance and its residues shall be conducted if there are reasons for concern. Such studies may include examination of pharmacological effects, effects in juvenile (prepubertal) animals, immunotoxicity or neurotoxicity.
3.2.2.7.Determination of No Observed Adverse Effect Levels (NOAEL)
The NOAEL is generally based on toxicological effects, but pharmacological effects might occasionally be more appropriate.
The lowest NOAEL shall be selected. All findings from previous sections together with all other relevant published data (including any relevant information on the effects of the active substance on human) and information, where appropriate, on chemicals having a closely related chemical structure shall be taken into consideration in identifying the lowest NOAEL, expressed as mg per kg body weight per day.
3.2.3.Assessment of consumer safety
Consumer safety is assessed by a comparison of the established ADI (Acceptable Daily Intake) and calculated theoretical intake of the additive or its metabolites from food. In the case of vitamins and trace elements, UL (Tolerable Upper Intake Level) can be used in place of ADI.
3.2.3.1.Proposal of the acceptable daily intake (ADI) for the active substance(s)
The acceptable daily intake (ADI) (expressed as mg of additive or additive related material per person per day) is derived by dividing the lowest NOAEL (mg per kg body weight) by an appropriate safety factor and multiplying by the average human body weight of 60 kg.
An ADI shall, where appropriate, be proposed. An ADI can also be ‘not specified’ because of low toxicity in animal tests. An ADI shall not be proposed if the substance shows genotoxic or carcinogenic properties relevant to humans.
The setting of an ADI normally requires the similarity of metabolic fate of the active substance in the target animals and laboratory animals (see 3.2.1.4 Bioavailability of residues) which ensures that consumers are exposed to the same residues as the laboratory animals used in toxicological studies. If not, additional studies in a second laboratory animal species or with the metabolites specific to the target species may still allow an ADI to be set.
The safety factor used to determine the ADI for a particular additive will take into consideration the nature of the biological effects and the quality of the data used to identify the NOAEL, the relevance of these effects to man and their reversibility and any knowledge of the direct effect(s) of the residues in human.
A safety factor of at least 100 in calculating the ADI (if a full toxicological package has been provided) shall be employed. Where data on the active substance are available for human, a lower safety factor may be acceptable. Higher safety factors might be applied to account for additional sources of uncertainty in data or where the NOAEL is set on the basis of a particular critical endpoint, such as teratogenicity.
3.2.3.2.Tolerable upper intake level (UL)
For some additives it may be more appropriate to base the safety assessment on the UL, which is the maximum level of total chronic daily intake of a nutrient (from all sources) judged (by national or international scientific bodies) to be unlikely to pose a risk of adverse health effects to consumers or to specific groups of consumers.
The dossier shall contain data to demonstrate that use of the additive would not lead to a situation in which the UL could be exceeded considering all possible sources of the nutrient.
If the resulting residue levels of the nutritional additive or its metabolite(s) in products of animal origin are higher than what is considered normal or expected for these products, this shall be clearly indicated.
3.2.3.3.Consumer exposure
The total intake of the additive and/or its metabolites from all sources by the consumer shall be below the ADI or UL.
Calculation of the theoretical intake from food of animal origin shall be performed considering the concentration (total residues as the arithmetic mean and the highest single value) measured in tissues and products at the termination of use of the additive. In addition, if necessary, at the different withdrawal times, the human daily food consumption values shall be determined following a worst case scenario.
For additives intended for multi-species, the exposure from tissues shall be independently calculated for mammals, birds and fish and the highest value taken. Where appropriate, exposure from milk and eggs shall be added to this figure. For example, where an additive is applied for lactating mammals and laying birds, the respective highest edible tissue values are added to those for milk and egg consumption. Where the additive is applied for fish and laying birds and lactating mammals, the respective highest edible tissue values are added to those for egg and milk consumption. Other combinations shall be envisaged in the same way.
In certain situations (e.g. some nutritional and sensory additives or additives intended for minor species) it may be appropriate to subsequently refine the human exposure assessment using more realistic consumption figures, but still keeping the most conservative approach. Where this is possible this shall be based on Community data.
Table 1
Theoretical daily human consumption figures (g tissues or products)
3.2.3.4.Proposal for maximum residue limits (MRLs)
Maximum residue limit means the maximum concentration of residues (expressed as μg marker residue per kg of edible wet tissue or product) which may be accepted by the Community to be legally permitted or recognised as acceptable in food. It is based on the type and amount of residue considered to be without any toxicological hazard for human health as expressed by the ADI. An MRL cannot be set in the absence of an ADI.
When establishing MRLs for feed additives, consideration is also given to residues that come from other sources (e.g., food of plant origin). Furthermore, the MRL may be reduced to be consistent with the conditions of use of feed additives and to the extent that practical analytical methods are available.
Where appropriate, individual MRLs (expressed as mg marker residue per kg of edible natural tissue or product) shall be set for different tissues or products of the target animal species. The individual MRLs in different tissues or products shall reflect the depletion kinetics and the variability of the residue levels within those tissues/products in the animal species intended for use. Variability shall normally be reflected by using the 95 % confidence limit of the mean. If the confidence limit cannot be calculated due to a low number of samples, variability is expressed by taking the highest individual value instead.
Studies concerning the Maximum Residue Limits of coccidiostats and histomonostats must be carried out following the appropriate rules in force for veterinary medicinal products (Volume 8 ‘The rules governing medicinal products in European Union — Notice to applicants and guidelines. Veterinary medicinal products. Establishment of maximum residue limits (MRLs) for residues of veterinary medicinal products in foodstuffs of animal origin’. October 2005).
The studies to establish maximum residue limits for additive categories other than coccidiostats and histomonostats, where necessary, shall be provided according to this Annex.
To determine the consumer exposure to the total residues (as calculated under 3.2.3.3.), the proposed MRLs for the different tissues or products shall take into account the ratio of marker residue to total residue (Table 2).
Table 2
Definitions used in deriving an MRL
| i-j | Individual tissues/products (liver, kidney, muscle, skin + fat, milk, eggs, honey) at different times |
| MRLi-j | Maximum residue limit in tissues/products (mg marker substance kg-1) |
| Qti-j | Daily human consumption of individual tissues/products (kg) set by Table 1 or its refinement |
| TRCi-j | Total residue concentration in individual tissues/products (mg kg-1) |
| MRCi-j | Marker residue concentration in individual tissues/products (mg kg-1) |
| RMTRi-j | Ratio MRCi-j to TRCi-j for individual tissues/products |
| DITRi-j | Dietary intake for individual tissues/products calculated from total residues (mg) DITRi-j = Qti-j x TRCi-j |
| DITRMRLi-j | Dietary intake calculated from MRLs (mg) of individual tissues/products DITRMRLi-j = Qti-j x MRLi-j x RMTRi- j -1 |
The measured values for TRC and MRC shall be inserted as appropriate in the template shown in Table 3, and the other values calculated. Where a full data set is not available because values fall below the limit of detection (LOD), an extrapolation of RMTR may be acceptable.
Deriving an MRL can only be performed if the sum of the individual DITRs is below the ADI. If the ADI is exceeded, an alternative would be to use data from a longer withdrawal time or lower dosages. A first proposal for an MRL can be obtained using the MRC value as a guide and taking into consideration the LOQ of the analytical method. The sum of the DITRMRL obtained from the proposed MRLs must be below the ADI and close to the sum of the individual DITRs. If the ADI is exceeded, then a lower MRL shall be proposed and the comparison repeated.
For certain additives, residues could arise below the MRL values in milk, eggs or meat which could nonetheless interfere with food quality in particular food processing procedures. For such additives, it may be appropriate to consider a ‘maximum (food product) processing compatible residue’ (MPCR) in addition to establishing MRL values.
Table 3
Template for deriving a MRL proposal
3.2.3.5.Proposal for a withdrawal period
The withdrawal time comprises the period after cessation of the administration of the additive which is necessary to enable the residue levels to fall below the MRLs.
3.3.Studies concerning the safety of use of the additive for users/workers
Workers can be exposed mainly by inhalation or topical exposure while manufacturing or handling or using the additive. For example, farm workers are potentially exposed when handling or mixing the additive. Additional information on how the substances are handled shall be provided.
An assessment of risk to workers shall be included. Where available, experience in the manufacturing plant is often an important source of information in evaluating the risks to workers from exposure to the additive itself by both airborne and topical routes. Of particular concern are additives/additive-treated feeds and/or animal excreta, which are in, or may give rise to, a dry powdery form, and feed additives which may have allergenic potential.
3.3.1.Toxicological risk assessment for user/worker safety
Risks to workers shall be assessed in a series of studies using the additive in the form for which the application has been submitted. Acute inhalation toxicity studies shall be performed unless the product is unlikely to form a respirable dust or mist. Studies on skin irritancy must be performed, and if these give negative results, mucous membrane (e.g. eye) irritancy shall be assessed. Allergenic potential/skin sensitisation potential shall also be assessed. The toxicity data generated to meet consumer safety (see 3.2.2) shall be used to assess the potential systemic toxicity of the additive. All these shall be assessed, if necessary, by direct measurement and specific studies.
3.3.1.1.Effects on the respiratory system
Evidence shall be provided that airborne levels of dust or mist of the additive will not constitute a hazard to the health of users/workers. This evidence shall include, where necessary:
inhalation tests in laboratory animals;
published epidemiological data and/or the applicants own data on its work plant and/or irritancy; and
respiratory system sensitisation tests.
Acute inhalation toxicity studies shall be performed if particles or droplets with a diameter of less than 50 μm constitute more than 1 % on a weight basis of the product.
Protocols for acute inhalation toxicity studies should be in line with OECD Guideline 403. If sub-chronic toxicity studies are considered necessary, they should follow OECD Guidelines 412 (Repeated Dose Inhalation Toxicity: 28-day or 14-day study) or 413 (Sub-chronic Inhalation Toxicity: 90-day study).
3.3.1.2.Effects on the eyes and skin
Where available, direct evidence of absence of irritancy and/or sensitisation shall be provided from known human situations. This shall be supplemented by findings from validated animal tests for skin and eye irritation, and for sensitisation potential using the appropriate additive. Allergic potential — skin sensitisation potential shall also be assessed. Protocols for these studies should be in line with OECD Guidelines 404 (Dermal Irritation/Corrosion), 405 (Eye Irritation/Corrosion), 406 (Skin Sensitisation), 429 (Skin Sensitisation — local lymph-node assay).
If corrosive properties are known, either from published data or specific in vitro tests, then further in vivo tests shall not be performed.
Dermal toxicity must be considered, if the additive is toxic by inhalation. Studies must be in line with OECD Guideline 402 (Acute Dermal Toxicity).
3.3.1.3.Systemic toxicity
The toxicity data generated to meet consumer safety and other requirements (including repeated dose toxicity, mutagenicity, carcinogenicity and reproductive testing and metabolic fate) shall be used to assess systemic toxicity.
3.3.1.4.Exposure assessment
Information shall be provided on how the use of the additive is likely to give rise to exposure by all routes (inhalation, through the skin or by ingestion). This information shall include a quantitative assessment, where available, such as typical airborne concentration, dermal contamination or ingestion. Where quantitative information is not available, sufficient information shall be given to enable an adequate assessment of exposure to be made.
3.3.2.Measures to control exposure
Using the information from the toxicology and exposure assessment, a conclusion shall be drawn about the risks to health of the users/workers (inhalation, irritancy, sensitisation and systemic toxicity). Precautionary measures may be proposed to reduce or eliminate exposure. However, use of personal protective devices shall only be regarded as a measure of last resort to protect against any residual risk once control measures are in place. It is preferable, for example, to consider reformulation of the product.
3.4.Studies concerning the safety of use of the additive for the environment
Consideration of the environmental impact of additives is important since administration of additives typically occurs over long periods, often involves large groups of animals and the active substance(s) may be excreted to a considerable extent either as the parent compound or its metabolites.
To determine the environmental impact of additives, a stepwise approach shall be followed. All additives have to be assessed through Phase I to identify those additives which do not need further testing. For the other additives a second phase (Phase II) assessment is needed to provide additional information, based upon which further studies may be considered necessary. These studies shall be conducted according to Directive 67/548/EEC.
3.4.1.Phase I assessment
The purpose of Phase I assessment is to determine if a significant environmental effect of the additive or its metabolites is likely and whether a Phase II assessment is necessary (see decision tree).
Exemption from Phase II assessment may be made on one of two criteria, unless there is scientifically-based evidence for concern:
(a)
the chemical nature and the biological effect of the additive and its conditions of use indicate that impact will be negligible, i.e. where the additive is:
a physiological or natural substance that will not result in a substantial increase of the concentration in the environment; or
intended for non-food producing animals;
(b)
the worst case Predicted Environmental Concentration (PEC) is too low to be of concern. The PEC shall be evaluated for each compartment of concern (see below), assuming that 100 % of the dose ingested is excreted as the parent compound.
If the applicant cannot demonstrate that the additive falls into one of these exemption categories, a Phase II assessment will be required.
3.4.1.1.Additives for terrestrial animals
When excreta from livestock are applied on land, the use of feed additives can lead to contamination of soil, ground water, and surface water (via drainage and run-off).
The worst case PEC for soil (PECsoil) would arise considering all excreted compounds being spread on land. If the PECsoil (default: 5 cm depth) is less than 10 μg/kg, no further assessment is required.
If the PEC for contamination of groundwater (PECgw) is less than 0,1 μg/l, no Phase II assessment of the environmental impact of the additive on groundwater is necessary.
3.4.1.2.Additives for aquatic animals
Feed additives used in aquaculture can result in contamination of sediment and water. The compartment of concern for the environmental risk assessment for fish farmed in cages is assumed to be the sediment. For fish farmed in land-based systems the effluent flowing to surface water is considered to pose the major environmental risk.
The worst case PEC for sediment (PECsediment) would arise considering all excreted compounds being deposited in the sediment. If the PECsediment (default: 20 cm depth) is less than 10 μg/kg wet weight, then no further assessment is required.
If the PEC in the surface water (PECsw) is less than 0,1 μg/l, no further assessment is required.
Phase I — Decision tree
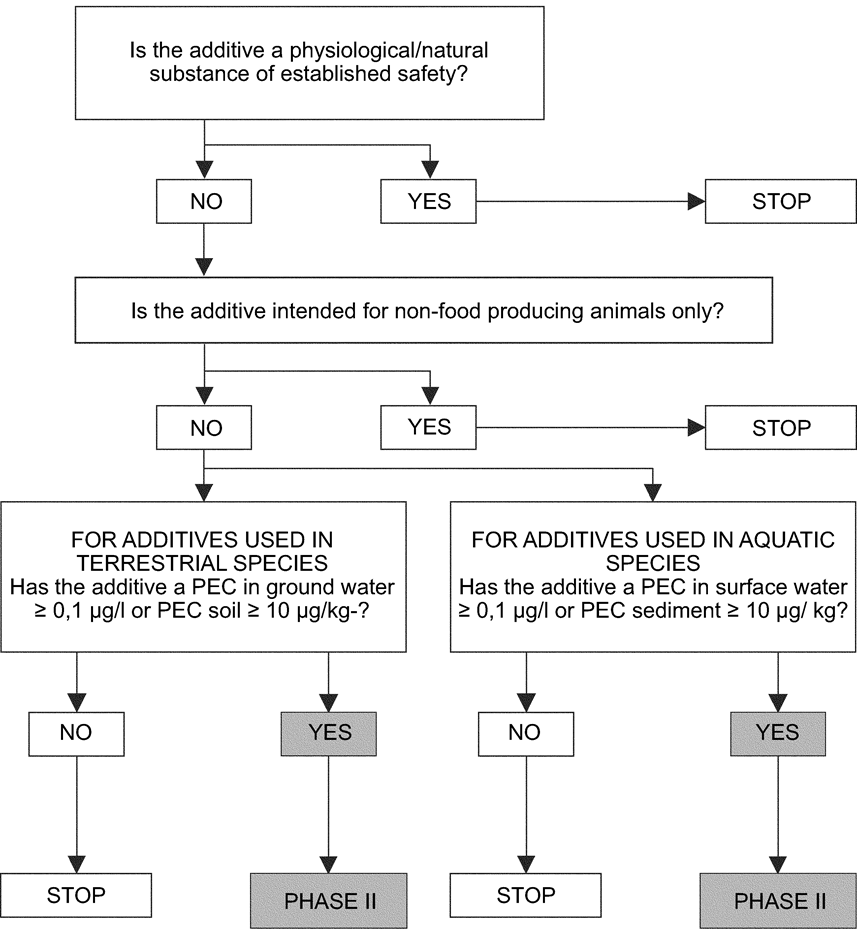
3.4.2.Phase II assessment
The aim of Phase II is to assess the potential for additives to affect non-target species in the environment, including both aquatic and terrestrial species or to reach groundwater at unacceptable levels. It is not practical to evaluate the effects of additives on every species in the environment that may be exposed to the additive following its administration to the target species. The taxonomic levels tested are intended to serve as surrogates or indicators for the range of species present in the environment.
The Phase II assessment is based on a risk quotient approach, where the calculated PEC and Predicted No Effect Concentration (PNEC) values for each compartment shall be compared. The PNEC is determined from experimentally determined endpoints divided by an appropriate assessment factor. The PNEC value shall be calculated for each compartment.
The Phase II assessment starts with a refinement of the PEC if possible, and uses a two-tiered approach to the environmental risk assessment.
The first tier, Phase IIA, makes use of a limited number of fate and effect studies to produce a conservative assessment of risk based on exposure and effects in the environmental compartment of concern. If the ratio of the PEC to the PNEC is lower than one (1), no further assessment is required, unless bioaccumulation is expected.
If the PEC/PNEC ratio predicts an unacceptable risk (ratio > 1), the applicant shall progress to Phase IIB to refine the environmental risk assessment.
3.4.2.1.Phase II A
In addition to the compartments considered in Phase I, the PEC for surface water has to be calculated considering runoff and drainage.
Based on data not considered in Phase I, a more refined PEC can be calculated for each environmental compartment of concern. In ascertaining the refined PEC, account shall be taken of:
(a)
the concentration of active substance(s)/metabolites of concern in manure/fish faeces following administration of the additive to animals at the proposed dose level. This calculation shall include consideration of dosage rates and amount of excreta produced;
(b)
the potential degradation of the excreted active substance(s)/metabolites of concern during normal manure processing practice and storage prior to its application to land;
(c)
the adsorption/desorption of the active substance(s)/metabolites of concern onto soil or sediment for aquaculture, preferentially determined by studies in soil/sediment (OECD 106);
(d)
degradation in soil and water/sediment systems (OECD 307 and 308, respectively); and
(e)
other factors such as hydrolysis, photolysis, evaporation, dilution through ploughing.
The highest value for the PEC obtained from these calculations for each environmental compartment of concern shall be adopted for Phase II risk assessment purposes.
If a high persistence in soil/sediment is anticipated (time to degradation of 90 % of original concentration of the compound: DT90 > 1 year), the potential for accumulation shall be considered.
The concentrations of additives (or metabolites) producing serious adverse effects for various trophic levels in the environmental compartments of concern shall be determined. These tests are mostly acute tests and should follow OECD or similar well-established guidelines. Studies for the terrestrial environment shall include: toxicity to earthworms; three terrestrial plants; and soil micro-organisms (e.g. effects on nitrogen fixation). Studies for the fresh water environment shall include: toxicity to fish; Daphnia magna; algae; and a sediment dwelling organism. In case of sea cages, three species of different taxa of sediment dwelling organisms shall be studied.
Calculation of the PNEC value shall be carried out for each compartment of concern. The PNEC is normally derived from the lowest toxicity value observed in the above tests and dividing by a safety factor of at least 100 depending on the endpoint and number of test species used.
The potential for bioaccumulation can be estimated from the value of the n-octanol/water partition coefficient, Log Kow. Values ≥ 3 indicate that the substance may bioaccumulated. In order to assess the risk for secondary poisoning it shall be considered whether to carry out a bioconcentration factor (BCF) study at Phase IIB.
3.4.2.2.Phase IIB (more detailed ecotoxicological studies)
For those additives where, following Phase IIA assessment, an environmental risk cannot be excluded, more information is required on the effects on biological species in the environmental compartment(s) in which Phase IIA studies indicate possible concern. In this situation, further tests are needed to determine the chronic and more specific effects on appropriate microbial, plant, and animal species. This additional information will allow the application a lower safety factor.
Suitable additional ecotoxicity tests are described in a number of publications, e.g. in OECD Guidelines. Careful choice of such tests is necessary to ensure that they are appropriate to the situation in which the additive and/or its metabolites may be released and dispersed in the environment. The refinement of the effect assessment for soil (PNECsoil) could be based on studies on the chronic effects on earthworms, additional studies on soil microflora and a number of relevant plant species, studies on grassland invertebrates (including insects) and feral birds.
The refinement of the effect assessment for water/sediment could be based on chronic toxicity tests on the most sensitive aquatic/benthic organisms identified in Phase IIA assessment.
Bioaccumulation studies, if necessary, should be performed according to OECD Guideline 305.
(1)
A non-exhaustive list is available in: www.efsa.europa.eu/en/science/feedap/feedap_opinion/993.html
Options/Help
Print Options
PrintThe Whole Regulation
PrintThe Whole Annex
PrintThis Division only
You have chosen to open the Whole Regulation
The Whole Regulation you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
The Schedules you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As adopted by EU): The original version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Opening Options
Different options to open legislation in order to view more content on screen at once
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the EU Official Journal
- lists of changes made by and/or affecting this legislation item
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.