Commission Implementing Regulation (EU) No 1348/2013
of 16 December 2013
amending Regulation (EEC) No 2568/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
THE EUROPEAN COMMISSION,
Having regard to the Treaty on the Functioning of the European Union,
Having regard to Council Regulation (EC) No 1234/2007 of 22 October 2007 establishing a common organisation of agricultural markets and on specific provisions for certain agricultural products (Single CMO Regulation)(1), and in particular Article 113(1)(a) and points (a) and (h) of the first paragraph of Article 121, in conjunction with Article 4 thereof,
Whereas:
(1) Commission Regulation (EEC) No 2568/91(2) defines the chemical and organoleptic characteristics of olive and olive-pomace oil and lays down methods of assessing those characteristics. Those methods and the limit values for the characteristics of oils should be updated on the basis of the opinion of chemical experts and in line with the work carried out within the International Olive Council (IOC).
(2) To ensure the implementation at Union level of the most recent international standards established by the IOC, certain methods of analysis as well as certain limit values for the characteristics of oils laid down in Regulation (EEC) No 2568/91 should be updated.
(3) Consequently, the limit values for stigmastadienes, waxes, myristic acid and fatty acids alkyl esters should be adapted and some decision trees for verifying whether on olive-oil sample is consistent with the category declared should be amended accordingly. Decision trees for campesterol and delta-7-stigmastenol accompanied by more restrictive parameters should be introduced in order to facilitate trade and guarantee olive oil authenticity, in the interest of consumer protection. The method of analysis relating to the composition and content of sterols and the determination of erythrodiol and uvaol should be replaced by a more reliable method which also covers triterpene dialcohols. It is also appropriate to review the organoleptic assessment of olive oil and to insert a method enabling the detection of extraneous vegetable oils in olive oils.
(4) In the light of the developments relating to the procedures for the conformity checks of oils, the method of sampling of olive oil and olive-pomace oils should be adapted accordingly.
(5) Regulation (EEC) No 2568/91 should therefore be amended accordingly.
(6) In order to allow a period of adjustment to the new rules, to give time for introducing the means of applying them and to avoid disturbance to commercial transactions, the amendments made by this Regulation should apply as from 1 March 2014. For the same reasons, provision should be made for olive oil and olive-pomace oils that are legally manufactured and labelled in the Union or legally imported into the Union and released for free circulation before that date to be marketed until all stocks are used up.
(7) The measures provided for in this Regulation are in accordance with the opinion of the Management Committee for the Common Organisation of Agricultural Markets.
HAS ADOPTED THIS REGULATION:
Article 1U.K.
Regulation (EEC) No 2568/91 is amended as follows:
Article 2 is replaced by the following:
‘Article 2
1.The characteristics of oils laid down in Annex I shall be determined in accordance with the following methods of analysis:
(a)for the determination of the free fatty acids, expressed as the percentage of oleic acid, the method set out in Annex II;
(b)for the determination of the peroxide index, the method set out in Annex III;
(c)for determination of the wax content, the method set out in Annex IV;
(d)for the determination of the composition and content of sterols and triterpene dialcohols by capillary-column gas chromatography, the method set out in Annex V;
(e)for the determination of the percentage of 2- glyceryl monopalmitate, the method set out in Annex VII;
(f)for spectrophotometric analysis, the method set out in Annex IX;
(g)for the determination of the fatty acid composition, the method set out in Annex X A and X B;
(h)for the determination of the volatile halogenated solvents, the method set out in Annex XI;
(i)for the evaluation of the organoleptic characteristics of virgin olive oil, the method set out in Annex XII;
(j)for the determination of stigmastadienes, the method set out in Annex XVII;
(k)for determining the content of triglycerides with ECN42, the method set out in Annex XVIII;
(l)for the determination of the aliphatic alcohol content, the method set out in Annex XIX;
(m)for the determination of the content of waxes, fatty acid methyl esters and fatty acid ethyl esters, the method set out in Annex XX.
In order to detect the presence of extraneous vegetable oils in olive oils, the method of analysis set out in Annex XXa shall be applied.
2.Verification by national authorities or their representatives of the organoleptic characteristics of virgin oils shall be effected by tasting panels approved by the Member States.
The organoleptic characteristics of an oil as referred to in the first subparagraph shall be deemed consonant with the category declared if a panel approved by the Member State confirms the grading.
Should the panel not confirm the category declared as regards the organoleptic characteristics, at the interested party's request, the national authorities or their representatives shall have carried out without delay two counter-assessments by other approved panels, at least one by a panel approved by the producer Member State concerned. The characteristics concerned shall be deemed consonant with the characteristics declared if at least two of the counter-assessments confirm the declared grade. If that is not the case, the interested party shall be responsible for the cost of the counter-assessments.
3.When the national authorities or their representatives verify the characteristics of the oil as provided for in paragraph 1, samples shall be taken in accordance with international standards EN ISO 661 on the preparation of test samples and EN ISO 5555 on sampling. However, notwithstanding point 6.8 of standard EN ISO 5555, in case of batches of such oils in immediate packaging, the sample shall be taken in accordance with Annex Ia to this Regulation. In case of bulk oils for which the sampling cannot be performed according to EN ISO 5555, the sampling shall be performed in accordance with instructions provided by the competent authority of the Member State.
Without prejudice to standard EN ISO 5555 and Chapter 6 of standard EN ISO 661, the samples taken shall be put in a dark place away from strong heat as quickly as possible and sent to the laboratory for analysis no later than the fifth working day after they are taken, otherwise the samples shall be kept in such a way that they will not be degraded or damaged during transport or storage before being sent to the laboratory.
4.For the purposes of the verification provided for in paragraph 3, the analyses referred to in Annexes II, III, IX, XII and XX and, where applicable, any counter-analyses required under national law, shall be carried out before the minimum durability date in case of packaged products. In case of sampling of bulk oils, those analyses shall be carried out no later than the sixth month after the month in which the sample was taken.
No time limit shall apply to the other analyses provided for in this Regulation.
Unless the sample was taken less than two months before the minimum durability date, if the results of the analyses do not match the characteristics of the category of olive oil or olive-pomace oil declared, the party concerned shall be notified no later than one month before the end of the period laid down in the first subparagraph.
5.For the purpose of determining the characteristics of olive oils by the methods provided for in the first subparagraph of paragraph 1, the analysis results shall be directly compared with the limits laid down in this Regulation.’
Annex I is replaced by the text set out in Annex I to this Regulation.
Annex Ia is replaced by the text set out in Annex II to this Regulation.
Annex Ib is replaced by the text set out in Annex III to this Regulation.
Annex V is replaced by the text set out in Annex IV to this Regulation.
Annex VI is deleted.
Annex XII is replaced by the text set out in Annex V to this Regulation.
Annex XXa, the text of which is set out in Annex VI to this Regulation, is inserted after Annex XX.
Article 2U.K.
Products which have been legally manufactured and labelled in the Union or legally imported into the Union and released for free circulation before 1 March 2014 may be marketed until all stocks are used up.
Article 3U.K.
This Regulation shall enter into force on the seventh day following that of its publication in the Official Journal of the European Union.
It shall apply from 1 March 2014.
This Regulation shall be binding in its entirety and directly applicable in all Member States.
Done at Brussels, 16 December 2013.
For the Commission
The President
José Manuel Barroso
ANNEX IU.K.
“ANNEX 1 OLIVE OIL CHARACTERISTICS
| a Total isomers which could (or could not) be separated by capillary column. | ||||||||||||
| b The olive oil has to be in conformity with the method set out in Annex XXa. | ||||||||||||
| c This limit applies to olive oils produced as from 1st March 2014 | ||||||||||||
| d Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be lampante olive oil if the total aliphatic alcohol content is less than or equal to 350 mg/kg or if the erythrodiol and uvaol content is less than or equal to 3,5 %. | ||||||||||||
| e Or where the median of defect is above 3,5 or the median of defect is less than or equal to 3,5 and the fruity median is equal to 0. | ||||||||||||
| f Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be crude olive-pomace oil if the total aliphatic alcohol content is above 350 mg/kg and if the erythrodiol and uvaol content is greater than 3,5 %. | ||||||||||||
| Category | Fatty acid ethyl esters (FAEEs) mg/kg (*) | Acidity (%) (*) | Peroxide índex mEq O2/kg (*) | Waxes mg/kg (**) | 2-glyceril monopalmitate (%) | Stigmastadienes mg/kga | Difference: ECN42 (HPLC) and ECN42b(theoretical calculation) | K232 (*) | K268 or K270 (*) | Delta-K (*) | Organoleptic evaluationMedian of defect (Md) (*) | Organoleptic evaluationFruity median (Mf) (*) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
1. Extra virgin olive oil | FAEEs ≤ 40 (2013-2014 crop year)c FAEEs ≤ 35 (2014-2015 crop year) FAEEs ≤ 30 (after 2015 crop years) | ≤ 0,8 | ≤ 20 |  | ≤ 0,9 if total palmitic acid % ≤ 14 % | ≤ 0,05 | ≤ |0,2| | ≤ 2,50 | ≤ 0,22 | ≤ 0,01 | Md = 0 | Mf > 0 |
| ≤ 1,0 if total palmitic acid % > 14 % | ||||||||||||
2. Virgin olive oil | — | ≤ 2,0 | ≤ 20 |  | ≤ 0,9 if total palmitic acid % ≤ 14 % | ≤ 0,05 | ≤ |0,2| | ≤ 2,60 | ≤ 0,25 | ≤ 0,01 | Md ≤ 3,5 | Mf > 0 |
| ≤ 1,0 if total palmitic acid % > 14 % | ||||||||||||
3. Lampante olive oil | — | > 2,0 | — |  d d | ≤ 0,9 if total palmitic acid % ≤ 14 % | ≤ 0,50 | ≤ |0,3| | — | — | — | Md > 3,5e | — |
| ≤ 1,1 if total palmitic acid % > 14 % | ||||||||||||
4. Refined olive oil | — | ≤ 0,3 | ≤ 5 |  | ≤ 0,9 if total palmitic acid % ≤ 14 % | — | ≤ |0,3| | — | ≤ 1,10 | ≤ 0,16 | — | — |
| ≤ 1,1 if total palmitic acid % > 14 % | ||||||||||||
5. Olive oil composed of refined and virgin olive oils | — | ≤ 1,0 | ≤ 15 |  | ≤ 0,9 if total palmitic acid % ≤ 14 % | — | ≤ |0,3| | — | ≤ 0,90 | ≤ 0,15 | — | — |
| ≤ 1,0 if total palmitic acid % > 14 % | ||||||||||||
6. Crude olive-pomace | — | — | — |  f f | ≤ 1,4 | — | ≤ |0,6| | — | — | — | — | — |
7. Refined olive-pomace oil | — | ≤ 0,3 | ≤ 5 |  | ≤ 1,4 | — | ≤ |0,5| | — | ≤ 2,00 | ≤ 0,20 | — | — |
8. Olive-pomace oil | — | ≤ 1,0 | ≤ 15 |  | ≤ 1,2 | — | ≤ |0,5| | — | ≤ 1,70 | ≤ 0,18 | — | — |
| a Other fatty acids content (%): palmitic: 7,50-20,00; palmitoleic: 0,30-3,50; heptadecanoic: ≤ 0,30; heptadecenoic: ≤ 0,30; stearic: 0,50-5,00; oleic: 55,00-83,00; linoleic: 3,50-21,00. | ||||||||||||||||
| b See the Appendix to this Annex. | ||||||||||||||||
| c App β-sitosterol: Delta-5,23-stigmastadienol+chlerosterol+beta-sitosterol+sitostanol+delta-5-avenasterol+delta-5,24-stigmastadienol. | ||||||||||||||||
| d Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be lampante olive oil if the total aliphatic alcohol content is less than or equal to 350 mg/kg or if the erythrodiol and uvaol content is less than or equal to 3,5 %. | ||||||||||||||||
| e Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be crude olive-pomace oil if the total aliphatic alcohol content is above 350 mg/kg and if the erythrodiol and uvaol content is greater than 3,5 %. | ||||||||||||||||
| Category | Fatty acid compositiona | Total transoleic isomers(%) | Total translinoleic + translinolenic isomers(%) | Sterols composition | Total sterols(mg/kg) | Erythrodiol and uvaol(%) (**) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Myristic(%) | Linolenic(%) | Arachidic(%) | Eicosenoic(%) | Behenic(%) | Lignoceric(%) | Cholesterol(%) | Brassicasterol(%) | Campesterolb(%) | Stigmasterol(%) | App β–sitosterol(%)c | Delta-7-stigmastenolb(%) | |||||
1. Extra virgin olive oil | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,40 | ≤ 0,20 | ≤ 0,20 | ≤ 0,05 | ≤ 0,05 | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | < Camp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 |
2. Virgin olive oil | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,40 | ≤ 0,20 | ≤ 0,20 | ≤ 0,05 | ≤ 0,05 | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | < Camp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 |
3. Lampante olive oil | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,40 | ≤ 0,20 | ≤ 0,20 | ≤ 0,10 | ≤ 0,10 | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | — | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5d |
4. Refined olive oil | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,40 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,30 | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | < Camp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 |
5. Olive oil composed of refined and virgin olive oils | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,40 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,30 | ≤ 0,5 | ≤ 0,1 | ≤ 4,0 | < Camp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 000 | ≤ 4,5 |
6. Crude olive-pomace oil | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,40 | ≤ 0,30 | ≤ 0,20 | ≤ 0,20 | ≤ 0,10 | ≤ 0,5 | ≤ 0,2 | ≤ 4,0 | — | ≥ 93,0 | ≤ 0,5 | ≥ 2 500 | > 4,5e |
7. Refined olive-pomace oil | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,40 | ≤ 0,30 | ≤ 0,20 | ≤ 0,40 | ≤ 0,35 | ≤ 0,5 | ≤ 0,2 | ≤ 4,0 | < Camp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 800 | > 4,5 |
8. Olive-pomace oil | ≤ 0,03 | ≤ 1,00 | ≤ 0,60 | ≤ 0,40 | ≤ 0,30 | ≤ 0,20 | ≤ 0,40 | ≤ 0,35 | ≤ 0,5 | ≤ 0,2 | ≤ 4,0 | < Camp. | ≥ 93,0 | ≤ 0,5 | ≥ 1 600 | > 4,5 |
Notes: U.K.
(a)The results of the analyses must be expressed to the same number of decimal places as used for each characteristic. The last digit must be increased by one unit if the following digit is greater than 4.U.K.
(b)If just a single characteristic does not match the values stated, the category of an oil can be changed or the oil declared impure for the purposes of this Regulation.U.K.
(c)If a characteristic is marked with an asterisk (*), referring to the quality of the oil, this means the following: - for lampante olive oil, it is possible for both the relevant limits to be different from the stated values at the same time, - for virgin olive oils, if at least one of these limits is different from the stated values, the category of the oil will be changed, although they will still be classified in one of the categories of virgin olive oil.U.K.
(d)If a characteristic is marked with two asterisks (**), this means that for all types of olive-pomace oil, it is possible for both the relevant limits to be different from the stated values at the same time.U.K.
Appendix Decision tree
Campesterol decision tree for virgin and extra virgin olive oils:

The other parameters shall comply with the limits fixed in this Regulation.
Delta-7-stigmastenol decision tree for:

The other parameters shall comply with the limits fixed in this Regulation.

ANNEX IIU.K.
“ANNEX Ia SAMPLING OF OLIVE OIL OR OLIVE-POMACE OIL DELIVERED IN IMMEDIATE PACKAGING
This method of sampling is applied to batches of olive oil or olive-pomace oil put up in immediate packaging. Different sampling methods apply, depending on whether the immediate packaging exceeds 5 litres or not.
“Batch” shall mean a set of sales units which are produced, manufactured and packed in circumstances such that the oil contained in each sales unit is considered to be homogenous in terms of all analytical characteristics. The individuation of a batch must be done in accordance with Directive 2011/91/EU of the European Parliament and of the Council(3).
“Increment” shall mean the quantity of oil contained in an immediate package and taken from a random point of the batch.
1.CONTENT OF PRIMARY SAMPLEU.K.
1.1. Immediate packaging not exceeding 5 litres U.K.
“Primary Sample” for immediate packaging not exceeding 5 litres shall mean the number of increments taken from a batch and in agreement with Table 1.
Table 1
Primary sample minimum size must comprise the following
The number of packs referred to in Table 1, which shall constitute a primary sample, can be increased by each Member State, according to their own needs (for example organoleptic assessment by a different laboratory from that which performed the chemical analyses, counter-analysis, etc.).
1.2. Immediate packaging exceeding 5 litres U.K.
“Primary Sample” for immediate packaging exceeding 5 litres shall mean a representative part of the total increments, obtained by a process of reduction and in agreement with Table 2. The primary sample must be composed of various examples.
“Example” of a primary sample shall mean each of the packages making up the primary sample.
Table 2
Minimum number of increments to be selected
| Number of packages in the lot | Minimum number of increments to be selected |
|---|---|
| Up to 10 | 1 |
| From … 11 to 150 | 2 |
| From … 151 to 500 | 3 |
| From … 501 to 1 500 | 4 |
| From … 1 501 to 2 500 | 5 |
| > 2 500 per 1 000 packages | 1 extra increment |
In order to reduce the volume of the sampling immediate packs, the content of the sampling increments is homogenised for the preparation of the primary sample. The portions of the different increments are poured into a common container for homogenisation by stirring, so that it will be best protected from air.
The content of the primary sample must be poured into a series of packages of the minimum capacity of 1,0 liter, each one of which constitutes an example of the primary sample.
The number of primary samples can be increased by each Member State, according to their own necessity (for example organoleptic assessment by a different laboratory from the one that performed the chemical analyses, counter-analysis, etc).
Each package must be filled in a way to minimise the air layer on top and then suitably closed and sealed to ensure the product is tamper-proof.
These examples must be labeled to ensure correct identification.
2.ANALYSES AND RESULTSU.K.
2.1.Each primary sample must be subdivided into laboratory samples, in accordance with point 2.5 of standard EN ISO 5555, and analysed according to the order shown in the decision tree set out in Annex Ib or in any other random order.U.K.
2.2.Where all the results of the analyses comply with the characteristics of the category of oil declared, the whole batch is to be declared to comply.U.K.
If a single result of the analyses does not comply with the characteristics of the category of oil declared, the whole batch is to be declared non compliant.
3.VERIFICATION OF THE CATEGORY OF BATCHU.K.
3.1.In order to verify the batch category, the competent authority may increase the number of primary samples taken at different points of the batch according to the following table:U.K.
| Table 3 | |
| Number of primary samples determined by the size of batch | |
| Size of batch (litres) | Number of primary samples |
|---|---|
| Less than 7 500 | 2 |
| From 7 500 to less than 25 000 | 3 |
| From 25 000 to less than 75 000 | 4 |
| From 75 000 to less than 125 000 | 5 |
| Equal to and more than 125 000 | 6 + 1 each 50 000 litres more |
Each increment constituting a primary sample must be taken from a continuous place in the batch; it is necessary to take note of the location of each primary sample and to identify it unambiguously.
The formation of each primary sample must be carried out according to the procedures referred to in points 1.1 and 1.2.
Each primary sample is then subjected to the analyses referred to in Article 2(1).
3.2.When one of the results of the analyses referred to in Article 2(1) of at least one primary sample does not comply with the characteristics of the declared category of oil, the whole sampling batch shall be declared non compliant.”U.K.
ANNEX IIIU.K.
“ANNEX Ib DECISION TREE FOR VERIFYING WHETHER AN OLIVE OIL SAMPLE IS CONSISTENT WITH THE CATEGORY DECLARED
Table 1 U.K.

Table 2 U.K.
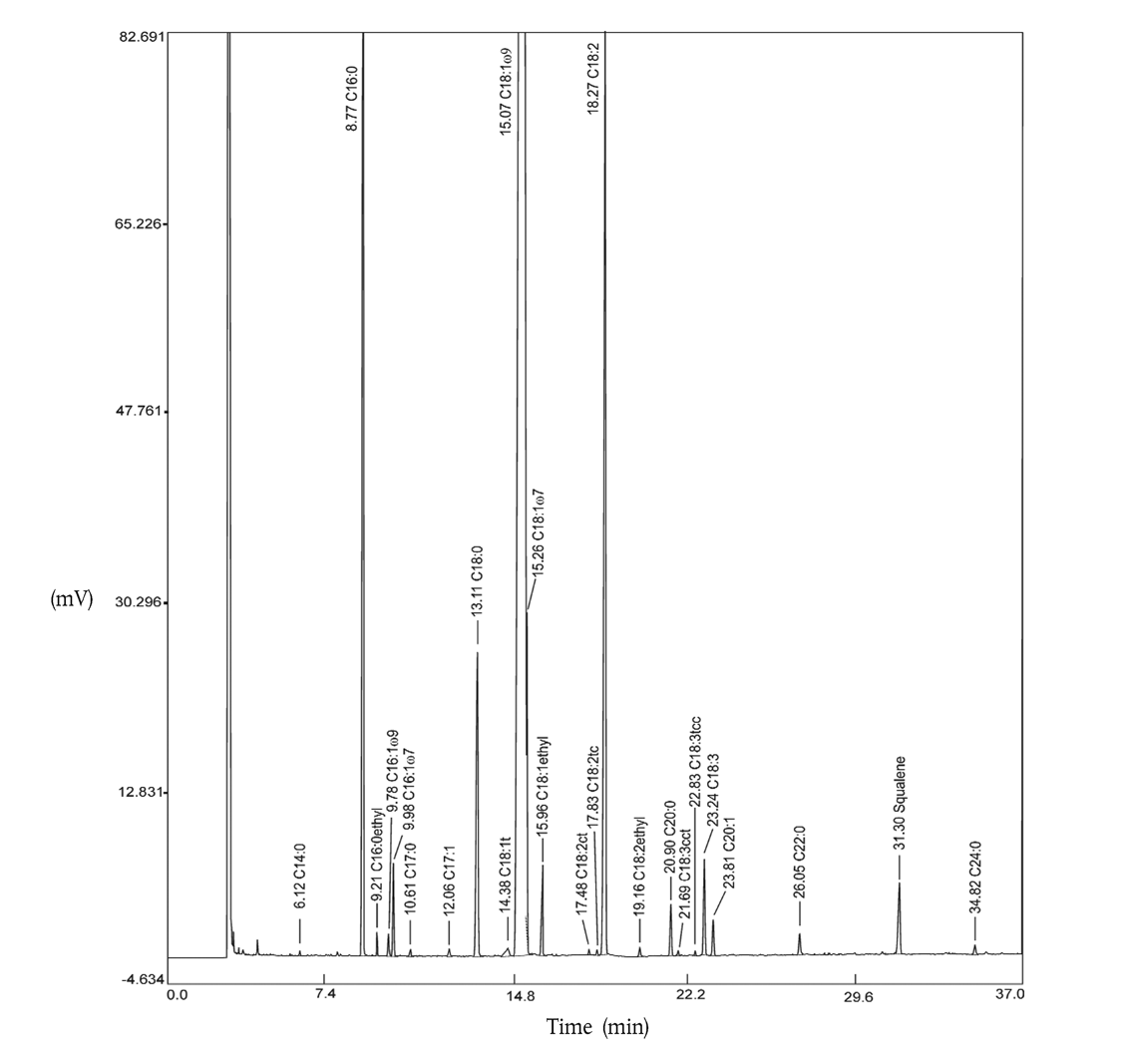
Table 3 U.K.

Appendix 1
Table of equivalence between the Annexes to this Regulation and the analyses specified in the decision tree
ANNEX IVU.K.
“ANNEX V DETERMINATION OF THE COMPOSITION AND CONTENT OF STEROLS AND TRITERPENES DIALCOHOLS BY CAPILLARY-COLUMN GAS CHROMATOGRAPHY
1.SCOPEU.K.
The method describes a procedure for determining the individual and total sterols and triterpene dialcohols content of olive oils and olive-pomace oils.
2.PRINCIPLEU.K.
The oil, with added α-cholestanol as an internal standard, is saponified with potassium hydroxide in ethanolic solution and the unsaponifiable matter is then extracted with ethyl ether.
The sterols and triterpene dialcohols fraction is separated from the unsaponifiable matter by thin-layer chromatography on a basic silica gel plate. The fractions recovered from the silica gel are transformed into trimethylsilyl ethers and then analysed by capillary column gas chromatography.
3.APPARATUSU.K.
The usual laboratory equipment and in particular the following:
250 ml flask fitted with a reflux condenser with ground-glass joints.
500 ml separating funnel.
250 ml flasks.
Complete apparatus for analysis by thin-layer chromatography using 20 x 20 cm glass plates.
Ultraviolet lamp with a wavelength of 254 or 366 nm.
100 μl and 500 μl microsyringes.
Cylindrical filter funnel with a G3 porous septum (porosity 15-40 μm) of diameter approximately 2 cm and a depth of 5 cm, suitable for filtration under vacuum with male ground-glass joint.
50 ml vacuum conical flask with ground-glass female joint, which can be fitted to the filter funnel (point 3.7).
10 ml test tube with a tapering bottom and a sealing glass stopper.
Gas chromatograph suitable for use with a capillary column with split injection system, consisting of:
A thermostatic chamber for columns capable of maintaining the desired temperature with an accuracy of ± 1°C;
A temperature-adjustable injection unit with a persilanised glass vaporising element and split system;
A flame ionisation detector (FID);
Data acquisition system suitable for use with the FID detector (point 3.10.3.), capable of manual integration.
Fused-silica capillary column of length 20 to 30 m, internal diameter 0,25 to 0,32 mm, coated with 5 % diphenyl - 95 % dimethylpolysiloxane (SE-52 or SE-54 stationary phase or equivalent), to a uniform thickness between 0,10 and 0,30 μm.
Microsyringe, of 10 ml capacity, for gas chromatography, with cemented needle suitable for split injection.
Calcium dichloride desiccator
4.REAGENTSU.K.
4.1.Potassium hydroxide minimum titre 85 %.U.K.
4.2.Potassium hydroxide ethanolic solution, approximately 2 N.U.K.
Dissolve 130 g of potassium hydroxide (point 4.1) with cooling in 200 ml of distilled water and then make up to one litre with ethanol (point 4.10). Keep the solution in well-stoppered dark glass bottles and stored maximum two days.
4.3.Ethyl ether, for analysis quality.U.K.
4.4.Potassium hydroxide ethanolic solution, approximately 0,2 N.U.K.
Dissolve 13 g of potassium hydroxide (point 4.1) in 20 ml of distilled water and make up to one litre with ethanol (point 4.10).
4.5.Anhydrous sodium sulphate, for analysis quality.U.K.
4.6.Glass plates (20x20 cm) coated with silica gel, without fluorescence indicator, thickness 0,25 mm (commercially available ready for use).U.K.
4.7.Toluene, for chromatography quality.U.K.
4.8.Acetone, for chromatography quality.U.K.
4.9.n-Hexane, for chromatography quality.U.K.
4.10.Ethyl ether, for chromatography quality.U.K.
4.11.Ethanol of analytical quality.U.K.
4.12.Ethyl acetate of analytical quality.U.K.
4.13.Reference solution for thin-layer chromatography: cholesterol or phytosterols, and erythrodiol 5 % solution in ethyl acetate (point 4.11).U.K.
4.14.2,7-dichlorofluorescein, 0,2 % in ethanolic solution. Make slightly basic by adding a few drops of 2 N alcoholic potassium hydroxide solution (point 4.2).U.K.
4.15.Anhydrous pyridine, for chromatography quality (see Note 5).U.K.
4.16.Hexamethyl disilazane of analytical quality.U.K.
4.17.Trimethylchlorosilane of analytical quality.U.K.
4.18.Sample solutions of sterol trimethylsilyl ethers.U.K.
To be prepared at the time of use from sterols and erythrodiol obtained from oils containing them.
4.19.α-cholestanol, purity more than 99 % (purity must be checked by GC analysis).U.K.
4.20.α-cholestanol internal standard solution, 0,2 % solution (m/V) in ethyl acetate (point 4.11).U.K.
4.21.Phenolphthalein solution, 10 g/L in ethanol (point 4.10).U.K.
4.22.Carrier gas: hydrogen or helium, gas-chromatographic purity.U.K.
4.23.Auxiliary gases: hydrogen, helium, nitrogen and air, of gas-chromatographic purity.U.K.
4.24.n-Hexane (point 4.9)/ethyl ether (point 4.10) mixture 65:35 (V/V).U.K.
4.25.Silylation reagent, consisting of a 9:3:1 (V/V/V) mixture of pyridine/hexamethyl disilazane/trimethylchlorosilane.U.K.
5.PROCEDUREU.K.
5.1.Preparation of the unsaponifiable matter.U.K.
5.1.1.Using a 500 μl microsyringe (point 3.6) introduce into the 250 ml flask (point 3.1) a volume of the α-cholestanol internal standard solution (point 4.20) containing an amount of cholestanol corresponding to approximately 10 % of the sterol content of the sample. For example, for 5 g of olive oil sample add 500 μl of the α-cholestanol solution (point 4.20) and 1 500 μl for olive-pomace oil. Evaporate until dryness with a gentle current of nitrogen in a warm water bath, after cooling the flask, weigh 5 ± 0,01 g of the dry filtered sample into the same flask.U.K.
Note 1:Animal or vegetable oils and fats containing appreciable quantities of cholesterol may show a peak having a retention time close to cholestanol. If this occurs, the sterol fraction will have to be analysed in duplicate with and without internal standard.U.K.
5.1.2.Add 50 ml of 2 N ethanolic potassium hydroxide solution (point 4.2) and some pumice, fit the reflux condenser and heat to gentle boiling until saponification takes place (the solution becomes clear). Continue heating for a further 20 minutes, then add 50 ml of distilled water from the top of the condenser, detach the condenser and cool the flask to approximately 30 °C.U.K.
5.1.3.Transfer the contents of the flask quantitatively into a 500 ml separating funnel (point 3.2) using several portions of distilled water (50 ml). Add approximately 80 ml of ethyl ether (point 4.10), shake vigorously for approximately 60 seconds, periodically releasing the pressure by inverting the separating funnel and opening the stopcock. Allow standing until there is complete separation of the two phases (Note 2).U.K.
Then draw off the soap solution as completely as possible into a second separating funnel. Perform two further extractions on the water-alcohol phase in the same way using 60 to 70 ml of ethyl ether (point 4.10).
Note 2: Any emulsion can be destroyed by adding small quantities of ethanol (point 4.11).U.K.
5.1.4.Combine the three ether extracts in one separating funnel containing 50 ml of water. Continue to wash with water (50 ml) until the wash water no longer gives a pink colour on the addition of a drop of phenolphthalein solution (point 4.21).U.K.
When the wash water has been removed, filter on anhydrous sodium sulphate (point 4.5) into a previously weighed 250 ml flask, washing the funnel and filter with small quantities of ethyl ether (point 4.10).
5.1.5.Evaporate the solvent by distillation on a rotary evaporator at 30 °C under vacuum. Add 5 ml of acetone and remove the volatile solvent completely in a gentle current of air. Dry the residue in the oven at 103±2 °C for 15 min. Cool in desiccators and weigh to the nearest 0,1 mg.U.K.
5.2.Separation of the sterol and triterpene dialcohols fraction (erythrodiol + uvaol)U.K.
5.2.1.Preparation of the basic thin layer chromatography plates. Immerse the silica gel plates (point 4.6) about 4 cm in the 0,2 N ethanolic potassium hydroxide solution (point 4.5) for 10 seconds, then allow to dry in a fume cupboard for two hours and finally place in an oven at 100 °C for one hour.U.K.
Remove from the oven and keep in a calcium chloride desiccator (point 3.13) until required for use (plates treated in this way must be used within 15 days).
Note 3:When basic silica gel plates are used to separate the sterol fraction there is no need to treat the unsaponifiable fraction with alumina. In this way all compounds of an acid nature (fatty acids and others) are retained on the spotting line and the sterols band is clearly separated from the aliphatic and triterpene alcohols band.U.K.
5.2.2.Place hexane/ethyl ether mixture (point 4.24) (Note 4) into the development chamber, to a depth of approximately 1 cm. Close the chamber with the appropriate cover and leave thus for at least half an hour, in a cool place, so that liquid-vapour equilibrium is established strips of filter paper dipping into the eluent may be placed on the internal surfaces of the chamber. This reduces developing time by approximately one-third and brings about more uniform and regular elution of the components.U.K.
Note 4:The developing mixture needs to be replaced for every test, in order to achieve perfectly reproducible elution conditions, alternative solvent 50:50 (V/V) n-hexane/ethyl ether may be used.U.K.
5.2.3.Prepare an approximately 5 % solution of the unsaponifiable (point 5.1.5) in ethyl acetate (point 4.12) and, using the 100 μl microsyringe, depose 0,3 ml of the solution on a narrow and uniform streak on the lower end (2 cm) of the chromatographic plate (point 5.2.1). In line with the streak, place 2 to 3 μl of the material reference solution (point 4.13), so that the sterol and triterpene dialcohols band can be identified after developing.U.K.
5.2.4.Place the plate in the developing chamber prepared as specified in point 5.2.2. The ambient temperature should be maintained between 15 and 20 °C (Note 5). Immediately close the chamber with the cover and allow eluting until the solvent front reaches approximately 1 cm from the upper edge of the plate. Remove the plate from the developing chamber and evaporate the solvent in a flow of hot air or by leaving the plate for a short while, under a hood.U.K.
Note 5: Higher temperature could worsen the separation.U.K.
5.2.5.Spray the plate lightly and uniformly with the 2,7-dichlorofluorescein solution (point 4.14) and then leave to dry. When the plate is observed under ultraviolet light, the sterols and triterpene dialcohols bands can be identified through being aligned with the spots obtained from the reference solution (point 4.13). Mark the limits of the bands along the edges of the fluorescence with a black pencil (see TLC plate figure 3).U.K.
5.2.6.By using a metal spatula, scrape off the silica gel of the marked area. Place the finely comminuted material removed into the filter funnel (point 3.7). Add 10 ml of hot ethyl acetate (point 4.12), mix carefully with the metal spatula and filter under vacuum, collecting the filtrate in the conical flask (point 3.8.) attached to the filter funnel.U.K.
Wash the residue in the flask three times with ethyl ether (point 4.3) (approximately 10 ml each time), collecting the filtrate in the same flask attached to the funnel, evaporate the filtrate to a volume of 4 to 5 ml, transfer the residual solution to the previously weighed 10 ml test tube (point 3.9), evaporate to dryness by mild heating, in a gentle flow of nitrogen, make up again using a few drops of acetone (point 4.8), evaporate again to dryness,
The residue contained in the test tube must consist of the sterol and triterpene dialchols fractions.
5.3.Preparation of the trimethylsilyl ethers.U.K.
5.3.1.Add the silylation reagent (point 4.25) (Note 6), in the ratio of 50 μl for every milligram of sterols and triterpene dialcohols, in the test tube containing the sterol and triterpene fraction, avoiding any uptake of moisture (Note 7).U.K.
Note 6:Ready for use solutions are available commercially. Other silylation reagents, such as, for example, bistrimethylsilyl trifluor acetamide + 1 % trimethylchlorosilane, which has to be diluted with an equal volume of anhydrous pyridine, are also available.U.K.
Pyridine can be replaced by the same amount of acetonitrile.
5.3.2.Stopper the test tube, shake carefully (without overturning) until the compounds are completely dissolved. Leave to stand for at least 15 minutes at ambient temperature and then centrifuge for a few minutes. The clear solution is ready for gas chromatographic analysis.U.K.
Note 7:The slight opalescence, which may form, is normal and does not cause any anomaly. The formation of a white flock or the appearance of a pink colour is indicative of the presence of moisture or deterioration of the reagent. If this occurs, the test must be repeated (only if hexamethyldisilazane/trimethylchlorosilane is used).U.K.
5.4.Gas chromatographic analysis.U.K.
5.4.1.Preliminary operations, capillary column conditioning.U.K.
5.4.1.1.Fit the column (point 3.11) in the gas chromatograph, by attaching the inlet end to the split injector and the outlet end to the detector.U.K.
Carry out general checks on the gas chromatograph unit (leaks from the gas circuits, detector efficiency, efficiency of the splitting system and recording system, etc.).
5.4.1.2.If the column is being used for the first time, it is recommended that it be subjected to conditioning: passing a gentle flow of gas through the column itself, then switch on the gas chromatography unit and begin a gradual heating, up to a temperature of at least 20 °C above the operating temperature (Note 8). Hold this temperature for at least two hours, then place the entire unit in operating mode (adjustment of gas flows and splitting, ignition of the flame, connection with the computing system, adjustment of the column, detector and injector temperature, etc.) and then record the signal with a sensitivity at least two times greater than that one intended for the analysis. The course of the base line must be linear, without peaks of any kind, and must not show drift.U.K.
A negative straight-line drift indicates leakage from the column connections; a positive drift indicates inadequate conditioning of the column.
Note 8:The conditioning temperature must always be at least 20 °C less than the maximum temperature specified for the stationary phase used.U.K.
5.4.2.Choice of operating conditions.U.K.
5.4.2.1.The operating conditions are as follows:U.K.
Column temperature: 260 ± 5 °C;
Injector temperature: 280-300 °C;
Detector temperature: 280-300 °C;
Linear velocity of the carrier gas: helium 20 to 35 cm/s; hydrogen 30 to 50 cm/s;
Splitting ratio: from 1:50 to 1:100;
Instrument sensitivity: from 4 to 16 times the minimum attenuation;
Recording sensitivity: 1 to 2 mV full scale;
Amount of substance injected: 0,5 to 1 μl of TMSE solution.
These conditions may be changed according to the characteristics of the column and gas chromatograph, so as to obtain chromatograms, which meet the following requirements:
The retention time for the ß-sitosterol peak should be at 20 ± 5 min;
The campesterol peak should be: for olive oil (mean content 3 %) 20 ± 5 % of full scale; for soybean oil (average content 20 %) 80 ± 10 % of full scale;
All the present sterols must be separated. In addition to being separated the peaks, they must also be completely resolved, i.e. the peak trace should return to the base line before leaving for the next peak. Incomplete resolution is, however, tolerated, provided that the peak at RRT 1,02 (Sitostanol) can be quantified using the perpendicular.
5.4.3.Analytical procedureU.K.
5.4.3.1.By using the 10 μl microsyringe, take 1 μl of hexane, draw in 0,5 μl of air and then 0,5 to 1 μl of the sample solution. Raise the plunger of the syringe further, so the needle is emptied. Push the needle through the membrane of the injector and after one to two seconds, inject rapidly, and then slowly remove the needle after around five seconds.U.K.
An automatic injector can be used as well.
5.4.3.2.Carry out the recording until the TMSE of the present triterpene dialcohols are completely eluted. The base line must continue to meet the requirements (point 5.4.1.2).U.K.
5.4.4.Peak identificationU.K.
Identify individual peaks on the basis of retention times and by comparison with the mixture of sterol and triterpene dialcohols TMSE, analysed under the same conditions (see Appendix).
The sterols and triterpene dialcohols are eluted in the following order: cholesterol, brassicasterol, ergosterol, 24-methylen-cholesterol, campesterol, campestanol, stigmasterol, Δ7-campesterol, Δ5,23-stigmastadienol, clerosterol, ß-sistosterol, sitostanol, Δ5-avenasterol, Δ5,24-stigmastadienol, Δ7-stigmastenol, Δ7-avenasterol, erythrodiol and uvaol.
The retention times for ß-sitosterol, for SE-52 and SE-54 columns, are shown in Table 1.
Figures 1 and 2 show typical chromatograms for some oils.
5.4.5.Quantitative evaluation.U.K.
5.4.5.1.Calculate the areas of the α-cholestanol and the sterol and triterpene dialcohols peaks by using the computing system. Ignore peaks for any compound which are not included (ergosterol must not be calculated) among those listed in Table 1. The response factor for α-cholestanol should be considered equal to 1.U.K.
5.4.5.2.Calculate the concentration of each individual sterol, in mg/kg of fatty material, as follows:U.K.

where:
=
peak area for sterol x, in computing system counts;
=
area of the α-cholestanol peak, in computing system counts;
=
mass of added α-cholestanol, in milligrams;
=
mass of the sample used for determination, in grams.
6.EXPRESSION OF THE RESULTSU.K.
6.1.Report individual sterol concentrations as mg/kg of fatty material and their sum as "total sterols".U.K.
The composition of each of the individual sterols and of the erythrodiol and uvaol should be expressed to one decimal point.
Total sterol composition must be expressed without any decimal point.
6.2.Calculate the percentage of each individual sterol from the ratio of the relevant peak area to the total peak area for sterols and erythrodiol and uvaol:U.K.

where:
=
peak area for x;
=
total peak area for sterols;
6.3.Apparent β-sitosterol: Δ5-23-stigmastadienol + clerosterol + β-sitosterol + sitostanol + Δ5-avenasterol + Δ5-24-stigmastadienol.U.K.
6.4.Calculate the percentage of erythrodiol and uvaol:U.K.

where
=
sum area for sterol in computing system counts;
=
area of Erythrodiol in computing system counts;
=
area of Uvaol in computing system counts;
Appendix Determination of the linear speed of the gas
With the gas chromatograph set to normal operating conditions, inject 1 to 3 μl of methane (or propane) and measure the time taken by the gas to pass through the column, from the time of injection to the time at which the peak appears (tM).
The linear speed in cm/s is given by L/tM, where L is the length of the column in centimetres and tM is the measured time, in seconds.
Table 1
Relative retention times for sterols
| Peak | Identification | Relative retention time | ||
|---|---|---|---|---|
| SE 54 column | SE 52 column | |||
| 1 | Cholesterol | Δ-5-cholesten-3ß-ol | 0,67 | 0,63 |
| 2 | Cholestanol | 5α-cholestan-3ß-ol | 0,68 | 0,64 |
| 3 | Brassicasterol | [24S]-24-methyl-Δ-5,22-cholestadien-3ß-ol | 0,73 | 0,71 |
| * | Ergosterol | [24S] 24 methy Δ5-7-22 cholestatrien 3ß-ol | 0,78 | 0,76 |
| 4 | 24-methylene-cholesterol | 24-methylene-Δ-5,24-cholestadien-3ß-o1 | 0,82 | 0,8 |
| 5 | Campesterol | (24R)-24-methyl-Δ-5-cholesten-3ß-ol | 0,83 | 0,81 |
| 6 | Campestanol | (24R)-24-methyl-cholestan-3ß-ol | 0,85 | 0,82 |
| 7 | Stigmasterol | (24S)-24-ethyl-Δ-5,22-cholestadien-3ß-ol | 0,88 | 0,87 |
| 8 | Δ-7-campesterol | (24R)-24-methyl-Δ-7-cholesten-3ß-ol | 0,93 | 0,92 |
| 9 | Δ-5,23-stigmastadienol | (24R,S)-24-ethyl-Δ-5,23-choIestadien-3ß-ol | 0,95 | 0,95 |
| 10 | Clerosterol | (24S)-24-ethyl-Δ-5,25-cholestadien-3ß-ol | 0,96 | 0,96 |
| 11 | ß-sitosterol | (24R)-24-ethyl-Δ-5-cholesten-3ß-ol | 1,0 | 1,0 |
| 12 | Sitostanol | 24-ethyl-cholestan-3ß-ol | 1,02 | 1,02 |
| 13 | Δ-5-avenasterol | (24Z)-24-ethylidene-Δ-cholesten-3ß-ol | 1,03 | 1,03 |
| 14 | Δ-5-24-stigmastadienol | (24R,S)-24-ethyl-Δ-5,24-cholestadien-3ß-ol | 1,08 | 1,08 |
| 15 | Δ-7-stigmastenol | (24R,S)-24-ethyl-Δ-7-cholesten-3ß-ol | 1,12 | 1,12 |
| 16 | Δ-7-avenasterol | (24Z)-24-ethylidene-Δ-7-cholesten-3ß-ol | 1,16 | 1,16 |
| 17 | Erythrodiol | 5α olean-12en-3ß28 diol | 1,41 | 1,41 |
| 18 | Uvaol | Δ12-ursen-3ß28 diol | 1,52 | 1,52 |
Figure 1
Gas chromatogram of the sterol and triterpene dialchols fraction of a lampante olive oil (spiked with internal standard)

Figure 2
Gas chromatogram of the sterol and triterpene dialchols fraction of a refined olive oil (spiked with internal standard)

Figure 3
TLC plate olive-pomace oil with the zone that must be scraped for sterols and triterpenic dialcohols determination

”
ANNEX VU.K.
“ANNEX XII THE INTERNATIONAL OLIVE COUNCIL’S METHOD FOR THE ORGANOLEPTIC ASSESSMENT OF VIRGIN OLIVE OIL
1.PURPOSE AND SCOPEU.K.
The purpose of this international method is to determine the procedure for assessing the organoleptic characteristics of virgin olive oil within the meaning of point 1 of Annex XVI to Regulation (EC) No 1234/2007 and to establish the method for its classification on the basis of those characteristics. It also provides indications for optional labelling.
The method described is applicable only to virgin olive oil and to the classification or labelling of such oils according to the intensity of the defects perceived and of the fruitiness, as determined by a group of tasters selected, trained and monitored as a panel.
It also provides for indications for optional labelling.
The IOC standards mentioned in this Annex are used in their last available version.
2.GENERAL BASIC VOCABULARY FOR SENSORY ANALYSISU.K.
Refer to the standard IOC/T.20/Doc. No 4 "Sensory Analysis: General Basic Vocabulary"
3.SPECIFIC VOCABULARYU.K.
3.1. Negative attributes U.K.
Fusty/muddy sediment: Characteristic flavour of oil obtained from olives piled or stored in such conditions as to have undergone an advanced stage of anaerobic fermentation, or of oil which has been left in contact with the sediment that settles in underground tanks and vats and which has also undergone a process of anaerobic fermentation.
Musty-humid-earthy: Characteristic flavour of oils obtained from fruit in which large numbers of fungi and yeasts have developed as a result of its being stored in humid conditions for several days or of oil obtained from olives that have been collected with earth or mud on them and which have not been washed.
Winey-vinegary-acid-sour: Characteristic flavour of certain oils reminiscent of wine or vinegar. This flavour is mainly due to a process of aerobic fermentation in the olives or in olive paste left on pressing mats which have not been properly cleaned and leads to the formation of acetic acid, ethyl acetate and ethanol.
Rancid: Flavour of oils which have undergone an intense process of oxidation.
Frostbitten olives (wet wood): Characteristic flavour of oils extracted from olives which have been injured by frost while on the tree.
3.2. Other negative attributes U.K.
Heated or: Characteristic flavour of oils caused by excessive and/or prolonged
Burnt: Heating during processing, particularly when the paste is thermally mixed, if this is done under unsuitable thermal conditions.
Hay–wood: Characteristic flavour of certain oils produced from olives that have dried out.
Rough: Thick, pasty mouth sensation produced by certain old oils.
Greasy: Flavour of oil reminiscent of that of diesel oil, grease or mineral oil.
Vegetable water: Flavour acquired by the oil as a result of prolonged contact with vegetable water which has undergone fermentation processes.
Brine: Flavour of oil extracted from olives which have been preserved in brine.
Metallic: Flavour that is reminiscent of metals. It is characteristic of oil which has been in prolonged contact with metallic surfaces during crushing, mixing, pressing or storage.
Esparto: Characteristic flavour of oil obtained from olives pressed in new esparto mats. The flavour may differ depending on whether the mats are made of green esparto or dried esparto.
Grubby: Flavour of oil obtained from olives which have been heavily attacked by the grubs of the olive fly (Bactrocera oleae).
Cucumber: Flavour produced when an oil is hermetically packed for too long, particularly in tin containers, and which is attributed to the formation of 2,6 nonadienal.
3.3. Positive attributes U.K.
Fruity: Set of olfactory sensations characteristic of the oil which depends on the variety and comes from sound, fresh olives, either ripe or unripe. It is perceived directly and/or through the back of the nose.
Bitter: Characteristic primary taste of oil obtained from green olives or olives turning colour. It is perceived in the circumvallate papillae on the “V” region of the tongue.
Pungent: Biting tactile sensation characteristic of oils produced at the start of the crop year, primarily from olives that are still unripe. It can be perceived throughout the whole of the mouth cavity, particularly in the throat.
3.4. Optional terminology for labelling purposes U.K.
Upon request, the panel leader may certify that the oils which have been assessed comply with the definitions and ranges corresponding to the following adjectives according to the intensity and perception of the attributes.
Positive attributes (fruity, bitter and pungent): According to the intensity of perception:
Intense, when the median of the attribute is more than 6;
Medium, when the median of the attribute is between 3 and 6;
Light, when the median of the attribute is less than 3.
Fruity: Set of olfactory sensations characteristic of the oil which depends on the variety of olive and comes from sound, fresh olives in which neither green nor ripe fruitiness predominates. It is perceived directly and/or through the back of the nose.
Greenly fruity: Set of olfactory sensations characteristic of the oil which is reminiscent of green fruit, depends on the variety of olive and comes from green, sound, fresh olives. It is perceived directly and/or through the back of the nose.
Ripely fruity: Set of olfactory sensations characteristic of the oil which is reminiscent of ripe fruit, depends on the variety of olive and comes from sound, fresh olives. It is perceived directly and/or through the back of the nose.
Well balanced: Oil which does not display a lack of balance, by which is meant the olfactory–gustatory and tactile sensation where the median of the bitter and/or pungent attributes is two points higher than the median of the fruitiness.
Mild oil: Oil for which the median of the bitter and pungent attributes is 2 or less.
4.GLASS FOR OIL TASTINGU.K.
Refer to the standard IOC/T.20/Doc. No 5, "Glass for Oil Tasting".
5.TEST ROOMU.K.
Refer to the standard IOC/T.20/Doc. No 6, "Guide for the Installation of a Test Room".
6.ACCESSORIESU.K.
The following accessories, which are required by tasters to perform their task properly, must be supplied in each booth and must be within easy reach:
glasses (standardised) containing the samples, code numbered, covered with a watch-glass and kept at 28 °C ± 2 °C;
profile sheet (see Figure 1) on hard copy, or on soft copy provided that the conditions of the profile sheet are met, together with the instructions for its use if necessary
pen or indelible ink
trays with slices of apple and/or water, carbonated water and/or rusks
glass of water at ambient temperature
sheet recalling the general rules listed in sections 8.4 and 9.1.1
spittoons.
7.PANEL LEADER AND TASTERSU.K.
7.1. Panel leader U.K.
The panel leader must be a suitably trained person with an expert knowledge of the kinds of oils which he or she will come across in the course of their work. They are the key figure in the panel and responsible for its organisation and running.
The work of the panel leader calls for basic training in the tools of sensory analysis, sensory skill, meticulousness in the preparation, organisation and performance of the tests and skill and patience to plan and execute the tests in a scientific manner.
They are the sole person responsible for selecting, training and monitoring the tasters in order to ascertain their level of aptitude. They are thus responsible for the appraisal of the tasters, which must always be objective and for which they must develop specific procedures based on tests and solid acceptance and rejection criteria. See standard IOC/T.20/Doc. No 14, "Guide for the selection, training and monitoring of skilled virgin olive oil tasters".
Panel leaders are responsible for the performance of the panel and hence for its evaluation, of which they must give reliable, objective proof. In any case, they must demonstrate at all times that the method and tasters are under control. Periodic calibration of the panel is recommended (IOC/T.20/Doc. No 14, § 5).
They hold ultimate responsibility for keeping the records of the panel. These records must always be traceable. They must comply with the assurance and quality requirements laid down in international sensory analysis standards and ensure the anonymity of the samples at all times.
They shall be responsible for inventorying and ensuring that the apparatus and equipment needed to comply with the specifications of this method is properly cleaned and maintained and shall keep written proof thereof, as well as of the compliance with the test conditions.
They shall be in charge of the reception and storage of the samples upon their arrival at the laboratory as well as of their storage after being tested. When doing so, they shall ensure at all times that the samples remain anonymous and are properly stored, for which purpose they must develop written procedures in order to ensure that the entire process is traceable and affords guarantees.
In addition, they are responsible for preparing, coding and presenting the samples to the tasters according to an appropriate experimental design in line with pre-established protocols, as well as for assembling and statistically processing the data obtained by the tasters.
They shall be in charge of developing and drafting any other procedures that might be necessary to complement this standard and to ensure that the panel functions properly.
They must seek ways of comparing the results of the panel with those obtained by other panels undertaking the analysis of virgin olive oil in order to ascertain whether the panel is working properly.
It is the duty of the panel leader to motivate the panel members by encouraging interest, curiosity and a competitive spirit among them. To do so, they are strongly recommended to ensure a smooth two-way flow of information with the panel members by keeping them informed about all the tasks they carry out and the results obtained. In addition, they shall ensure that their opinion is not known and shall prevent possible leaders from asserting their criteria over the other tasters.
They shall summon the tasters sufficiently in advance and shall answer any queries regarding the performance of the tests, but shall refrain from suggesting any opinion to them on the sample.
7.2. Tasters U.K.
The people acting as tasters in organoleptic tests carried out on olive oils must do so voluntarily, with all the ensuing consequences of such a voluntary act in terms of obligations and the absence of financial payment. It is therefore advisable for candidates to submit an application in writing. Candidates shall be selected, trained and monitored by the panel leader in accordance with their skills in distinguishing between similar samples; it should be borne in mind that their accuracy will improve with training.
Tasters must act like real sensory observers, setting aside their personal tastes and solely reporting the sensations they perceive. To do so, they must always work in silence, in a relaxed, unhurried manner, paying the fullest possible sensory attention to the sample they are tasting.
Between 8 and 12 tasters are required for each test, although it is wise to keep some extra tasters in reserve to cover possible absences.
8.TEST CONDITIONSU.K.
8.1. Presentation of the sample U.K.
The oil sample for analysis shall be presented in standardised tasting glasses conforming to the standard IOC/T.20/Doc. No 5 ‘Glass for oil tasting’.
The glass shall contain 14–16 ml of oil, or between 12,8 and 14,6 g if the samples are to be weighed, and shall be covered with a watch-glass.
Each glass shall be marked with a code made up of digits or a combination of letters and digits chosen at random. The code will be marked by means of an odourfree system.
8.2. Test and sample temperature U.K.
The oil samples intended for tasting shall be kept in the glasses at 28 °C ± 2 °C throughout the test. This temperature has been chosen because it makes it easier to observe organoleptic differences than at ambient temperature and because at lower temperatures the aromatic compounds peculiar to these oils volatilise poorly while higher temperatures lead to the formation of volatile compounds peculiar to heated oils. See the standard IOC/T.20/Doc. No 5 “Glass for Oil Tasting” for the method which has to be used for heating the samples when in the glass.
The test room must be at a temperature between 20 ° and 25 °C (see IOC/T.20/Doc. No 6).
8.3. Test times U.K.
The morning is the best time for tasting oils. It has been proved that there are optimum perception periods as regards taste and smell during the day. Meals are preceded by a period in which olfactory–gustatory sensitivity increases, whereas afterwards this perception decreases.
However, this criterion should not be taken to the extreme where hunger may distract the tasters, thus decreasing their discriminatory capacity; therefore, it is recommended to hold the tasting sessions between 10.00 in the morning and 12 noon.
8.4. Tasters: general rules of conduct U.K.
The following recommendations apply to the conduct of the tasters during their work.
When called by the panel leader to participate in an organoleptic test, tasters should be able to attend at the time set beforehand and shall observe the following:
They shall not smoke or drink coffee at least 30 minutes before the time set for the test.
They must not have used any fragrance, cosmetic or soap whose smell could linger until the time of the test. They must use an unperfumed soap to wash their hands which they shall then rinse and dry as often as necessary to eliminate any smell.
They shall fast at least one hour before the tasting is carried out.
Should they feel physically unwell, and in particular if their sense of smell or taste is affected, or if they are under any psychological effect that prevents them from concentrating on their work, the tasters shall refrain from tasting and shall inform the panel leader accordingly.
When they have complied with the above, the tasters shall take up their place in the booth allotted to them in an orderly, quiet manner.
They shall carefully read the instructions given on the profile sheet and shall not begin to examine the sample until fully prepared for the task they have to perform (relaxed and unhurried). If any doubts should arise, they should consult the panel leader in private.
They must remain silent while performing their tasks.
They must keep their mobile phone switched off at all times to avoid interfering with the concentration and work of their colleagues.
9.PROCEDURE FOR THE ORGANOLEPTIC ASSESSMENT AND CLASSIFICATION OF VIRGIN OLIVE OILU.K.
9.1. Tasting technique U.K.
9.1.1.The tasters shall pick up the glass, keeping it covered with the watch-glass, and shall bend it gently; they shall then rotate the glass fully in this position so as to wet the inside as much as possible. Once this stage is completed, they shall remove the watch-glass and smell the sample, taking slow deep breaths to evaluate the oil. Smelling should not exceed 30 seconds. If no conclusion has been reached during this time, they shall take a short rest before trying again.U.K.
When the olfactory test has been performed, the tasters shall then evaluate the buccal sensations (overall retronasal olfactory, gustatory and tactile sensations). To do so, they shall take a small sip of approximately 3 ml of oil. It is very important to distribute the oil throughout the whole of the mouth cavity, from the front part of the mouth and tongue along the sides to the back part and to the palate support and throat, since it is a known fact that the perception of tastes and tactile sensations varies in intensity depending on the area of the tongue, palate and throat.
It should be stressed that it is essential for a sufficient amount of the oil to be spread very slowly over the back of the tongue towards the palate support and throat while the taster concentrates on the order in which the bitter and pungent stimuli appear. If this is not done, both of these stimuli may escape notice in some oils or else the bitter stimulus may be obscured by the pungent stimulus.
Taking short, successive breaths, drawing in air through the mouth, enables the taster not only to spread the sample extensively over the whole of the mouth but also to perceive the volatile aromatic compounds via the back of the nose by forcing the use of this channel.
The tactile sensation of pungency should be taken into consideration. For this purpose it is advisable to ingest the oil.
9.1.2.When organoleptically assessing a virgin olive oil, it is recommended that FOUR SAMPLES at the most be evaluated in each session with a maximum of three sessions per day, to avoid the contrast effect that could be produced by immediately tasting other samples.U.K.
As successive tastings produce fatigue or loss of sensitivity caused by the preceding samples, it is necessary to use a product that can eliminate the remains of the oil from the preceding tasting from the mouth.
The use of a small slice of apple is recommended which, after being chewed, can be disposed of in the spittoon. Then rinse out the mouth with a little water at ambient temperature. At least 15 minutes shall lapse between the end of one session and the start of the next.
9.2. Use of the profile sheet by tasters U.K.
The profile sheet intended for use by tasters is detailed in Figure 1 of this Annex.
Each taster on the panel shall smell and then taste(4) the oil under consideration. They shall then enter the intensity with which they perceive each of the negative and positive attributes on the 10-cm scale shown in the profile sheet provided.
Should the tasters perceive any negative attributes not listed in section 4, they shall record them under the "others" heading, using the term or terms that most accurately describes the attributes.
9.3. Use of the data by the panel leaders U.K.
The panel leader shall collect the profile sheets completed by each taster and shall review the intensities assigned to the different attributes. Should they find any anomaly, they shall invite the taster to revise his or her profile sheet and, if necessary, to repeat the test.
The panel leader shall enter the assessment data of each panel member in a computer program like that provided by the standard IOC/T.20/Doc. No 15) with a view to statistically calculating the results of the analysis, based on the calculation of their median. See sections 9.4 and Appendix to this Annex. The data for a given sample shall be entered with the aid of a matrix comprising 9 columns representing the 9 sensory attributes and n lines representing the n panel members used.
When a defect is perceived and entered under the "others" heading by at least 50 % of the panel, the panel leader shall calculate the median of the defect and shall arrive at the corresponding classification.
The value of the robust coefficient of variation which defines classification (defect with the strongest intensity and fruity attribute) must be no greater than 20 %.
If the opposite is the case, the panel leader must repeat the evaluation of the specific sample in another tasting session.
If this situation arises often, the panel leader is recommended to give the tasters specific additional training (IOC/T.20/Doc. No 14, § 5) and to use the repeatability index and deviation index to check panel performance (IOC/T.20/Doc. No 14, § 6).
9.4. Classification of the oil U.K.
The oil is graded as follows in line with the median of the defects and the median for the fruity attribute. The median of the defects is defined as the median of the defect perceived with the greatest intensity. The median of the defects and the median of the fruity attribute are expressed to one decimal place.
The oil is graded by comparing the median value of the defects and the median for the fruity attribute with the reference ranges given below. The error of the method has been taken into account when establishing the limits of these ranges, which are therefore considered to be absolute. The software packages allow the grading to be displayed as a table of statistics or a graph.
Extra virgin olive oil: the median of the defects is 0 and the median of the fruity attribute is above 0;
Virgin olive oil: the median of the defects is above 0 but not more than 3,5 and the median of the fruity attribute is above 0;
Lampante olive oil: the median of defect is above 3,5 or the median of the defect is less than or equal to 3,5 and the fruity median is equal to 0.
Note 1:U.K.
When the median of the bitter and/or pungent attribute is more than 5,0, the panel leader shall state so on the test certificate.
Figure 1
PROFILE SHEET FOR VIRGIN OLIVE OIL
| a Delete as appropriate | ||||
| Intensity of perception of defects | ||||
|---|---|---|---|---|
| Fusty/muddy sedimenta | ||||
| Musty/humid/earthya | ||||
| Winey/vinegary acid/soura | ||||
| Frostbitten olives (wet wood) | ||||
| Rancid | ||||
| Other negative attributes: | ||||
| Descriptor: | Metallic Hay Grubby Rough Brine Heated or burnt Vegetable water Esparto Cucumber Greasy | |||
| Intensity of perception of positive attributes | ||||
| Fruity | ||||
| Green | Ripe | |||
| Bitter | ||||
| Pungent | ||||
| Name of taster: | Taster code: | |||
| Sample code: | Signature: | |||
Appendix Method for calculating the median and the confidence intervals
Median U.K.

The median is defined as the real number Xm characterised by the fact that the probability (p) that the distribution values (X) are below this number (Xm), is less than and equal to 0,5 and that simultaneously the probability (p) that the distribution values (X) are below or equal to Xm is greater than and equal to 0,5. A more practical definition is that the median is the 50th percentile of a distribution of numbers arranged in increasing order. In simpler terms, it is the midpoint of an ordered set of odd numbers, or the mean of two midpoints of an ordered set of even numbers.
Robust standard deviation U.K.
In order to arrive at a reliable estimate of the variability around the mean it is necessary to refer to the robust standard deviation as estimated according to Stuart and Kendall (4). The formula gives the asymptotic robust standard deviation, i.e. the robust estimate of the variability of the data considered where N is the number of observations and IQR is the interquartile range which encompasses exactly 50% of the cases of a given probability distribution:
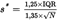
The interquartile range is calculated by calculating the magnitude of the difference between the 75th and 25th percentile.

Where the percentile is the value Xpc characterised by the fact that the probability (p) that the distribution values are less than Xpc is less than and equal to a specific hundreth and that simultaneously the probability (p) that the distribution values are less than or equal to Xpc is greater than and equal to that specific hundredth. The hundredth indicates the distribution fractile chosen. In the case of the median it is equal to 50/100.

For practical purposes, the percentile is the distribution value corresponding to a specific area subtended from the distribution or density curve. To give an example, the 25th percentile represents the distribution value corresponding to an area equal to 0,25 or 25/100.
In this method percentiles are computed on the basis of the real values which appear in the data matrix (percentiles computing procedure).
Robust coefficient of variation (%) U.K.
The CVr% represents a pure number which indicates the percentage variability of the set of numbers analysed. For this reason it is very useful for checking the reliability of the panel assessors.

Confidence intervals of the median at 95% U.K.
The confidence intervals at 95% (value of the error of the first kind equal to 0,05 or 5%) represent the interval within which the value of the median could vary if it were possible to repeat an experiment an infinite number of times. In practice, it indicates the interval of variability of the test in the operating conditions adopted starting from the assumption that it is possible to repeat it many times. As with the CVr%, the interval helps to assess the reliability of the test.


where C = 1,96 for the confidence interval at the 95% level.
An example of the calculation sheet is presented in Annex I to the standard IOC/T 20/Doc. No 15.
References U.K.
(1)Wilkinson, L. 1990. Systat: The system for statistics. Evanston, IL.SYSTAT Inc.U.K.
(2)Cicchitelli, G. 1984. Probabilità e Statistica. Maggioli Editore, Rimini.U.K.
(3)Massart, D.L.; Vandeginste, B.G.M.; Deming, Y.; Michotte, L. 1988. Chemometrics. A textbook. Elsevier. Amsterdam.U.K.
(4)Kendall, M.G.; Stuart, A. 1967. The advanced theory of statistics. Vol. 1. Hafner Publishing Co.U.K.
(5)McGill, R.; Tukey, J.W.; Larsen, W.A. 1978. Variation of Box Plots. The American Statistician, 32, (2), 12-16.U.K.
(6)IOC/T.28/Doc. No 1 September 2007, Guidelines for the accreditation of sensory testing laboratories with particular reference to virgin olive oil according to standard ISO/IEC 17025:2005.U.K.
(7)IOC/T.20/Doc. No 14.U.K.
(8)IOC/T.20/Doc. No 15.U.K.
(9)ISO/IEC 17025:05.”U.K.
ANNEX VIU.K.
“ANNEX XXa METHOD FOR THE DETECTION OF EXTRANEOUS OILS IN OLIVE OILS
1.SCOPEU.K.
This method is used to detect the presence of extraneous vegetable oils in olive oils. High linoleic vegetable oils (soybean, rapeseed, sunflower, etc.), and some high oleic vegetable oils - such as hazelnut, high oleic sunflower and olive-pomace oils - can be detected in olive oils. The level detected depends on the type of extraneous oil and the variety of olive. For hazelnut oil, a detection level between 5 and 15 % is common. The method is unable to identify the type of extraneous oil detected, and only indicates if the olive oil is genuine or non-genuine.
2.PRINCIPLEU.K.
The oil is purified by solid phase extraction (SPE) on silica gel cartridges. The triacylglycerol (TAG) composition is determined by reverse phase high resolution liquid chromatography using a refractive index detector and propionitrile as the mobile phase. Fatty acid methyl esters (FAMEs) are prepared from purified oil by methylation with a cold solution of KOH in methanol (Annex X B) and then the esters are analysed by capillary gas chromatography using high polar columns (Annex X A). The theoretical triacylglycerol composition is calculated from the fatty acid composition by a computer program assuming a 1,3-random, 2-random distribution of fatty acids in the triacylglycerol, with restrictions for saturated fatty acids in the 2-position. The calculation method is a modification of the procedure described in Annex XVIII. Several mathematical algorithms are calculated from theoretical and experimental (HPLC) triacylglycerol compositions, and the resulting values are compared with those contained in a database built from genuine olive oils.
3.MATERIAL AND REAGENTSU.K.
3.1. Oil purification U.K.
3.1.1.25-ml conical flasks.U.K.
3.1.2.5-ml screw top glass tubes and caps fitted with PTFE joint.U.K.
3.1.3.Silica gel cartridges, 1 g (6 ml), for solid phase extraction (for example, Waters, Massachusetts, USA).U.K.
3.1.4. n-hexane, analytical grade.U.K.
3.1.5.Solvent mixture of hexane/diethyl ether (87:13, v/v).U.K.
3.1.6. N-heptane, analytical grade.U.K.
3.1.7.Acetone, analytical grade.U.K.
3.2. HPLC analysis of triacylglycerols U.K.
3.2.1.Micro syringes (50 μL) and needles for HPLC injection.U.K.
3.2.2.Propionitrile, super purity or HPLC grade (for example, ROMIL, Cambridge, United Kingdom), used as mobile phase.U.K.
3.2.3.HPLC column (25 cm × 4 mm internal diameter), packed with RP-18 phase (4 μm particle size).U.K.
3.3. Preparation of fatty acid methyl esters U.K.
(See Annex X B)
3.3.1.Methanol containing not more than 0,5 % water.U.K.
3.3.2.Heptane, analytical grade.U.K.
3.3.3.A 2N solution of potassium hydroxide in methanol. Dissolve 1,1 g of potassium hydroxide in 10 ml of methanol.U.K.
3.3.4.5-ml screw top glass tubes and caps provided with PTFE joint.U.K.
3.4. GC analysis of FAMEs U.K.
(See method for the determination of trans-unsaturated fatty acids by capillary column gas chromatography set out in Annex X A).
3.4.1Micro syringes (5 μL) and needles for GC injection.U.K.
3.4.2Hydrogen or helium as carrier gas.U.K.
3.4.3Hydrogen and oxygen for FID detector.U.K.
3.4.4Nitrogen or helium as auxiliary carrier gas.U.K.
3.4.5.Fused silica capillary column (50-60 m × 0,25 – 0,30 mm internal diameter) coated with cyanopropylpolysiloxane or cyanopropylphenylsiloxane phases (SP-2380 or similar) with 0,20-0,25 μm of film thickness.U.K.
4.APPARATUSU.K.
4.1.Vacuum apparatus for solid phase extraction.U.K.
4.2.Rotary evaporator.U.K.
4.3.HPLC equipment composed of:U.K.
Degasser for the mobile phase.
Rheodyne injector valve with a 10 μL loop.
High pressure pump unit.
Thermostatic oven for the HPLC column capable of maintaining sub-ambient temperatures (15-20 °C), (for example, Peltier type).
Refractive index detector.
Computerised data acquisition system provided with an integration program.
4.4Capillary gas chromatography equipment described in Annex X A, provided with:U.K.
Split injector.
Flame ionisation detector (FID).
Oven with programmable temperature.
Computerised data acquisition system provided with an integration program.
4.5.Computer with Microsoft EXCEL program.U.K.
5.ANALYTICAL PROCEDUREU.K.
5.1. Oil purification U.K.
An SPE silica gel cartridge is placed in a vacuum elution apparatus and washed under vacuum with 6 ml of hexane. The vacuum is released to prevent the column from drying and a conical flask is placed under the cartridge. A solution of the oil (0,12 g, approximately) in 0,5 ml of hexane is loaded into the column and the solution is pulled through and then eluted with 10 ml of the solvent mixture (3.1.5) of hexane-diethyl ether (87:13 v/v) under vacuum. The eluted solvent is homogenised and approximately half of the volume is poured into another conical flask. Both solutions are separately evaporated to dryness in a rotary evaporator under reduced pressure at room temperature. For triacylglycerol analysis, one of the residues is dissolved in 1 ml of acetone (See first paragraph of point 5.2) and poured into a 5-ml screw top glass tube. The other residue is dissolved in 1 ml of n-heptane and poured into a second 5-ml screw top glass tube for preparing the fatty acid methyl esters.
Note: Oil purification may be done using a silica gel column, as described in IUPAC method 2.507.U.K.
5.2. HPLC analysis of triacylglycerols U.K.
Set up the HPLC system, maintaining the column temperature at 20 °C and using propionitrile as the mobile phase at a flow rate of 0,6 ml/min. When the baseline is stable run a solvent injection; if the base line appears disturbed in the region from 12 to 25 min, use another type of acetone or a mixture of propionitrile/acetone (25:75) to dissolve the sample.
Note: Some types of acetone produce disturbances of the baseline in the above-mentioned region.U.K.
Inject a 10 μl aliquot of the solution of purified oil in acetone (5 %). The run takes approximately 60 min. Oven temperature and/or flow rate must be adjusted to achieve a chromatogram similar to that depicted in Figure 1 where trilinolein (peak 1) elutes at 15,5 min and the resolutions between the pairs LLL/OLLn (peaks 1 and 2) and OLL/OOLn (peaks 4 and 5) are good.
The height of peak 2 (OLLn+PoLL) must reach at least 3 % of the full scale.
5.3. Preparation of fatty acid methyl esters U.K.
Add 0,1 mL of a 2N solution of potassium hydroxide in methanol to the solution of purified oil in 1 mL of n-heptane. Cap the tube and screw tight. Shake the tube vigorously for 15 seconds and leave to stratify until the upper layer becomes clear (5 minutes). The n-heptane solution is ready to be injected into the gas chromatograph. The solution may be left at room temperature for a maximum of 12 hours.
5.4. GC analysis of fatty acid methyl esters U.K.
The procedure described in the method for the determination of trans-unsaturated fatty acids must be used (see Annex X A).
The GC system is set up at an oven temperature of 165 °C. The recommended oven temperature is isothermal at 165 °C for 10 min, then raising it to 200 °C at 1,5 °C/min. An injector temperature between 220 °C and 250 °C is recommended to minimise the formation of trans-fatty acids (see Annex X A). Detector temperature 250 °C. Hydrogen or helium must be used as the carrier gas at a column head pressure of 130 kPa, approximately. Injection volume 1μL in split injection mode.
A GC profile similar to that shown in Figure 2 must be obtained. Special attention must be paid to the resolution between C18:3 and C20:1 (the C18:3 peak must appear before the C20:1). To achieve these conditions, the initial temperature and/or the column head pressure must be optimised. Adjust the injector conditions (temperature, split ratio and volume injection) to minimise the discrimination of palmitic and palmitoleic acid.
The height of the C20:0 peak must be about 20 % of full scale to quantify the trans isomers. If the C18:0 peak appears distorted, reduce the sample amount.
6.INTEGRATION OF CHROMATOGRAPHIC PEAKSU.K.
6.1. HPLC chromatogram U.K.
Figure 1 shows a typical HPLC chromatogram of the triacylglycerols of a purified olive oil. For peak integration, three baselines must be traced: the first between the start of peak 1 and the end of peak 3; the second between the start of peak 4 and the valley before peak 8; the third between the valley preceding peak 8 and the end of peak 18.
The total area is the sum of the areas of all the peaks (identified and not identified) from peak 1 to peak 18. The percentage of each peak is given by

The percentages have to be given to two decimal figures.
6.2. GC chromatogram U.K.
Figure 2 shows a GC chromatogram of fatty acid alkyl esters obtained from a purified olive oil. Percentages of the following fatty acids must be calculated:
| Palmitic; | P (C16:0) | = | methyl ester + ethyl ester |
| Stearic; | S (C18:0) | = | methyl ester |
| Palmitoleic; | Po (C16:1) | = | sum of methyl esters of the two cis-isomers |
| Oleic; | O (C18:1) | = | sum of methyl esters of the two cis-isomers + ethyl ester + trans-isomers |
| Linoleic; | L (C18:2) | = | methyl ester+ ethyl ester + trans-isomers |
| Linolenic; | Ln (C18:3) | = | methyl ester + trans-isomers |
| Arachidic; | A (C20:0) | = | methyl ester |
| Eicosenoic (gondoic); | G (C20:1) | = | methyl ester |
Ethyl and trans-isomers esters may be absent in the GC chromatogram.
Total area (AT) is the sum of all the peaks appearing in the chromatogram from C14:0 to C24:0, except that corresponding to squalene. The percentage of each peak is calculated as follows:
The results have to be expressed to two decimal places.
For the calculations of the computer programs, it is not necessary to normalise to 100 because this is done automatically.
Figure 1
HPLC chromatogram of TAGs of a “Chamlali” virgin olive oil. Main components of chromatographic peaks

Table 1
Repeatability data of the determination of virgin olive oil TAGs by HPLC at a column temperature of 20 °C and using propionitrile as mobile phase
| n = 3 replicates RSDr = Relative Standard Deviation of the repeatability | ||||||||||||
| ECN | HPLC peaks | TAGs | Sample 1 | Sample 2 | Sample 3 | Sample 4 | Sample 5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean (%) | RSDr (%) | Mean (%) | RSDr (%) | Mean (%) | RSDr (%) | Mean (%) | RSDr (%) | Mean (%) | RSDr (%) | |||
| 42 | 1 | LLL | 0,02 | 7,23 | 0,066 | 5,18 | 0,095 | 4,1 | 0,113 | 0,95 | 0,34 | 1,05 |
| 2 | OLLn+ PoLL | 0,085 | 7,44 | 0,24 | 1,78 | 0,26 | 2,25 | 0,35 | 2,02 | 0,5 | 2,83 | |
| 3 | PLLn | 0,023 | 15,74 | 0,039 | 5,51 | 0,057 | 5,62 | 0,082 | 4,35 | 0,12 | 6,15 | |
| 44 | 4 | OLL | 0,47 | 1,52 | 1,53 | 0,42 | 2,62 | 0,98 | 3,35 | 1,05 | 4,37 | 1,13 |
| 5 | OOLn+ PoOL | 1,07 | 2,01 | 1,54 | 0,46 | 1,61 | 0,71 | 1,72 | 1,07 | 1,77 | 2,4 | |
| 6 | PLL+ PoPoO | 0,11 | 12,86 | 0,24 | 4,37 | 0,65 | 1,32 | 1,35 | 0,73 | 2,28 | 1,24 | |
| 7 | POLn+ PpoPo+ PpoL | 0,42 | 5,11 | 0,49 | 2,89 | 0,55 | 2,01 | 0,85 | 1,83 | 1,09 | 1,96 | |
| 46 | 8 | OOL+ LnPP | 6,72 | 0,63 | 8,79 | 0,31 | 11,21 | 0,42 | 13,25 | 0,33 | 15,24 | 0,23 |
| 9 | PoOO | 1,24 | 2,86 | 1,49 | 0,95 | 1,63 | 0,85 | 2,12 | 0,45 | 2,52 | 0,56 | |
| 10 | SLL+ PLO | 2,7 | 0,65 | 4,05 | 0,7 | 6,02 | 0,65 | 9,86 | 0,53 | 11,53 | 0,31 | |
| 11 | PoOP+ SpoL+ SOLn+ SpoPo | 0,64 | 4,42 | 0,69 | 3,02 | 0,79 | 1,23 | 1,53 | 0,89 | 1,7 | 1,66 | |
| 48 | 12+13 | OOO+ PLP+ PoPP | 49,6 | 0,07 | 48,15 | 0,06 | 42,93 | 0,06 | 33,25 | 0,1 | 24,16 | 0,06 |
| 14 | SOL | 0,82 | 1,72 | 0,92 | 1,56 | 1,05 | 1,32 | 1,25 | 1,05 | 1,6 | 1,77 | |
| 15 | POO | 22,75 | 0,25 | 21,8 | 0,2 | 21,05 | 0,3 | 20,36 | 0,35 | 20,17 | 0,14 | |
| 50 | 16 | POP | 3,05 | 0,46 | 4,56 | 0,42 | 4,98 | 0,52 | 5,26 | 0,41 | 5,57 | 0,38 |
| 17 | SOO | 6,87 | 0,21 | 5,56 | 0,33 | 4,86 | 0,43 | 4,12 | 0,72 | 3,09 | 0,69 | |
| 18 | POS+ SLS | 1,73 | 1,23 | 1,65 | 1,1 | 1,54 | 0,99 | 1,49 | 1,1 | 1,41 | 1,0 | |
Figure 2
GC chromatogram of fatty acid alkyl esters obtained from an olive-pomace oil by transesterification with a cold solution of KOH in methanol

7.DETECTION OF EXTRANEOUS OILS IN OLIVE OILSU.K.
The calculation method of the detection of extraneous oils in olive oils by means of a comparison of mathematical algorithms with a data base built from genuine olive oils is set out in Annex I to standard IOC/T.20/Doc. No 25.”
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis (OJ L 248, 5.9.1991, p. 1).
Directive 2011/91/EU of the European Parliament and of the Council of 13 December 2011 on indications or marks identifying the lot to which a foodstuff belongs (OJ L 334, 16.12.2011, p. 1).
They may refrain from tasting an oil when they notice any extremely intense negative attribute by direct olfactory means, in which case they shall record this exceptional circumstance in the profile sheet.