- Latest available (Revised)
- Original (As adopted by EU)
Commission Implementing Regulation (EU) No 503/2013Show full title
Commission Implementing Regulation (EU) No 503/2013 of 3 April 2013 on applications for authorisation of genetically modified food and feed in accordance with Regulation (EC) No 1829/2003 of the European Parliament and of the Council and amending Commission Regulations (EC) No 641/2004 and (EC) No 1981/2006 (Text with EEA relevance)
You are here:

What Version
Advanced Features
- Show Geographical Extent(e.g. England, Wales, Scotland and Northern Ireland)
- Show Timeline of Changes
More Resources
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


This item of legislation originated from the EU
Legislation.gov.uk publishes the UK version. EUR-Lex publishes the EU version. The EU Exit Web Archive holds a snapshot of EUR-Lex’s version from IP completion day (31 December 2020 11.00 p.m.).
Changes over time for: ANNEX II
Changes to legislation:
There are outstanding changes not yet made to Commission Implementing Regulation (EU) No 503/2013. Any changes that have already been made to the legislation appear in the content and are referenced with annotations.
Changes to Legislation
Revised legislation carried on this site may not be fully up to date. Changes and effects are recorded by our editorial team in lists which can be found in the ‘Changes to Legislation’ area. Where those effects have yet to be applied to the text of the legislation by the editorial team they are also listed alongside the legislation in the affected provisions. Use the ‘more’ link to open the changes and effects relevant to the provision you are viewing.
Changes and effects yet to be applied to the whole legislation item and associated provisions
- Signature words omitted by S.I. 2019/705 reg. 53
- Annex 1 Pt. 7 para. 1.1(a) omitted by S.I. 2019/705 reg. 54(e)(iii) (This amendment not applied to legislation.gov.uk. Reg. 54(e) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(b))
- Annex 1 Pt. 7 para. 1 substituted by S.I. 2019/705, reg. 54(e)(iv) (as substituted) by S.I. 2020/1504 reg. 17(10)(b)
- Annex 1 Pt. 1 para. 2 words substituted by S.I. 2019/705 reg. 54(a)(i) (This amendment not applied to legislation.gov.uk. Reg. 54(a)(b) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(a))
- Annex 1 Pt. 1 para. 4(c) words substituted by S.I. 2019/705 reg. 54(a)(ii) (This amendment not applied to legislation.gov.uk. Reg. 54(a)(b) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(a))
- Annex 1 Pt. 1 para. 8 words substituted by S.I. 2019/705 reg. 54(a)(iii) (This amendment not applied to legislation.gov.uk. Reg. 54(a)(b) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(a))
- Annex 1 Pt. 2 para. 1.1(b) words substituted by S.I. 2019/705 reg. 54(b)(i) (This amendment not applied to legislation.gov.uk. Reg. 54(a)(b) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(a))
- Annex 1 Pt. 2 para. 1.1(e)(v) words substituted by S.I. 2019/705 reg. 54(b)(ii) (This amendment not applied to legislation.gov.uk. Reg. 54(a)(b) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(a))
- Annex 1 Pt. 2 para. 1.1(e)(vi) words substituted by S.I. 2019/705 reg. 54(b)(iii) (This amendment not applied to legislation.gov.uk. Reg. 54(a)(b) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(a))
- Annex 1 Pt. 2 para. 7 words substituted by S.I. 2019/705 reg. 54(b)(iv) (This amendment not applied to legislation.gov.uk. Reg. 54(a)(b) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(a))
- Annex 1 Pt. 7 para. 1.8(h) words substituted by S.I. 2019/705 reg. 54(e)(iv) (This amendment not applied to legislation.gov.uk. Reg. 54(e) substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(10)(b))
- Annex 1 Pt. 1 para. 2 words substituted by S.I. 2019/705, reg. 54(a)(i) (as substituted) by S.I. 2020/1504 reg. 17(10)(a)
- Annex 1 Pt. 1 para. 4 words substituted by S.I. 2019/705, reg. 54(a)(ii) (as substituted) by S.I. 2020/1504 reg. 17(10)(a)
- Annex 1 Pt. 1 para. 8 words substituted by S.I. 2019/705, reg. 54(a)(iii) (as substituted) by S.I. 2020/1504 reg. 17(10)(a)
- Annex 1 Pt. 2 para. 1 words substituted by S.I. 2019/705, reg. 54(b)(i) (as substituted) by S.I. 2020/1504 reg. 17(10)(a)
- Annex 1 Pt. 2 para. 1 words substituted by S.I. 2019/705, reg. 54(b)(ii) (as substituted) by S.I. 2020/1504 reg. 17(10)(a)
- Annex 1 Pt. 2 para. 1 words substituted by S.I. 2019/705, reg. 54(b)(iii) (as substituted) by S.I. 2020/1504 reg. 17(10)(a)
- Annex 1 Pt. 2 para. 7 words substituted by S.I. 2019/705, reg. 54(b)(iii) (as substituted) by S.I. 2020/1504 reg. 17(10)(a)
- Annex 1 Pt. 7 para. 1 words substituted by S.I. 2019/705, reg. 54(e)(iii) (as substituted) by S.I. 2020/1504 reg. 17(10)(b)
- Annex 3 para. 1 words omitted by S.I. 2019/705 reg. 56(a)(ii)
- Annex 3 point C para. 3.1(4) words omitted by S.I. 2019/705 reg. 56(c)
- Annex 3 para. 1(3) words substituted by S.I. 2019/705 reg. 56(a)(i)
- Annex 2 s. 2 para. 1.3.1 words omitted by S.I. 2019/705 reg. 55(a)(ii)
- Annex 2 s. 2 para. 2 words omitted by S.I. 2019/705 reg. 55(a)(ix)(aa)
- Annex 2 s. 2 para. 1.1.2(b) words substituted by S.I. 2019/705 reg. 55(a)(i)
- Annex 2 s. 2 para. 1.3.2.1 words substituted by S.I. 2019/705 reg. 55(a)(iii)
- Annex 2 s. 2 para. 1.3.2.2 words substituted by S.I. 2019/705 reg. 55(a)(iv)
- Annex 2 s. 2 para. 2 words substituted by S.I. 2019/705 reg. 55(a)(ix)(bb)
- Annex 2 s. 2 para. 1.4.2 words substituted by S.I. 2019/705 reg. 55(a)(v)
- Annex 2 s. 2 para. 1.4.4.1 words substituted by S.I. 2019/705 reg. 55(a)(vi)
- Annex 2 s. 2 para. 1.5 words substituted by S.I. 2019/705 reg. 55(a)(vii)
- Annex 2 s. 2 para. 1.6.4 words substituted by S.I. 2019/705 reg. 55(a)(viii)
- Art. 4(1)(b) words substituted by S.I. 2019/705 reg. 49 (This amendment not applied to legislation.gov.uk. Reg. 49 substituted immediately before IP completion day by S.I. 2020/1504, regs. 1(2), 17(9))
- Art. 4(1)(b) words substituted by S.I. 2019/705, reg. 49 (as substituted) by S.I. 2020/1504 reg. 17(9)
ANNEX IIU.K. SCIENTIFIC REQUIREMENTS FOR THE RISK ASSESSMENT OF GENETICALLY MODIFIED FOOD AND FEED
I.INTRODUCTIONU.K.
1.DEFINITIONSU.K.
For the purpose of this Annex, the following definitions shall apply:
1.
‘hazard identification’ means the identification of biological, chemical and physical agents capable of causing adverse health effects and which may be present in a particular food and feed or group of foods and feeds;
2.
‘hazard characterisation’ means the qualitative and/or quantitative evaluation of the nature of the adverse health effects associated with biological, chemical and physical agents which may be present in food and feed;
3.
‘risk characterisation’ means the qualitative and/or quantitative estimation, including attendant uncertainties, of the probability of occurrence and severity of known or potential adverse health effects in a given population based on hazard identification, hazard characterisation and exposure assessment.
2.SPECIFIC CONSIDERATIONSU.K.
2.1. Insertion of marker genes and other nucleic acid(s) sequences not essential to achieve the desired trait U.K.
In order to facilitate the risk assessment, the applicant shall endeavour to minimise the presence of inserted nucleic acid(s) sequences not essential to achieve the desired trait.
During the process of genetic modification of plants and other organisms, marker genes are often used to facilitate the selection and identification of genetically modified cells, containing the gene of interest inserted into the genome of the host organism, among the vast majority of untransformed cells. The applicant shall carefully select such marker genes and Article 4(2) of Directive 2001/18/EC shall be respected. Against that background, the applicant shall therefore aim to develop GMOs without the use of antibiotic resistance marker genes.
2.2. Risk assessment of genetically modified food and feed containing stacked transformation events U.K.
For the risk assessment of genetically modified food and feed containing stacked transformation events obtained by conventional crossing of genetically modified plants containing one or several transformation event(s), the applicant shall provide a risk assessment of each single transformation event or, in accordance with Article 3(6) of this Regulation, refer to already submitted application(s). The risk assessment of genetically modified food and feed containing stacked transformation events shall also include an assessment of the following aspects:
(a)
stability of the transformation events;
(b)
expression of the transformation events;
(c)
potential synergistic or antagonistic effects resulting from the combination of the transformation events shall be subject to an assessment in accordance with Sections 1.4 (Toxicology), 1.5 (Allergenicity) and 1.6 (Nutritional assessment).
For genetically modified food and feed containing, consisting of or produced from genetically modified plants, whose cultivation is associated with the production of genetically modified material containing various subcombinations of transformation events (segregating crops), the application shall include all subcombinations independently of their origin which have not yet been authorised. In such a case, the applicant shall provide a scientific rationale justifying that there is no need to provide experimental data for the concerned subcombinations or, in the absence of such scientific rationale, provide the experimental data.
For genetically modified food and feed containing, consisting of or produced from genetically modified plants, the cultivation of which does not lead to the production of genetically modified material containing various combinations of transformation events (non-segregating crops), the application shall only cover the combination which is to be placed on the market.
The rules set out in this Section apply mutatis mutandis to transformation events which are combined by other means such as co- and retransformation.
II.SCIENTIFIC REQUIREMENTSU.K.
1.HAZARD IDENTIFICATION AND CHARACTERISATIONU.K.
1.1. Information relating to the recipient or (where appropriate) parental plants U.K.
1.1.1.The applicant shall provide comprehensive information relating to the recipient or (where appropriate) the parental plants in order to:U.K.
(a)
evaluate all issues of potential concern, such as the presence of natural toxins or allergens;
(b)
identify the need for specific analyses.
1.1.2.For the purposes referred to in point 1.1.1, the applicant shall provide the following information:U.K.
(a)
Complete name:
(i)
family name;
(ii)
genus;
(iii)
species;
(iv)
subspecies;
(v)
cultivar/breeding line or strain;
(vi)
common name.
(b)
Geographical distribution and cultivation of the plant, including its distribution within the Union.
(c)
Information on the recipient or parental plants relevant to their safety, including any known toxicity or allergenicity.
(d)
Data on the past and present use of the recipient plant. This information shall include the history of safe use for consumption as food or feed, information on how the plant is typically cultivated, transported and stored, whether special processing is required to make the plant safe to eat, and describe the normal role of the plant in the diet (such as which part of the plant is used as a food or feed source, whether its consumption is important in particular subgroups of the population, what important macro- or micro-nutrients it contributes to the diet).
1.2. Molecular Characterisation U.K.
1.2.1. Information relating to the genetic modification U.K.
The applicant shall provide sufficient information on the genetic modification:
(a)
to identify the nucleic acid(s) intended for transformation and related vector sequences potentially delivered to the recipient plant;
(b)
to characterise the nucleic acid(s) actually inserted in the plant.
1.2.1.1.Description of the methods used for the genetic modificationU.K.
The applicant shall provide information on the following elements:
(a)
the method of genetic transformation including relevant references;
(b)
the recipient plant material;
(c)
the species and the strain of Agrobacterium and other microbes, if used during the genetic transformation process;
(d)
the helper plasmids, if used during the genetic transformation process;
(e)
the source of carrier nucleic acid(s), if used during the genetic transformation process.
1.2.1.2.Nature and source of vector usedU.K.
The applicant shall provide the following information:
(a)
a physical map of the functional elements and other plasmid/vector components together with the relevant information needed for the interpretation of the molecular analyses (such as restriction sites, the position of primers used in polymerase chain reaction (PCR), the location of probes used in Southern analysis). The region intended for insertion shall be clearly indicated;
(b)
a table identifying each component of the plasmid/vector (including the region intended for insertion), its size, its origin and its intended function.
1.2.1.3.Source of nucleic acid(s) used for transformation, size and intended function of each constituent fragment of the region intended for insertionU.K.
The applicant shall provide information on the donor organism(s) and on the nucleic acid(s) sequence(s) intended to be inserted in order to determine whether the nature of the donor organism(s) or the nucleic acid sequence(s) may trigger any safety issue.
Information regarding the function of the nucleic acid region(s) intended for insertion shall comprise the following elements:
(a)
the complete sequence of the nucleic acid(s) intended to be inserted, including information on any deliberate alteration(s) to the corresponding sequence(s) in the donor organism(s);
(b)
the history of safe use of the gene product(s) arising from the regions intended for insertion;
(c)
data on the possible relationship of the gene products with known toxins, anti-nutrients and allergens.
Information regarding each donor organism shall comprise:
taxonomic classification;
history of use regarding food and feed safety.
1.2.2. Information relating to the genetically modified plant U.K.
1.2.2.1.General description of the trait(s) and characteristics which have been introduced or modifiedU.K.
Information provided under this point may be limited to a general description of the introduced trait(s) and the resulting changes to the phenotype and metabolism of the plant.
For example, when the introduced trait is herbicide tolerance, the applicant shall provide information on the mode of action of the active substance and its metabolism in the plant.
1.2.2.2.Information on the sequences actually inserted/deletedU.K.
The applicant shall provide the following information:
(a)
the size and copy number of all detectable inserts, both complete and partial; this is typically determined by Southern analysis.
Probe/restriction enzyme combinations used for this purpose shall provide complete coverage of sequences that could be inserted into the genetically modified plant, such as any parts of the plasmid/vector or any carrier or foreign nucleic acid(s) remaining in the genetically modified plant.
The Southern analysis shall span the entire transgenic locus(i) as well as flanking sequences and include all appropriate controls.
For the determination of copy number of the insert, complementary methods may also be used (such as real-time PCR);
(b)
the organisation and sequence of the inserted genetic material at each insertion site in a standardised electronic format, with the aim of identifying changes in the inserted sequences compared to the sequence intended for insertion;
(c)
in the case of deletion(s), size and function of the deleted region(s), whenever possible;
(d)
subcellular location(s) of insert(s) (nucleus, chloroplasts, mitochondria, or maintained in a non-integrated form) and methods for its/their determination;
(e)
sequence information in a standardised electronic format for both 5’ and 3’ flanking regions at each insertion site, with the aim of identifying interruptions of known genes.
Bioinformatic analyses shall be conducted using up-to-date databases with the aim of performing both intraspecies and interspecies similarity searches.
In the case of genetically modified plants containing stacked transformation events, the safety of potential interactions between any unintended modifications at each insertion site shall be assessed;
(f)
Open Reading Frames (hereafter referred to as ‘ORFs’ and defined as any nucleotide sequence that contains a string of codons that is uninterrupted by the presence of a stop codon in the same reading frame) created as a result of the genetic modification either at the junction sites with genomic DNA or due to internal rearrangements of the insert(s).
The ORFs shall be analysed between stop codons, not limiting their lengths. Bioinformatic analyses shall be conducted to investigate possible similarities with known toxins or allergens using up-to-date databases.
The characteristics and versions of the databases shall be provided.
Depending on the information gathered, further analyses (such as transcription analysis) may be needed to complete the risk assessment.
1.2.2.3.Information on the expression of the insert(s)U.K.
The applicant shall provide information:
to demonstrate whether the inserted/modified sequence results in intended changes at the protein, RNA and/or metabolite levels;
to characterise the potential unintended expression of new ORFs identified under point 1.2.2.2(f) as raising a safety concern.
For those purposes, the applicant shall provide the following information:
(a)
The method(s) used for expression analysis together with their performance characteristics;
(b)
Information on developmental expression of the insert during the life cycle of the plant.
The requirement for information on developmental expression shall be considered on a case-by-case basis taking into account the promoter used, the intended effect(s) of the modification and scope of the application;
(c)
Parts of the plant where the insert/modified sequences are expressed;
(d)
Potential unintended expression of new ORFs identified under point 1.2.2.2(f) which raise a safety concern;
(e)
Protein expression data, including the raw data, obtained from field trials and related to the conditions in which the crop is grown.
Data on expression levels from those parts of the plant used for food and feed purposes shall be provided in all cases.
In addition, information shall also be provided on the expression of target genes in other parts of the plant when tissue-specific promoters have been used and when this is relevant for the safety assessment. The minimum requirement for protein expression shall be data provided from three growing sites or from one site over three seasons. Permutations of the sites and seasons shall be acceptable provided that the minimum requirement is met. When justified by the nature of the insert (such as silencing approaches or where biochemical pathways have been intentionally modified), specific RNA(s) or metabolite(s) shall be analysed.
For silencing approaches by RNAi expression, potential ‘off target’ genes should be searched by in silico analysis to assess if the genetic modification could affect the expression of other genes which raise safety concerns;
(f)
With regard to the stacking of transformation events by conventional crossing, expression data shall be provided to assess the potential interactions between the events which may raise any additional safety concerns over protein and trait expression compared with the single transformation events. The comparison shall be carried out with data obtained from plants grown in the same field trials. On a case-by-case basis, and where concerns arise, additional information may be necessary.
1.2.2.4.Genetic stability of the insert and phenotypic stability of the genetically modified plantU.K.
The applicant shall provide information:
(a)
to demonstrate the genetic stability of the transgenic locus(i) and the phenotypic stability and inheritance pattern(s) of the introduced trait(s);
(b)
in case of stacked transformation events, to establish that each of the transformation events stacked in the plant has the same molecular properties and characteristics as in the plants with the single transformation events.
For the purposes of that information, applicants shall provide data which demonstrate stability over multiple (normally five) generations or vegetative cycles for plant containing single transformation events. Data from the first and the last generations of vegetative cycles are sufficient. The source of the material used for the analysis shall be specified. Data shall be analysed using appropriate statistical methods.
For stacked transformation events comparisons between the original transformation events and the stacked transformation events shall be carried out using plant materials representative of those designed for commercial production. The applicant shall provide adequate justification for the plant materials used. Comparisons shall include comparisons of sequences of the inserts and the flanking regions obtained from GM plants containing single events and plants containing stacked transformation events.
To assess genetic stability of the transformation event(s), applicants shall use appropriate molecular approaches referred to in Section 1.2.2.2.
1.2.2.5.Potential risk associated with horizontal gene transferU.K.
The applicant shall assess the probability of horizontal gene transfer from the product to humans, animals and micro-organisms and any potential associated risk when intact and functional nucleic acid(s) remains in the genetically modified food and feed.
1.2.3. Conclusions of the molecular characterisation U.K.
The molecular characterisation shall provide data on the structure and expression of the insert(s), and on the stability of the intended trait(s). This shall also apply to situations where transformation events have been stacked by conventional breeding.
It shall be specifically indicated whether the molecular characterisation of the genetic modification(s) raises safety concerns with regard to the interruption of endogenous genes or regulatory sequences.
The molecular characterisation shall also aim to identify whether the genetic modification(s) raise(s) any issues regarding the potential for producing proteins/substances other than those intended and in particular new toxins or allergens.
The potential unintended changes identified in this Section shall be addressed in the relevant complementary part(s) of the safety assessment.
1.3. Comparative analysis U.K.
The comparative analysis of composition and agronomic as well as phenotypic characteristics shall constitute, together with the molecular characterisation, the starting point to structure and conduct the risk assessment of a new genetically modified food and feed.
It shall aim at identifying similarities and differences:
(a)
in composition, agronomic performance and phenotypic characteristics (intended and unintended alterations) between the genetically modified plant and its conventional counterpart;
(b)
in composition between the genetically modified food and feed and its conventional counterpart.
Where no appropriate conventional counterpart can be identified, a comparative safety assessment cannot be made and consequently a safety and nutritional assessment of the genetically modified food or feed shall be carried out as for novel foods falling within the scope of Regulation (EC) No 258/97 of the European Parliament and of the Council(1) that do not have conventional counterparts (such as where the genetically modified food or feed is not closely related to a food or feed with a history of safe use or where a specific trait or specific traits are introduced with the intention of bringing complex changes in the composition of the genetically modified food or feed).
1.3.1. Choice of the conventional counterpart and additional comparators U.K.
In the case of vegetatively propagated crops, the conventional counterpart shall, in principle, be the near-isogenic variety used to generate the transgenic line.
In the case of crops that reproduce sexually, the conventional counterpart shall have a genetic background comparable to the genetically modified plant. When the GM plant has been developed using back-crossing, a conventional counterpart with a genetic background that is as close as possible to the genetically modified plant shall be selected.
In addition, the applicant may include a comparator having a closer genetic background to the genetically modified plant than the conventional counterpart (such as a negative segregant).
In the case of herbicide tolerant genetically modified plants and in order to assess whether the expected agricultural practices influence the expression of the studied endpoints, three test materials shall be compared: the genetically modified plant exposed to the intended herbicide; the conventional counterpart treated with conventional herbicide management regimes; and the genetically modified plant treated with the same conventional herbicide management regimes.
In the case of stacked transformation events, it is not always possible to use a conventional counterpart with a genetic background as close to the genetically modified plant as with conventional counterpart normally used for single transformation events. In such circumstances, the applicant shall provide reasoned justification on the choice of the conventional counterpart and assess its limitations for the risk assessment. In addition, single parental genetically modified lines or genetically modified lines containing a subcombination of the stacked transformation events for which an application has been submitted or negative segregants derived from these genetically modified lines may also be included as additional comparators. The applicant shall provide detailed information justifying the choice of additional comparators.
In all cases, the applicant shall provide information on the breeding scheme (pedigree) in relation to the genetically modified plant, the conventional counterpart and, where appropriate, additional comparators together with an adequate justification of their selection. The history of safe use of the conventional counterpart shall be adequately supported by both qualitative and quantitative data.
More detailed guidance for the application of the requirements of this Section is available in the EFSA scientific opinion ‘Guidance on selection of comparators for the risk assessment of genetically modified plants and derived food and feed’(2).
1.3.2. Experimental design and statistical analysis of data from field trials for comparative analysis U.K.
1.3.2.1.Description of the protocols for the experimental designU.K.
(a)Principles of experimental designU.K.
Field trials used for the production of material for the comparative analysis shall be performed in order to determine whether the genetically modified plant and/or the genetically modified food and feed is different from its conventional counterpart and/or equivalent to non-genetically modified reference varieties with a history of safe use.
For each endpoint, the comparative analysis shall involve the following two approaches:
(i)
a test of difference, to verify whether the genetically modified plant is different from its conventional counterpart and might therefore be considered a hazard depending on the type of the identified difference, and on the magnitude and type of exposure;
(ii)
a test of equivalence to verify whether the genetically modified plant is equivalent or not to non-genetically modified reference varieties, apart from the introduced trait(s).
In testing for difference, the null hypothesis shall be that there is no difference between the GMO and its conventional counterpart against the alternative hypothesis that a difference exists.
Where additional comparator(s) are used for the risk assessment, a test of difference shall be carried out between the genetically modified plant and each of the additional comparator(s) following the requirements set out in Section 1.3.2.2 for the test of difference between the genetically modified plant and its conventional counterpart.
In testing for equivalence, the null hypothesis shall be that the difference between the GMO and the set of reference varieties is at least as great as a specified minimum size (see Section 1.3.2.2) against the alternative hypothesis that there is no difference or a smaller difference than the specified minimum between the GMO and the set of reference varieties.
Rejection of the null hypothesis shall be required in order to conclude that the GMO and the set of reference varieties are unambiguously equivalent for the endpoint considered. The equivalence limits used for the test of equivalence shall represent appropriately the range of natural variation expected for reference varieties with a history of safe use.
(b)Specific protocols for experimental designU.K.
Natural variation may have several sources: variation within a variety arises due to environmental factors and variation between varieties arises due to a combination of both genetic and environmental factors. In order to identify and estimate differences attributable only to genotypes, it is essential to control environmental variability. Therefore, non-genetically modified reference varieties shall be included in the experimental design of the field trials and in sufficient numbers to ensure an adequate estimate of the variability required to set the equivalence limits. All test materials consisting of genetically modified plants, conventional counterpart, reference varieties and, where appropriate, additional comparator(s) shall be randomised to plots within a single field at each site, usually in a completely randomised or randomised block experimental design. The different sites selected for the field trials shall reflect the different meteorological and agronomic conditions under which the crop is to be grown; the choice shall be explicitly justified. The choice of non-genetically modified reference varieties shall be appropriate for the chosen sites and shall be justified explicitly. In the case the sites cover a restricted range of growing conditions, the applicant shall replicate the field trials over more than one year.
Within each site the test materials consisting of genetically modified plants, conventional counterpart and, where appropriate, additional comparator(s) shall be identical for all replicates. In addition, unless there is explicit justification for not doing so, at each site there shall be at least three appropriate non-genetically modified reference varieties of the crop that have a known history of safe use, which shall also be identical between replicates. The replication at each site is the number of results obtained for each test material; the replication should never be less than four at any site. However, if only two appropriate reference varieties are available at a particular site, then the replication shall be six at that site; if only one is available then the replication shall be eight.
Each field trial shall be replicated at a minimum of eight sites, chosen to be representative of the range of likely receiving environments where the plant is to be grown. The field trials may be conducted in a single year, or spread over multiple years. The non-genetically modified reference varieties may vary between sites and at least six different reference varieties shall be used over the entire set of field trials.
When the genetically modified plant is tested together with other genetically modified plants of the same crop species (such as Zea mays) the production of material for the comparative assessment of these different genetically modified plant may be produced simultaneously at the same site and within the same field trial by the placing of the different genetically modified plants and their appropriate comparator(s) in the same randomised block. This shall be subject to the following two strict conditions:
(i)
the conventional counterpart and, where appropriate, additional comparator(s) shall always occur together with the genetically modified plant in the same block;
(ii)
all the different genetically modified plants and their comparator(s) and all the non-genetically modified reference varieties used to test equivalence with those genetically modified plants shall be fully randomised within each block.
If the number of plots per block required for such a field trial were to exceed 16, then a partially balanced incomplete block design may be used, to reduce the number of plots per block, by excluding some of the genetically modified plants and their appropriate comparator(s) from each block. This shall be subject to the following two strict conditions:
(i)
the conventional counterpart shall always occur together with its particular genetically modified plant in the same block;
(ii)
all of the non-genetically modified reference varieties shall appear in each of the incomplete blocks and be fully randomised with the plants and their comparator(s).
The field trials shall be adequately described, giving information on important parameters such as management of the field before sowing, date of sowing, soil type, herbicide use, climatic and other cultivation/environmental conditions during growth and time of harvest, as well as the conditions during storage of the harvested material.
More detailed guidance for the application of the requirements of this Section is available in the EFSA opinion on ‘Statistical considerations for the safety evaluation of GMOs’(3).
1.3.2.2.Statistical analysisU.K.
Analysis of data shall be presented in a clear format, using standardised scientific units. The raw data and the programming code used for the statistical analysis shall be given in an editable form.
Data transformation may be necessary to ensure normality and to provide an appropriate scale on which statistical effects are additive. For many endpoint response variables, a logarithmic transformation is expected to be appropriate. In such cases, any difference between the genetically modified material and any other test material shall be interpreted as a ratio on the natural scale. However, when logarithmic transformation does not provide appropriate results, the natural scale or another scale shall be considered.
The total variability of each endpoint observed in the field trials shall be estimated and partitioned using appropriate statistical models in order to derive two sets of confidence limits and to set a lower and upper equivalence limit based on the variability observed among the reference varieties. One set of confidence limits shall be used in the test of difference; the other set and the equivalence limits shall be used in the test of equivalence.
A linear mixed statistical model shall be used for calculation of the confidence limits for both tests (that is to say, the difference and equivalence tests); a slightly different model shall be used to estimate the equivalence limits to be used in the equivalence test.
Denote by I an indicator variable (uncentered in the mixed model) such that I = 1 for a field plot having any of the non-GM reference varieties, and I = 0 otherwise. Then the random factors for model 1 should be, but not necessarily be restricted to, those representing the variation: (i) between the test materials (a set that includes the GM plant, its conventional counterpart, each of the non-GM reference varieties and any additional comparators); (ii) in the interaction between the test materials and I; (iii) between sites; and (iv) between blocks within sites. Model 2 should be identical to model 1 except that the random factor representing the interaction between the test materials and I is omitted.
The fixed factor for both models should have as many levels as there are test materials and represent the contrasts between the means of the test materials. The test materials are as defined above: the GM plant; its conventional counterpart; the set of non-GM reference varieties; and any additional test materials. The set of non-GM reference varieties is considered as a single level of the fixed factor. For the difference test, the component of the fixed factor of interest is the single degree-of-freedom contrast between the GM plant and its conventional counterpart. For the equivalence test, the component of the fixed factor of interest is the single degree-of-freedom contrast between the GM plant and the set of non-GM reference varieties.
Both the difference test and the equivalence test shall be implemented using the correspondence between hypothesis testing and the construction of confidence limits. In the case of equivalence testing, the approach used shall follow the two one-sided tests (TOST) methodology by rejecting the null hypothesis of non-equivalence when the both confidence limits fall between the equivalence limits. The choice of 90 % confidence limits corresponds to the customary 95 % level for statistical testing of equivalence.
The results of the difference and equivalence tests shall be represented visually for all the endpoints simultaneously, on a single graph or a few graphs.
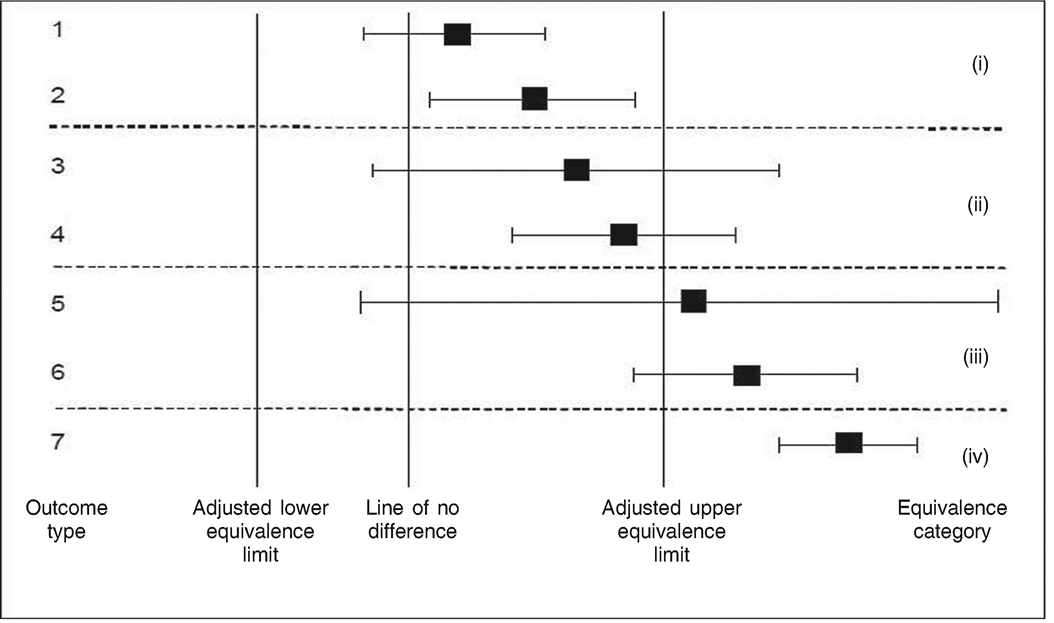
The graph(s) shall show the line of zero difference between the genetically modified material and its conventional counterpart, and for each endpoint: the lower and upper adjusted equivalence limits; the mean difference between the genetically modified material and its conventional counterpart; and the confidence limits for this difference (see the set of possible outcomes for a single endpoint in the graph in Figure 1).
When, in addition to the conventional counterpart, another test material is used as comparator, the mean difference between the genetically modified material and that comparator, its confidence limits and its adjusted equivalence limits shall be displayed on the graph(s), for all such additional comparators, by referring this to the same zero baseline as defined by the conventional counterpart. The line of zero difference on the logarithmic scale corresponds to a multiplicative factor of unity on the natural scale. The horizontal axis shall be labelled with values that specify the change on the natural scale. In the case of logarithmic transformation, changes of 2x and ½x will appear equally spaced on either side of the line of zero difference.
Despite the expected proportion of spurious significant differences, the applicant shall report and discuss all significant differences observed between the genetically modified crop, its conventional counterpart and, where applicable, any other test material, focusing on their biological relevance (see Section 3 on Risk Characterisation).
For reporting, full details shall be given for each endpoint analysed, listing:
(a)
the assumptions underlying the analysis;
(b)
full specification of the mixed models chosen, including fixed and random effects;
(c)
results of any test of interaction between the test materials and sites;
(d)
fixed effects, together with the appropriate estimated residual variation with which they are compared, and variance components for the random factors;
(e)
estimated degrees of freedom;
(f)
any other relevant statistics.
A discussion on the likely impact of other growing conditions not tested in the field trial shall be provided.
Figure 1: simplified version of a graph for comparative assessment showing the seven outcome types possible for each single endpoint. After adjustment of the equivalence limits, a single confidence limit (for the difference) serves visually for assessing the outcome of both tests (difference and equivalence). Here, only the upper adjusted equivalence limit is considered. Shown are: the mean of the genetically modified crop on an appropriate scale (square); the confidence limits (whiskers) for the difference between the genetically modified crop and its conventional counterpart (bar shows confidence interval); a vertical line indicating no difference (for test of difference); and vertical lines indicating adjusted equivalence limits (for test of equivalence). For outcome types 1, 3 and 5, the null hypothesis of no difference cannot be rejected: for outcomes 2, 4, 6 and 7, the genetically modified crop is different from its conventional counterpart. Regarding interpretation of equivalence, four categories (i) to (iv) are identified: in category (i), the null hypothesis of non-equivalence is rejected in favour of equivalence; in categories (ii), (iii) and (iv), non-equivalence cannot be rejected.
A.
Regarding test of difference, each outcome from the graph shall be categorised as follows and the respective appropriate conclusion shall be drawn.
(i)
Outcome types 1, 3 and 5: the confidence interval bar overlaps with the line of no-difference. The null hypothesis of no difference cannot be rejected and the appropriate conclusion is that there is insufficient evidence that the genetically modified crop and its conventional counterpart differ.
(ii)
Outcome types 2, 4, 6 and 7: the confidence interval bar does not overlap with the line of no-difference. The null hypothesis of no difference must be rejected and the appropriate conclusion is that the genetically modified crop is significantly different from its conventional counterpart.
B.
Regarding test of equivalence, each outcome from the graph shall be categorised as follows, and the respective appropriate conclusion shall be drawn.
(i)
Outcome types 1 and 2 (category (i), Figure 1): both confidence limits lie between the adjusted equivalence limits and the null hypothesis of non-equivalence is rejected. The appropriate conclusion is that the genetically modified crop is equivalent to the set of non-genetically modified reference varieties.
(ii)
Outcome types 3 and 4 (category (ii), Figure 1): the mean of the genetically modified crop lies between the adjusted equivalence limits, but the confidence interval bar overlaps at least one of the adjusted equivalence limits on the graph. Non-equivalence cannot be rejected and the appropriate conclusion is that equivalence between the genetically modified crop and the set of non-genetically modified reference varieties is more likely to be the case than lack of equivalence.
(iii)
Outcome types 5 and 6 (category (iii), Figure 1): the mean of the genetically modified crop lies outside the adjusted equivalence limits, but the confidence interval bar overlaps with at least one of the adjusted equivalence limits. Non-equivalence cannot be rejected and the appropriate conclusion is that lack of equivalence between the genetically modified crop and the set of non-genetically modified reference varieties is more likely to be the case than equivalence.
(iv)
Outcome type 7 (category (iv), Figure 1): both confidence limits lie outside the adjusted equivalence limits. The appropriate conclusion is that there is lack of equivalence between the genetically modified crop and the set of non-genetically modified reference varieties.
In the case of significant difference and/or lack of equivalence for any particular endpoint, further statistical analysis shall be carried out to assess whether there are interactions between any of the test materials and site, possibly using a simple standard ANOVA approach. Whatever approach is adopted, details shall be given, for each endpoint analysed, listing: (a) the assumptions underlying the analysis, and, when appropriate: (b) degrees of freedom; (c) the estimated residual variation for each source of variation, and variance components; (d) any other relevant statistics. These additional analyses are intended to aid the interpretation of any significant differences found and to study potential interactions between test materials and other factors.
More detailed guidance for the application of the requirements of this Section is available in the EFSA opinion on ‘Statistical considerations for the safety evaluation of GMOs’(4).
1.3.3. Selection of material and compounds for analysis U.K.
Analysis of the composition of plant material is crucial when comparing the genetically modified food and feed with its conventional counterpart. The material to be used for the comparative assessment shall be selected while taking into account the uses of the genetically modified plant and the nature of the genetic modification. In the case of herbicide tolerant genetically modified plants, three test materials shall be used: the genetically modified plant exposed to the intended herbicide; the conventional counterpart treated with conventional herbicide management regimes; and the genetically modified plant treated with the same conventional herbicide management regimes. Unless duly justified, analysis shall be carried out on the raw agricultural commodity, as this usually represents the main point of entry of the material into the food and feed production and processing chain. Additional analysis of processed products (such as food and feed, food ingredients, feed materials, food and feed additives or food flavourings) shall be conducted, where appropriate and on a case-by-case basis (see also Section 1.3.6). The sampling, analysis and preparation of the tested material shall be carried out according to appropriate quality standards.
1.3.4. Comparative analysis of composition U.K.
In addition to the analysis on the level of the newly expressed proteins (see Section 1.2.2.3), the compositional analysis shall be carried out on an appropriate range of compounds. In each case, the applicant shall provide at least analysis on proximates (including moisture and total ash), key macro- and micro-nutrients, anti-nutritional compounds, natural toxins, and already identified allergens, as well as other secondary plant metabolites characteristic for specific crop plant species, as referred to in the Organisation for Economic Cooperation and Development (OECD) consensus documents on compositional considerations for new plant varieties (OECD consensus documents)(5). The vitamins and minerals selected for analysis shall be those which are present at levels which are nutritionally significant and/or which make nutritionally significant contributions to the diet at the levels at which the plant is consumed. The specific analyses required shall depend on the plant species examined, but shall include a detailed assessment appropriate to the intended effect of the genetic modification, the considered nutritional value and use of the plant. The applicant shall pay particular attention to key nutrients such as proteins, carbohydrates, lipids/fats, fibre, vitamins and minerals. For example, a fatty acid profile shall be included for oil-rich plants (main individual saturated, mono-unsaturated and poly-unsaturated fatty acids) and an amino acid profile (individual protein amino acids and main non-protein amino acids) for plants used as an important protein source. Analysis of plant cell wall components are also required for the vegetative parts of plants used for feed purposes.
The applicant shall also provide an analysis on key toxins inherently present in the recipient plant which may adversely affect human/animal health depending on their toxic potency and levels. The concentrations of such compounds shall be assessed according to plant species and the proposed use of the food and feed product. Similarly, anti-nutritional compounds, such as digestive enzyme inhibitors, and already identified allergens shall be studied.
The characteristics of the introduced trait may trigger further analysis of specific compounds including metabolites of potentially modified metabolic pathways. The applicant shall consider when appropriate, the inclusion of compounds other than the key nutrients, key toxins, anti-nutrients and allergens identified by the OECD consensus documents and justify the selection of these compounds.
1.3.5. Comparative analysis of agronomic and phenotypic characteristics U.K.
The applicant shall provide a comparison between the genetically modified plant and its conventional counterpart. This comparison shall enable the applicant to identify unintended effects resulting from the genetic modification and shall address also plant biology and agronomic traits, including common breeding parameters (such as yield, plant morphology, flowering time, day degrees to maturity, duration of pollen viability, response to plant pathogens and insect pests, sensitivity to abiotic stress). The protocols of these field trials shall follow the specifications set out in Section 1.3.2.
Where transformation events are stacked by conventional crossing, there may also be changes to agronomic and phenotypic characteristics. Possible differences in phenotypic characteristics and agronomic properties of stacked transformation events shall be assessed in field trials. Where appropriate, the applicant shall provide additional information on agronomic traits of the stacked transformation events from additional field trials.
1.3.6. Effects of processing U.K.
The applicant shall assess whether or not the processing and/or preserving technologies applied are likely to modify the characteristics of genetically modified end products compared with their respective conventional counterpart. The applicant shall provide a description of the different processing technologies in sufficient detail, paying special attention to the steps which may lead to significant changes in the product content, quality or purity.
Genetic modification can target metabolic pathways resulting in changes in the concentration of non-protein substances or in new metabolites (such as in nutritionally enhanced foods). Processed products may be assessed together with the assessment of the genetically modified plant for the safety of the genetic modification, or a processed product may be assessed separately. The applicant shall provide the scientific rationale for the risk assessment of these products. On a case-by-case basis, the submission of additional experimental data shall be considered by the applicant.
When appropriate, depending on the product, information shall be necessary on the composition, level of undesirable substances, nutritional value and metabolism, as well as on the intended use.
When appropriate, depending on the nature of the newly expressed protein(s), it shall be necessary to assess the extent to which the processing steps lead to the concentration or to the elimination, denaturation and/or degradation of these protein(s) in the final product.
1.3.7. Conclusion U.K.
The conclusion of the comparative analysis shall clearly state:
(a)
whether agronomic and phenotypic characteristics of the genetically modified plant are, except for the introduced trait(s), different to the characteristics of its conventional counterpart and/or equivalent to the reference varieties, taking into account natural variation;
(b)
whether compositional characteristics of the genetically modified food and feed are, taking into account natural variation, different to the characteristics of its conventional counterpart and/or equivalent to the reference varieties, except for the introduced trait(s);
(c)
characteristics for which the genetically modified plant or the genetically modified food and feed are different to the characteristics of its conventional counterpart and/or not equivalent to the reference varieties taking into account natural variation, which need further investigation;
(d)
whether, in the case of transformation events stacked by conventional crossing, there are indications of interactions between the combined transformation events.
1.4. Toxicology U.K.
The toxicological impact of any changes on the whole genetically modified food/feed resulting from the genetic modification such as the introduction of new genes, gene silencing or over-expression of an endogenous gene shall be assessed.
Toxicological assessment shall be performed in order to:
(a)
demonstrate that the intended effect(s) of the genetic modification has no adverse effects on human and animal health;
(b)
demonstrate that unintended effect(s) of the genetic modification(s) identified or assumed to have occurred based on the preceding comparative molecular, compositional or phenotypic analyses, have no adverse effects on human and animal health;
(c)
identify potential adverse effects of new constituents and determine their highest dose level that does not result in adverse effects. From data obtained from an appropriate animal study, an acceptable daily intake (ADI) of single compounds by humans may be derived by using uncertainty or safety factors that take into account differences between test animal species and humans, and inter-individual variations among humans;
(d)
identify potential adverse effects on the whole genetically modified food/feed or address remaining uncertainties through the performance of 90-day feeding studies.
The applicant shall consider the nature of toxicological testing to be conducted on new constituents and whole genetically modified food/feed based on the outcome of the molecular and comparative analysis referred to in Sections 1.2 and 1.3, namely the differences identified between the genetically modified product and its conventional counterpart, including intended as well as unintended changes. The applicant shall also assess the results of the performed toxicological tests to consider the need to conduct additional testing on new constituents or on whole genetically modified food/feed as set out in Sections 1.4.4.2 and 1.4.4.3.
The applicant shall take into account the presence of: newly expressed proteins, the potential presence of other new constituents and/or possible changes in the level of natural constituents beyond normal variation. The specific information requirements and testing strategies are set out in Sections 1.4.1 to 1.4.4.
As regards applications of which the scope include or is restricted to genetically modified food and feed produced from genetically modified plants, toxicological studies with the processed products shall be provided except if the applicant provides a risk assessment of the genetically modified plant (or relevant parts of it) demonstrating its safety and there are no indications that the processed genetically modified food and feed would be any different from their respective conventional counterpart. The applicant shall provide adequate justification in this regard.
Toxicology studies designed to evaluate risks to human and/or animal health shall complement each other. Most studies required for the assessment of the safety of the genetically modified food are also valid for the assessment of genetically modified feed.
Besides the exposure of consumers and animals through intake of food and feed, any adverse effect(s) on individuals that could be due to their exposure to genetically modified food and feed material as part of their professional activities, for example, farming, seed processing, shall be reported by the applicant. Appropriate studies shall be performed to further characterise these indications of potential adverse effects.
The applicant shall use internationally agreed protocols and test methods for toxicity testing (see Tables 1 and 2 of Section 1.7). Adaptations of these protocols or use of any methods that differ from such protocols shall be justified in the application.
1.4.1. Testing of newly expressed proteins U.K.
The applicant shall provide an evaluation of all newly expressed proteins. The studies required to investigate the potential toxicity of a newly expressed protein shall be selected on a case-by-case basis, depending on the knowledge available with respect to the protein’s source, function or activity and history of human or animal consumption. As regards proteins expressed in the genetically modified plant, in the case where the history of safe use for consumption as food and/or feed of both the plant and the newly expressed proteins is duly documented, specific toxicity testing as provided for in this Section shall not be required. In such case, the applicant shall provide the necessary information regarding the history of safe use of the proteins.
Where specific testing is required, the tested protein shall be equivalent to the newly expressed protein as it is expressed in the genetically modified plant. If, due to the lack of sufficient amount of test materials from the plant, a protein produced by micro-organisms is used, the structural, biochemical and functional equivalence of this microbial substitute to the newly expressed plant protein shall be demonstrated. In particular, comparisons of the molecular weight, amino acid sequence, post-translational modification, immunological reactivity and, in the case of enzymes, the enzymatic activity are needed to provide evidence for the equivalence. In case of differences between the plant expressed protein and its microbial substitute, the significance of these differences for the safety studies shall be evaluated.
To demonstrate the safety of newly expressed proteins, the applicant shall provide:
(a)
A molecular and biochemical characterisation of the newly expressed protein, including determination of the primary structure, molecular weight (for example using mass spectrometry), studies on post-translational modifications and a description of its function. In the case of newly expressed enzymes, information on the enzyme activities including the temperature and pH range for optimum activity, substrate specificity, and possible reaction products shall also be provided. The potential interaction with other plant constituents shall also be evaluated.
(b)
An up-to-date search for homology to proteins known to cause adverse effects, such as toxic proteins. A search for homology to proteins exerting a normal metabolic or structural function may also contribute valuable information. The database(s) and the methodology used to carry out the search shall be specified.
(c)
A description of the stability of the protein under relevant processing and storage conditions and the expected treatment of the food and feed. The influences of temperature and pH changes shall be examined and potential modification(s) of the proteins (such as denaturation) and/or production of stable protein fragments generated through such treatments shall be characterised.
(d)
Data concerning the resistance of the newly expressed protein to proteolytic enzymes (such as pepsin), such as by in vitro investigations using appropriate and standardised tests. Stable breakdown products shall be characterised and evaluated with regard to the potential to cause adverse health effects linked to their biological activity.
(e)
A repeated-dose 28-day oral toxicity study with the newly expressed protein in rodents. When appropriate depending on the outcome of the 28-day toxicity study, further targeted investigations shall be provided, including an analysis of immunotoxicity.
Acute toxicity testing of the newly expressed proteins of genetically modified plants is of little additional value for the risk assessment of the repeated human and animal consumption of genetically modified food and feed and shall not be provided as part of the studies performed under this point.
The applicant shall perform studies with combined administration of proteins when the genetic modification results in the expression of two or more proteins in the genetically modified plant and when, based on scientific knowledge, a possibility of synergistic or antagonistic interactions of safety concerns is identified.
1.4.2. Testing of new constituents other than proteins U.K.
The applicant shall provide a risk assessment of identified new constituents other than proteins. This shall include, on a case-by-case basis, an evaluation of their toxic potency and of the need of toxicological testing as well as a determination of their concentration in the genetically modified food and feed. To establish the safety of new constituents having no history of safe use for consumption in food and feed, the applicant shall provide information analogous to that described in the ‘Guidance for submissions for food additive evaluations by the EFSA Panel on Food Additives and Nutrient Sources added to Food’ of 16 August 2012(6) and Commission Regulation (EC) No 429/2008 of 25 April 2008 on detailed rules for the implementation of Regulation (EC) No 1831/2003 as regards the preparation and the presentation of applications and the assessment and the authorisation of feed additives(7). This shall include the submission of information on a core set of studies such as on metabolism/toxicokinetics, subchronic toxicity, genotoxicity, chronic toxicity, carcinogenicity and reproduction and developmental toxicity, accompanied by any other appropriate type of study. For specific guidelines for animal tests, see Table 1 of Section 1.7 of this Annex. Genotoxicity test protocols are provided for in Table 2 of Section 1.7 of this Annex.
1.4.3. Information on altered levels of food and feed constituents U.K.
This Section shall apply only in the case where the intended or unintended effect of the genetic modification would result in an alteration of the levels of food and feed constituents beyond the natural variation.
To demonstrate the safety of the altered levels of food and feed constituents such as macro- and micro-nutrients, anti-nutrients, and natural toxins as well as other secondary plant metabolites, the applicant shall submit a detailed risk assessment based on the knowledge of the physiological function and/or toxic properties of these constituents.
The result of that risk assessment shall determine if, and to what extent, the applicant shall provide additional toxicological tests to the 90-day feeding study in rodents with whole genetically modified food/feed on selected food and feed constituents.
1.4.4. Testing of the whole genetically modified food and feed U.K.
The applicant shall primarily base its risk assessment of the genetically modified food and feed on molecular characterisation, comparative agronomic, phenotypic and comprehensive compositional analysis, and the toxicological evaluation of the identified intended and unintended effects, including a 90-day feeding study in rodents with whole genetically modified food/feed as specified in Section 1.4.4.1. Under the circumstances set out in points 1.4.4.2 and 1.4.4.3 of this Section, additional specific toxicological studies with the whole genetically modified food and feed shall be carried out.
1.4.4.1.90-day feeding study in rodents with whole genetically modified food/feedU.K.
The applicant shall include a 90-day feeding study with whole food and feed in rodents for the assessment of food and feed containing, consisting of or produced from genetically modified plants with a single transformation event or with stacked transformation events which are not obtained by conventional crossing of genetically modified plants containing a single transformation event.
In the case of stacked transformation events obtained by conventional crossing of genetically modified plants containing one or several transformation event(s), a 90-day feeding study with whole food and feed in rodents shall be included for each of the genetically modified plant with a single transformation event which was used. An additional 90-day feeding study with whole food and feed in rodents with the genetically modified plant with the stacked transformation events shall be included where indications of potential adverse effects are identified during the assessment of: (i) the stability of the inserts; (ii) the expression of the inserts; and (iii) the potential synergistic or antagonistic effects resulting from the combination of the transformation events.
The design of the toxicity study with genetically modified food and feed should be performed according to the ‘subchronic oral toxicity test repeated-dose 90-day oral toxicity study in rodents’ (see Table 1) following an adapted protocol. In principle a minimum of two test doses and a negative control shall be used. The highest dose shall be the maximum achievable without causing nutritional imbalance; the lowest dose shall contain the tested food and/or feed in an amount always above the anticipated human/target animal intake level. The genetically modified food and feed analysed should be relevant to the product to be consumed. In the case of herbicide tolerant genetically modified plants, the tested material should come from the genetically modified plant exposed to the intended herbicide. Whenever possible, information on natural variation of test parameters shall be derived from historical background data rather than from the inclusion of reference varieties, consisting of commercially available food and feed derived from non-GM plants with a history of safe use, in the experiments. The statistical analysis shall focus on the detection of possible differences between the test material and its control. A power analysis to estimate a sample size capable of detecting a pre-specified biologically relevant effect size with a specified power and significance level should be used. More detailed guidance for performing this study is provided in the EFSA Guidance on conducting repeated-dose 90-day oral toxicity study in rodents on whole food/feed(8).
1.4.4.2.Animal studies with respect to reproductive and developmental toxicity testingU.K.
When information required in Sections 1.4.1, 1.4.2 and 1.4.3 on the genetically modified food and feed suggest the potential for reproductive, developmental or chronic toxicity or in case of indications of adverse effects from the 90-day feeding study in rodents (such as functional and/or histological modifications of nervous, endocrine, reproductive or immunological tissues/organs), appropriate testing shall be performed. Protocols for reproductive, developmental and chronic toxicity testing (see Table 1 of Section 1.7) may be adapted for the purposes of testing the whole genetically modified food and feed.
Given that the 90-day feeding study in rodents is only designed to detect effects on adult reproductive organ weights and histopathology and that it does not detect other effects on reproduction or development, testing of the whole food and feed beyond a 90-day rodent feeding study shall be conducted where hazards in this respect have been identified.
1.4.4.3.Other animal studies to examine the safety and the characteristics of genetically modified food and feed (see also Sections 1.6.1 and 1.6.2)U.K.
Feeding studies with target animal species shall be submitted when indications of adverse effects are drawn from information required in Sections 1.4.1, 1.4.2 and 1.4.3 on the genetically modified food and feed or from the outcome of the 90-day feeding study in rodents. Their focus shall be on the safety of new constituents (newly expressed proteins and other new constituents), on the identification and characterisation of unintended effects, and on the nutritional impact of any intentional, substantial, compositional modifications of the genetically modified plant (see also Section 1.6).
Studies of that type shall be limited to plant materials suitable for inclusion in their diets and which can be nutritionally matched to a suitable control diet.
1.4.4.4.Interpretation of relevance of animal studiesU.K.
Relevant effects observed in the animal trials shall be evaluated in order to identify potential consequences for the health of humans and animals and to assess their relevance for the safety of the genetically modified food and feed. This evaluation may be supported by additional information and considerations. Attention should be paid to the fact that certain effects may be specific for the test animal, but not for humans due to interspecies differences.
The applicant shall, in particular, consider dose-response relationships in parameters that have been changed (that is to say, commensurate increases in changes at increased doses) since they provide a strong indication for an effect of the tested compound. When a difference is noted only at the highest dose applied, other factors shall be considered to determine whether there is a relationship with treatment. Information on the background variability in a given parameter may be obtained by the applicant from data from other animals of the same species/strain tested in the same or other experiments, or from internationally harmonised databases.
In tests where animals of both genders are used, changes occurring in animals of one gender only may still be relevant indicators of an effect, depending on the parameter being changed and the mechanism by which the change may have been caused. For example, animals of one gender may be more or even specifically prone to changes caused by a given constituent than animals of the other gender, such as in the case of endocrine effects.
The applicant shall also identify possible inter-relationships between observed changes in single parameters which may strengthen the indication that an effect has occurred. For example, liver damage, which may be observed in the liver itself as a change in histopathology, gross pathology, and organ weights, may also be evident from the changed levels of certain liver-derived compounds, such as enzymes or bilirubin in serum.
With regard to the potential cause for an observed effect, the likelihood of causality shall be taken into account, not only for the test compound, but also for other factors that may have also influenced the outcomes (such as body weight decrease due to reduced intake of less palatable diet). Supportive data for a hypothesis of causality between the test compound and effects in test animals may include, for example, predictive data for plausible effects from in vitro and in silico experiments and dose-response relationships observed in the animal test.
1.4.5. Conclusion of the toxicological assessment U.K.
The conclusion of the toxicological assessment shall indicate whether:
(a)
potential adverse effects identified in other parts of the safety assessment have been confirmed or discarded;
(b)
the available information on the newly expressed protein(s) and other new constituents resulting from the genetic modification gives indications of potential adverse effects in particular, whether and at which dose levels adverse effects were identified in specific studies;
(c)
the information on natural constituents of which the levels are different from those in its conventional counterpart provides indications of potential adverse effects, in particular, whether and at which dose levels adverse effects were identified in specific studies;
(d)
adverse effects have been identified from the studies made on the whole genetically modified food and feed and at which dose levels.
The applicant shall evaluate the result of the toxicological assessment in the light of anticipated intake of the genetically modified food and feed (see Section 2).
1.5. Allergenicity U.K.
Food allergy is an adverse reaction to food and represents an important public health problem. Food allergy is different from toxic reactions and intolerance. Allergy is a pathological deviation of the immune response to a particular substance, which affects only some individuals where a combined effect of variations in the environment and genetic predisposition has resulted in allergic sensitisation.
In allergic individuals, sometimes minute amounts of a food that is well tolerated by the vast majority of the population can cause serious symptoms and death. It is not the allergen per se, but the allergic person’s abnormal reaction to the allergen that causes the adverse health effect.
Food allergy can be caused by various immune mechanisms. However, IgE-mediated food allergy represents the main form of food allergy, that causes the most severe reactions and the only form causing life-threatening reactions. This IgE-mediated food allergy has been the focus in the risk assessment of allergenicity of GMOs. Importantly, food allergy consists of two separate phases: first sensitisation where no symptoms occur while the capacity of the immune system to react increases dramatically, and later elicitation (provocation) with clinical manifestations.
When ingested, the allergen(s), that is to say, the sensitising food or food constituent is to some extent degraded by digestive enzymes, absorbed by the gut mucosa (small amounts even by the oral mucosa), processed in specialised cells of the immune system and then presented to the reactive immune cells that produce an immune response. Sensitisation can also occur if the food allergen comes into contact with the skin or is inhaled.
The majority of the constituents that are responsible for allergenicity of foods, as well as of pollens, are proteins. Some protein breakdown products, that is to say, peptide fragments, may conserve part of the allergenicity of the native protein and thus can also be considered as allergens.
The specific allergy risk of GMOs is associated with: (i) exposure to newly expressed protein(s) that can be present in edible parts of the plants or in the pollen; this point is related to the biological source of the transgene; and (ii) alterations to the allergenicity of the whole plant and its products, for example, due to over-expression of natural endogenous allergens as an unintended effect of the genetic modification; this point is related to the biology of the recipient plant itself.
More detailed guidance for the application of the requirements of this Section is available in the EFSA scientific opinion on the assessment of allergenicity of GM plants and micro-organisms and derived food and feed adopted on 30 June 2010(9).
1.5.1. Assessment of allergenicity of the newly expressed protein U.K.
Allergenicity is not an intrinsic, fully predictable property of a given protein but is a biological activity requiring an interaction with individuals with a pre-disposed genetic background. Allergenicity therefore depends upon the genetic diversity and variability in atopic humans. Frequency, severity and specificity of allergic reactions also depend upon geographic and environmental factors. Given this lack of complete predictability, it is necessary to consider several aspects in the assessment of allergenicity to obtain a cumulative body of evidence which minimises any uncertainty with regard to the protein(s) in question.
When studying the structural characteristics and the biological and physicochemical properties of a newly expressed protein, it is essential that the tested protein is equivalent with respect to structure and activity to the newly expressed protein in the genetically modified plant. Studies carried out using purified target proteins prepared by expression in organisms such as Escherichia coli shall be acceptable provided that the properties of the microbial substitute protein are identical to those of the protein expressed in the plant, thus taking into account all post-translational modifications that specifically occur in the plant.
The applicant shall verify whether the source of the transgene is allergenic. When the introduced genetic material is obtained from wheat, rye, barley, oats or related cereal grains, the applicant shall also assess the newly expressed proteins for a possible role in the elicitation of gluten-sensitive enteropathy or other enteropathies which are not IgE-mediated. Where transformation events have been stacked, the applicant shall provide an assessment of any potential for increased allergenicity to humans and animals on a case-by-case approach. These potential effects may arise from additive, synergistic or antagonistic effects of the gene products.
The applicant shall follow an integrated, case-by-case approach, that is to say, a weight-of-evidence approach, in the assessment of possible allergenicity of newly expressed proteins. This approach shall include:
(a)
Amino acid sequence homology comparison between the newly expressed protein and known allergens
In every case, a search for sequence homologies and/or structural similarities between the expressed protein and known allergens shall be performed to identify potential IgE cross-reactivity between the newly expressed protein and known allergens. The applicant shall ensure that the quality and the comprehensiveness of the databases are state of the art. The alignment-based criterion involving 35 % sequence identity to a known allergen over a window of at least 80 amino acids is considered a minimal requirement. All sequence alignment parameters used in the analysis shall be provided including calculation of percent identity (PID). The calculation of PID shall be performed on a window of 80 amino acids with gaps so that inserted gaps are treated as mismatches. In some cases, for assessing short peptidic fragments such as ORFs, a search for sequences of contiguous identical or chemically similar amino acid residue can be conducted. However, this search shall not be carried out routinely for the identification of potential linear IgE binding epitopes because of its poor sensitivity or specificity.
(b)
Specific serum screening
When there is an indication of sequence homology or structure similarities, an important procedure for assessing the potential that exposure to the newly expressed proteins might elicit an allergic reaction in individuals already sensitised to cross-reactive proteins, is based on in vitro tests that measure the capacity of specific IgE from serum of allergic patients to bind the test protein(s). There is inter-individual variability in the specificity and affinity of the human IgE response. In particular, the specificity of the IgE antibodies to the different allergens present in a given food/source and/or to the different epitopes present on a given protein may vary amongst allergic individuals. In order to optimise the sensitivity of the test, individual sera from well-characterised allergic individuals shall be used. The applicant shall perform specific serum screening in the following cases:
(i)
the source of the introduced gene is considered allergenic, even if no sequence homology of the newly expressed protein to a known allergen is demonstrated; or
(ii)
the source is not known to be allergenic, but there are indications of a relationship between the newly expressed protein and a known allergen, based on sequence homology or structure similarity.
Specific serum screening shall be undertaken with individual sera from individuals with a proven and well-characterised allergy to the source or to the potentially cross-reacting allergen using relevant immunochemical tests. IgE-binding assays (such as Radio or Enzyme Allergosorbent Assay (RAST or EAST), Enzyme Linked Immunosorbent Assay (ELISA) and electrophoresis followed by immunoblotting with specific IgE-containing sera) are adequate methods.
(c)
Pepsin resistance and in vitro digestibility tests
Stability to digestion by proteolytic enzymes has long been considered a characteristic of allergenic proteins. Although it has been established that no absolute correlation exists, resistance of proteins to pepsin digestion is an additional criterion to be considered in the weight-of-evidence approach for the assessment of allergenicity. The pepsin resistance test is generally performed under quite standardised conditions, at low pH values and high pepsin:protein ratios. It is recognised that the pepsin resistance test does not reflect the physiological conditions of the digestion. The digestibility of the newly expressed proteins in specific segments of the population, such as infants and individuals with impaired digestive functions, may be assessed using in vitro digestibility tests using different conditions. Also, since the protein encoded by the newly introduced genes will be present in the product as a complex matrix, the impact of the possible interaction between the protein and other components of the matrix, as well as the effects of the processing, shall be taken into account in additional in vitro digestibility tests. Depending on the outcome of the in vitro digestibility test, a comparison of the intact, the heat-denatured and the pepsin-digested proteins for IgE binding shall be assessed since an altered digestibility may impact on the allergenicity of the newly expressed protein.
(d)
Additional tests
Although additional tests including in vitro cell based assays or in vivo tests on animal models have not been validated to date for regulatory purposes, they may provide useful additional information, for example, on the potential of the newly expressed protein for de novo sensitisation.
1.5.2. Assessment of allergenicity of the genetically modified food or feed U.K.
When the recipient plant is known to be allergenic, the applicant shall assess any potential change in the allergenicity of the genetically modified food or feed by comparison of the allergen repertoire with that of its conventional counterpart. The potential over-expression of natural endogenous allergen(s) in the genetically modified plant shall, in particular, be investigated.
The applicant shall follow a case-by-case approach depending on the available information on the allergenic potential of the recipient plant. It is generally performed by analytical methodologies such as proteomics in association with the use of allergic human sera as probes. Sera from clinically well-characterised allergic individuals that are the reference material for IgE binding studies may be available in limited number and quantity. In order to minimise the use of human sera, preliminary important information on the likelihood of an unintended alteration of the overall allergenicity of the genetically modified plant can be obtained by using sera of animals experimentally sensitised in well-defined conditions and by including relevant identified endogenous allergens in the comparative compositional analysis.
In addition, the applicant shall provide, where available, information on the prevalence of allergy in persons working with, coming into contact with or in the vicinity of genetically modified plant cultivation.
1.5.3. Adjuvanticity U.K.
Adjuvants are substances that, when co-administered with an antigen, increase the immune response to the antigen and therefore might increase as well the allergic response. In cases when known functional aspects of the newly expressed protein or structural similarity to known strong adjuvants may indicate possible adjuvant activity, the applicant shall assess the possible role of these proteins as adjuvants. As for allergens, interactions with other constituents of the food matrix and/or processing may alter the structure and bioavailability of an adjuvant and thus modify its biological activity.
1.5.4. Conclusion of the allergenicity assessment U.K.
The conclusion of the allergenicity assessment shall indicate:
(a)
whether the novel protein(s) is likely to be allergenic;
(b)
whether the genetically modified food or feed is likely to be more allergenic than its conventional counterpart.
When there is a likelihood of increased allergenicity due to the genetic modification, the genetically modified food or feed shall be further characterised in the light of its anticipated intake (see Section 2). The applicant shall propose appropriate conditions for placing on the market (such as post-market monitoring and labelling).
1.6. Nutritional assessment U.K.
1.6.1. Objectives of the nutritional assessment U.K.
The applicant shall provide a nutritional evaluation to demonstrate that:
(a)
the introduction of the genetically modified food and feed into the market is not nutritionally disadvantageous to humans and animals, respectively. This evaluation shall include the relevance for the nutrition of newly expressed proteins, other new constituents, and changes in the levels of food and feed constituents, as well as potential alterations in the total diet of the consumer or the animal;
(b)
unintended effects of the genetic modification that were identified or that may be assumed to have occurred based on the preceding molecular, compositional or phenotypic analyses, in accordance with Sections 1.2 and 1.3, have not adversely affected the nutritional value of the genetically modified food and feed.
For stacked transformation events combined by conventional crossing, an assessment of the potential changes in nutritional value that might arise from synergistic or antagonistic effects of the gene products including compositional changes shall be provided by the applicant. This may be particularly relevant where the combined expression of the newly introduced genes has unexpected effects on biochemical pathways.
1.6.2. Points to consider for the nutritional assessment of genetically modified food and feed U.K.
The nutritional assessment of genetically modified food and feed shall consider:
(a)
the composition of the genetically modified food and feed with regard to the levels of nutrients and anti-nutrients (see compositional studies as described in Section 1.3);
(b)
the bioavailability and biological efficacy of nutrients in the food and feed taking into account the potential influences of transport, storage and expected treatment of the food and feed;
(c)
the anticipated dietary intake of the food and feed (see Section 2) and resulting nutritional impact.
When the comparative analysis has identified compositional characteristics of the genetically modified food and feed that are different from its conventional counterpart and/or not equivalent to the characteristics of the reference varieties, their nutritional relevance shall be assessed on the basis of current scientific knowledge. If that assessment does conclude to the nutritional equivalence between the genetically modified food and feed and its conventional counterpart, no further studies shall be carried out. By contrast if, on the basis of the assessment of the information obtained from the comparative analysis, it is not possible to conclude on the nutritional equivalence, further nutritional studies shall be carried out. Comparative growth studies shall be conducted with young rapidly growing animal species (such as broiler chicks as animal model for non-ruminants; lambs for ruminants; or other rapidly growing species).
1.6.3. Nutritional studies of genetically modified food U.K.
The applicant shall determine the necessity and design of nutritional studies on the basis of the introduced trait(s), the outcome of the comparative analysis, and of the 90-day feeding study, where available. Supplementary information regarding the nutritional value may be obtained from comparative growth performance studies conducted with other animal species, such as broiler chickens, addressing the nutritional assessment of genetically modified feed. When nutritional studies are conducted, the control diet(s) shall include the conventional counterpart and where appropriate additional comparator(s). In the case of herbicide tolerant genetically modified plants, the tested material should come from the genetically modified plant exposed to the intended herbicide.
Genetically modified foods modified to provide additional health benefits to the consumer as compared to conventional foods, may benefit specific populations or subpopulations while others may be at risk from the same food. In cases where an altered bioavailability needs to be established and may raise concern for subpopulation(s), the level of the nutrient in the food shall be determined, taking into account all the different forms of the compound. The methods to test for bioavailability shall be selected on a case-by-case basis depending on the nutrient or other constituent, the food containing these constituents, as well as the health, nutritional status and dietary practices of the specific population(s) anticipated to consume the food.
1.6.4. Nutritional studies of genetically modified feed U.K.
The applicant shall determine the necessity and design of further nutritional studies on the basis of the introduced trait(s), the outcome of the comparative analysis, and the 90-day feeding study, where available. Supplementary information regarding the nutritional value may be obtained from comparative growth performance studies conducted with other animal species, such as broiler chickens, addressing the nutritional assessment of genetically modified feed. When nutritional studies are conducted, the control diet(s) shall include the conventional counterpart and where appropriate additional comparator(s).
In the case of genetically modified feed with improved nutritional characteristics, feeding studies with target animal of food producing species shall be conducted to assess the impact on the feed. In the case of genetically modified plants modified for improved content and bioavailability of nutrients, studies with target food producing animal species shall be conducted to determine the bioavailability of individual nutrients in the genetically modified plant compared to its conventional counterpart. In the case of genetically modified plants specifically modified with traits to enhance animal performance through increased nutrient density (such as increased oil content) or an enhanced level of a specific nutrient (such as an essential amino acid or a vitamin), an appropriate control diet using its conventional counterpart shall be formulated by supplementing it with the specific nutrient to the extent of the change effected in the genetically modified plant. Regarding co-products (such as oilseeds meals), from which the ingredient targeted by the genetic modification has been extracted, these may be compared with co-products produced from the conventional counterpart.
Target animal feeding studies shall span either the growing and/or finishing period to slaughter for chickens, pigs, and cattle for fattening or a major part of a lactation cycle for dairy cows, or laying cycle for laying hens or quails. For feedstuffs intended only for aquaculture, growth studies with aquatic species such as carp, catfish, salmonidae or typical herbivores shall be chosen.
When appropriate, tests with various experimental designs shall be provided to demonstrate that the nutritionally improved genetically modified plant fulfils the expected nutritional value. The exact experimental design and statistical approaches of feeding experiments in food producing animals to test the nutritional value of genetically modified feed, modified for enhanced nutritional characteristics, shall depend on the targeted animal species, type of plant trait(s) studied and the size of the expected effect. The experimental diets shall be formulated in such a way that the key measured endpoints are responsive to a difference in the quantity and/or availability of the nutrient in question. Endpoint measurements shall vary with the target species used in the study, but shall include feed intake, body weight, animal performance and bioavailability of nutrients.
More detailed guidance for the application of the requirements of this Section is available in the Report of the EFSA GMO Panel Working Group on Animal Feeding Trials(10).
1.6.5. Conclusion of the nutritional assessment U.K.
The conclusion of the nutritional assessment of genetically modified food and feed shall indicate whether the genetically modified food and feed is nutritionally equivalent to its conventional counterpart, taking natural variations into account.
The applicant shall evaluate the result of the nutritional assessment in the light of anticipated intake of the genetically modified food and feed (see Section 2).
1.7. Standardised guidelines for toxicity tests U.K.
The applicant shall use for toxicity testing internationally agreed guidelines and test methods described by Commission Regulation (EC) No 440/2008 of 30 May 2008 laying down test methods pursuant to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH)(11) (see Tables 1 and 2). A non-exhaustive list of validated test methods which, where necessary, shall be used in a possibly adapted form for GMO toxicological testing is provided in Tables 1 and 2.
The performance of test methods depends on the type of genetically modified food and feed, type of the genetic modification and resulting intended and unintended alterations, intended use and exposure/intake, and the available knowledge. Some of the tests were developed for the assessment of risks at the workplace (see Sections 1.4 and 1.5).
Table 1
Non-exhaustive list of validated test methods for chemicals as provided by Regulation (EC) No 440/2008 and which may be used in a possibly adapted form for GMO toxicological testing
| Title | Reference of the method in Part B of the Annex to Regulation (EC) No 440/2008 |
|---|---|
| ACUTE TOXICITY (DERMAL) | B.3. |
| SKIN SENSITISATION | B.6. |
| REPEATED-DOSE (28 DAYS) TOXICITY (ORAL) | B.7. |
| REPEATED-DOSE (28 DAYS) TOXICITY (DERMAL) | B.9. |
| SUBCHRONIC ORAL TOXICITY TEST REPEATED-DOSE 90-DAY ORAL TOXICITY STUDY IN RODENTS | B.26. |
| CHRONIC TOXICITY TEST | B.30. |
| CARCINOGENICITY TEST | B32. |
| COMBINED CHRONIC TOXICITY/CARCINOGENICITY TEST | B.33. |
| ONE-GENERATION REPRODUCTION TOXICITY TEST | B.34. |
| TWO-GENERATION REPRODUCTION TOXICITY STUDY | B.35. |
| TOXICOKINETICS | B.36. |
| NEUROTOXICITY STUDY IN RODENTS | B.43. |
Table 2
Genotoxicity tests as provided by Regulation (EC) No 440/2008
| Title | Reference of the method in Part B of the Annex to Regulation (EC) No 440/2008 |
|---|---|
| MUTAGENICITY — IN VIVO MAMMALIAN BONE MARROW CHROMOSOME ABERRATION TEST | B.11. |
| MUTAGENICITY — IN VIVO MAMMALIAN ERYTHROCYTE MICRONUCLEUS TEST | B.12. |
| MUTAGENICITY: REVERSE MUTATION TEST USING BACTERIA | B.13/14. |
| MUTAGENICITY TESTING AND SCREENING FOR CARCINOGENICITY GENE MUTATION — SACCHAROMYCES CEREVISIAE | B.15. |
| MITOTIC RECOMBINATION — SACCHAROMYCES CEREVISIAE | B.16. |
| DNA DAMAGE AND REPAIR — UNSCHEDULED DNA SYNTHESIS — MAMMALIAN CELLS IN VITRO | B.18. |
| MUTAGENICITY — IN VITRO MAMMALIAN CELL GENE MUTATION TEST | B.17. |
| SISTER CHROMATID EXCHANGE ASSAY IN VITRO | B.19. |
| IN VITRO MAMMALIAN CELL TRANSFORMATION TESTS | B.21. |
| MAMMALIAN SPERMATOGONIAL CHROMOSOME ABERRATION TEST | B.23. |
2.EXPOSURE ASSESSMENT — ANTICIPATED INTAKE/EXTENT OF USEU.K.
An estimate of the expected intake shall be an essential element in the risk assessment of genetically modified food and feed and shall also be required for the nutritional evaluation. Information shall be provided by the applicant on the intended function, the dietary role, and the expected level of use of the genetically modified food and feed in the EU. In addition, the expected range of concentrations of newly produced proteins or existing plant proteins deliberately modified in the genetically modified food(s) and feed(s) to be placed on the market shall be provided.
On the basis of representative consumption data for products obtained from the respective conventional plants, the applicant shall estimate the anticipated average and maximum intake of the genetically modified food and feed. Probabilistic methods may be used to determine ranges of plausible values rather than single values or point estimates. The applicant shall identify and consider particular groups of the EU population with an expected higher exposure and shall consider this higher exposure within the risk assessment. Any assumptions made in the exposure assessment shall be described. Recent developments in methodologies and appropriate consumption data shall be used. Data on import and production quantities may provide additional information for the intake assessment.
The applicant shall determine by appropriate methods the concentrations of the newly expressed proteins, other new constituents and endogenous food and feed constituents, of which the levels have been altered as a result of the genetic modification (for example, due to changes in metabolic pathways) in those parts of the genetically modified plant intended for food or feed use. Expected intake of these constituents shall be estimated taking into account the influences of processing, storage and expected treatment of the food and feed in question, for example, potential accumulation or reduction. In cases where the genetic modification has resulted in an altered level of a natural constituent, or if a new constituent occurs naturally in other food and feed products, the anticipated change in total intake of this constituent shall be assessed considering realistic as well as worst case intake scenarios.
The applicant shall provide information on known or anticipated human/animal intake of analogous genetically modified food and feed and on other routes of exposure to the respective new and natural constituents, including amount, frequency and other factors influencing exposure.
3.RISK CHARACTERISATIONU.K.
3.1. Introduction U.K.
The applicant shall base its risk characterisation of genetically modified plants and foods and feed on data from hazard identification, hazard characterisation, and on exposure/intake data. The applicant shall ensure that the risk characterisation is comprehensive by considering all the available evidence from several analysis including molecular analysis, phenotypic, agronomical and compositional analysis, toxicity and allergenicity testing. The applicant shall consider indications resulting from the risk characterisation that may require specific activities for post-market monitoring of genetically modified food and feed.
In performing its risk characterisation, the applicant shall demonstrate that the hazard identification and hazard characterisation are complete. The applicant shall discuss the quality of existing data and information. The discussion shall clearly indicate how this body of information has been taken into account in the determination of the final risk characterisation.
The applicant shall provide estimations of the uncertainties associated to each test as well as to the different stages of the risk assessment. The applicant shall quantify them to the possible extent. A distinction shall be made between uncertainties that reflect natural variations in biological parameters (including variations in susceptibility in populations), and variation amongst different species’ responses.
Depending on the issue to be addressed and the available data, the applicant shall perform a qualitative and, where possible, quantitative risk characterisation. The conditions for the estimated risk, and associated uncertainties, shall be as precise as possible.
3.2. Issues to be considered for risk characterisation U.K.
When appropriate and depending on the type of genetic modification, the applicant shall carry out a risk assessment of genetically modified plants in an integrative manner in accordance with Section 3.1. This risk assessment shall be performed on a case-by-case basis depending on the modified plant and the type of genetic modification, the cultivation practices of the genetically modified plant and uses of the genetically modified food and feed. The applicant shall take into account the different issues considered in hazard identification and hazard characterisation and exposure steps. The outcomes of these issues shall be considered by the applicant together in the risk characterisation step. The list of issues provided in this Section shall not be exhaustive.
3.2.1. Molecular characterisation U.K.
Evaluation of the characteristics and previous use of the donor and the recipient plant shall be a key element to identify the need for specific analyses, such as the occurrence of specific toxins, or allergens in the unmodified recipient plant which may be unintentionally increased as result of the genetic modification.
Transformation protocols, molecular characterisation strategies and the specificity and sensitivity of the methods used shall be discussed by the applicant in relation to the intentional and possibly unintentional insertion and expression of gene sequences.
Where sequence analysis has identified a potential hazard, the applicant shall demonstrate how approaches like bioinformatic analysis, compositional/agronomical analysis and possibly animal feeding trials with the whole genetically modified food and feed contribute to the safety assessment. The value of the results obtained shall be evaluated in the light of the available knowledge on the structure and function of genomic databases of the crop species in question or related species.
In cases of GM plants containing stacked transformation events, the additional risks possibly arising from the combined effects of the stacked genes shall be evaluated.
3.2.2. Comparative analysis U.K.
The first goal of the comparative analysis is to identify possible differences between the GM plant and its conventional counterpart and, where appropriate, additional comparators. The second goal of the comparative analysis is to identify possible lack of equivalences between the GM plant and its reference varieties. These differences and/or lack of equivalences should be assessed with respect to their possible impact on food and feed safety and nutritional properties, taking into account natural variation. The estimated risk and associated uncertainties should be as precise as possible and taken into account.
The applicant shall demonstrate that the comparative analysis between the genetically modified plant and its conventional counterpart with respect to agronomic, morphological and compositional characteristics has been carried out in accordance with the requirements of this Regulation. The selection of the conventional counterpart and, where appropriate, additional comparators shall be justified.
3.2.3. Food and feed safety in relation to intake U.K.
The applicant shall evaluate the data generated to estimate possible short-term and long-term risks to human or animal health associated with the consumption of genetically modified food or feed with respect to the expression of new proteins/metabolites, as well as significantly altered levels of original plant proteins/metabolites in genetically modified foods/feed. This evaluation shall include a thorough analysis of the relevance and limitations of each test as well as of the whole information.
The applicant shall consider the range of levels observed for the compounds known to occur in the conventional counterpart and in reference varieties. This variability may be caused by differences that are genotype-dependent, environmentally dependent, or caused by genotype x environment interactions. In addition, the range of levels observed in a broad spectrum of food and feed representative for the human and animal diet may be taken into account given that it reflects the levels of the specific compound to which consumers may be exposed.
If single constituents and/or whole genetically modified food and feed are found to induce adverse effects in specific studies, information on dose response relationships, threshold levels, delayed onset of adverse effects, risks for certain groups in the population, and use of uncertainty factors in extrapolation of animal data to humans shall be submitted.
The applicant shall consider data on the characteristics of the new compounds present in the genetically modified plant including potential biological effects in humans and animals. If the compounds have known adverse health effects and maximum levels for the presence of these compounds in the plant or its products were laid down in specific legislation, these maximum levels shall be taken into account. Otherwise, reference values for acceptable or tolerable levels of intake, such as the acceptable daily intake (ADI) or tolerable upper intake level (UL), shall be considered in relation to the anticipated intake. In cases where the compound has been safely consumed in food, the intake levels of consumers from a conventional diet shall be considered as safe.
The applicant shall evaluate the information on the effects of processing on the new compounds. Potential accumulation/depletion in food and feed products entering the human or animal diet shall be considered. The applicant shall also evaluate the relevance of differences resulting from chemical reactions known to occur under processing conditions.
In cases where more complex genetic modifications are produced, for example, via transfer of multiple genes in a single construct, retransformation of pre-existing genetically modified lines, and stacking of transformation events through conventional breeding of genetically modified parents, the applicant shall discuss strategies for the assessment of any risk(s) associated with possible interactions between the newly expressed proteins, new metabolites and original plant constituents. The assessment shall take into account all available information including the mode of action of the newly expressed proteins, the molecular and compositional/agronomical characteristics of the genetically modified plant, and the outcome of animal toxicity studies and feeding trials.
The applicant shall evaluate data obtained to assess the allergenic potential of newly expressed proteins in genetically modified plants with respect to introduction of new allergenic proteins into the food and feed plants, a possible provocation of allergic reactions of susceptible individuals, as well as information to demonstrate that the genetic modification does not cause unintended changes in the characteristics and/or levels of expression of endogenous allergenic proteins in the genetically modified food. In particular, the choice of the test models shall be justified with respect to specificity, predictability and validation status.
With respect to intake estimations of genetically modified foods, the applicant shall evaluate the applied methodologies with respect to uncertainties associated with the prediction of long-term intake. Specific attention shall be paid to those genetically modified plants which are aimed at modifying nutritional characteristics of the food and feed. For those genetically modified products, the requirement for post-market monitoring shall be discussed as a mechanism for determining actual changes to overall dietary intake patterns of the genetically modified food, to what extent this has occurred and whether or not the product induces known (side) effects or unexpected side effects. If the performance of post-market monitoring is deemed necessary, the reliability, sensitivity and specificity of the proposed methods shall be provided.
3.3. The result of risk characterisation U.K.
In accordance with the requirements of Articles 4 and 16 of Regulation (EC) No 1829/2003, the applicant shall ensure that the final risk characterisation clearly demonstrates that:
(a)
the genetically modified food and feed has no adverse effects on human and animal health;
(b)
the genetically modified food does not differ from the food which it is intended to replace to such an extent that its normal consumption would be nutritionally disadvantageous for the consumer;
(c)
the genetically modified food does not mislead the consumer;
(d)
the genetically modified feed does not harm or mislead the consumer by impairing the distinctive features of the animal products;
(e)
the genetically modified feed does not differ from the feed which it is intended to replace to such an extent that its normal consumption would be nutritionally disadvantageous for animals or humans.
The applicant shall clearly indicate what assumptions have been made during the risk assessment in order to predict the probability of occurrence and severity of adverse effect(s) in a given population, and the nature and magnitude of uncertainties associated with establishing these risks.
The applicant shall also include detailed information justifying the inclusion or not of a proposal for labelling in the application, in accordance with Article 13(2)(a), Article 13(3), Article 25(2)(c) and Article 25(3) of Regulation (EC) No 1829/2003.
(2)
EFSA Journal 2011; 9(5):2149.
(3)
EFSA Journal 2010; 8(1):1250.
(4)
EFSA Journal 2010; 8(1):1250.
(5)
http://www.oecd.org/document/15/0,3746,en_2649_34385_46726799_1_1_1_1,00.html
(6)
http://www.efsa.europa.eu/en/efsajournal/pub/2760.htm
(8)
EFSA Journal 2011; 9(12):2438.
(9)
EFSA Journal 2010; 8(7):1700.
(10)
EFSA, 2008 Report of the EFSA GMO Panel Working Group on Animal Feeding Trials, 2008. Safety and nutritional assessment of GM plants and derived food and feed. The role of animal feeding trials. Food and Chemical Toxicology 46 (2008) S2-S70.
Options/Help
Print Options
PrintThe Whole Regulation
PrintThis Annex only
You have chosen to open the Whole Regulation
The Whole Regulation you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
The Schedules you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As adopted by EU): The original version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
See additional information alongside the content
Geographical Extent: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Show Timeline of Changes: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Opening Options
Different options to open legislation in order to view more content on screen at once
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the EU Official Journal
- lists of changes made by and/or affecting this legislation item
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
Timeline of Changes
This timeline shows the different versions taken from EUR-Lex before exit day and during the implementation period as well as any subsequent versions created after the implementation period as a result of changes made by UK legislation.
The dates for the EU versions are taken from the document dates on EUR-Lex and may not always coincide with when the changes came into force for the document.
For any versions created after the implementation period as a result of changes made by UK legislation the date will coincide with the earliest date on which the change (e.g an insertion, a repeal or a substitution) that was applied came into force. For further information see our guide to revised legislation on Understanding Legislation.
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.