Regulation 6
SCHEDULE 2METHODS OF ANALYSIS
PART IGENERAL PROVISIONS
Introduction
1.—(a) In general a single method of analysis applies for the determination of the presence or quantity of a substance in feeding stuffs. Where two or more methods are prescribed the choice between them shall, except where otherwise indicated, be left to the agricultural analyst concerned; the method used must however be indicated in the certificate of analysis.
(b)the result given in the analysis report shall be the average value obtained from at least two independent determinations, carried out on separate portions of the sample, and of satisfactory repeatability.
(c)The result shall be expressed, in the manner laid down in the method of analysis, to an appropriate number of significant figures and shall be corrected, if necessary, to the moisture content of the final sample prior to preparation (see paragraph 3(d) below).
Reagents and Apparatus
2.—(a) Unless otherwise specified in the method of analysis concerned, all reagents must be analytically pure. The purity of the reagents, especially when determining trace elements, must be checked by a blank test. Depending upon the results obtained, further purification of the reagents may be required.
(b)Where any operation involves preparation of solutions, dilution, rinsing or washing, as part of a method of analysis, water must be used unless the specification of the method indicates otherwise.
(c)Water should, in the absence of good reason to the contrary, be demineralized or distilled. Where indicated in the method of analysis concerned it must be subjected to special purification procedures.
(d)All instruments or apparatus used must be clean, especially when very small amounts of substances have to be determined.
Preparation of the sample for analysis
3.—(a) Samples must be prepared in such a way that the amounts weighed out, as provided for in the methods of analysis, are homogeneous and representative of the final sample.
(b)All the necessary operations must be performed in such a way as to avoid, as far as possible, any change in, or contamination of, the sample. Grinding, mixing and sieving should be carried out as quickly as possible, with minimal exposure of the sample to air and light. Overgrinding is to be avoided. Mills and grinders likely to heat the sample appreciably should not be used. Nevertheless, where some loss or gain of moisture is unavoidable, allowance should be made for such changes (see sub-paragraph (d) below). Manual grinding is recommended for feeding stuffs which are particularly sensitive to heat. Care should also be taken to ensure that the apparatus itself is not a source of contamination by trace elements.
(c)If the final sample as received consists of unopened packages or containers then, immediately prior to the preparation of the sample for analysis, all the contents shall be thoroughly mixed together.
(d)If the sample is appreciably moist, or if for any reason the preparation cannot be carried out without significant changes in the moisture content of the sample, determine the moisture content before and after preparation, using the method specified in columns 2 and 3 of Annex I to Part II of this Schedule, appearing opposite to the reference to “Moisture” in column 1 of that Annex.
(e)When a microscopial examination for the presence of undesirable substances is required then, in the absence of good reason to the contrary–
(i)the sample should be crushed and ground only to such an extent as facilitates the examination, and
(ii)grinding to pass 1 mm should not be used where it could lead to difficulties in identifying the undesirable substances listed in Schedule 5 of the Feeding Stuffs Regulations 1995(1).
(f) Procedure
Mix the sample thoroughly either mechanically or manually. Divide the sample into two equal portions (the quartering method should be used where applicable). Preliminary crushing and/or grinding may be necessary, if the sample is in a coarse condition, to facilitate division. Keep one of the portions in a suitable container, i.e. non-corrodible, clean and dry and fitted with an air-tight stopper, and prepare the other portion or a representative part of it, of at least 100 g, as indicated below.
(i)Feeding stuffs which can be ground as such
Unless otherwise specified in the method of analysis concerned, sieve the whole sample through a sieve having apertures of 1 mm square(2)(3), in accordance with recommendation ISO R565, after grinding, if necessary.
Mix the sieved sample and collect it in a suitable container, i.e. non-corrodible, clean and dry and fitted with an air-tight stopper. Mix again, immediately before weighing out the amounts for analysis.
(ii)Feeding stuffs which can be ground only after drying
Unless otherwise specified in the method of analysis concerned, dry the sample to reduce its moisture content to a level of 8–12%, in accordance with the preliminary drying procedure specified in point 4.3 of the method referred to in sub-paragraph (d) above, until grinding enables the sample to be passed wholly through a sieve having apertures of 1 mm square(2)(3). Then proceed as indicated in sub-paragraph (f)(i) above.
(iii)Liquid or semi-liquid feeding stuffs
Collect the sample in a suitable container, i.e. non-corrodible, clean and dry and fitted with an air-tight stopper. Mix thoroughly immediately before weighing out the amount for analysis.
(iv)Other feeding stuffs
A sample which cannot be prepared according to any of the above procedures should be treated by any other procedure which ensures that the amounts weighted out for analysis are homogeneous and representative of the final sample.
(g) Storage of samples
Samples must be stored at such a temperature as will cause no compositional changes. A sample intended for the analysis of vitamins, or substances which are particularly sensitive to light, should be placed in a brown glass container.
PART IIMETHODS OF ANALYSIS
The methods of analysis for the purposes of this Schedule are–
(a)Community methods of analysis, as specified in Annex I to this Part of this Schedule;
(b)the method for determining uric acid, as specified in Annex II to this Part of this Schedule; and
(c)the method for determining isobutylidenediurea, as specified in Annex III to this Part of this Schedule.
ANNEX ICOMMUNITY METHODS OF ANALYSIS
| Column 1 | Column 2 | Column 3 |
|---|---|---|
| Substance | Community Provision | Official Journal Reference |
(1) Where the one-dimensional thin layer chromatographic method is the appropriate one. | ||
(2) Where the high performance liquid chromatographic method is the appropriate one. | ||
(3) Where the pancreatic method is the appropriate one. | ||
(4) Where the polarimctric method is the appropriate one. | ||
| Aflatoxin B1 | Parts A and C of the Annex to Directive 76/372/EEC (Part A was replaced in part by paragraph I of the Annex to Directive 92/95/EEC. Part C was replaced entirely by the Annex to Directive 94/14/EC)(1)(1) | OJ No. L102, 15.4.76, p. 8. OJ No. L327, 13.11.92, p. 54. OJ No. L94, 13.4.94, p.30. |
| Aflatoxin B1 | Parts B and C of the Annex to Directive 76/372/EEC (Part B was replaced entirely by paragraph II of the Annex to Directive 92/95/EEC. Part C was replaced entirely by the Annex to Directive 94/14/EC)(2)(2) | OJ No. L102, 15.4.76, p. 8. OJ No. L327, 13.11.92, p. 54. OJ No. L94, 13.4.94, p. 30. |
| Amino Acids | Part A of the Annex to Directive 98/64/EC | OJ No. L257, 19.98.98, p. 14. |
| Ammonia and volatile nitrogenous bases | Part II of the Annex to Directive 71/393/EEC | OJ No. L279, 20.12.71, p. 7 (OJ/SE 1971(III), p. 987). |
| Ash | Point 5 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Ash insoluble in hydrochloric acid | Point 6 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Calcium | Point 3 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Carbonates | Point 4 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Fibre | Point 3 of Annex I to Directive 73/46/EEC (as replaced entirely by the Annex to Directive 92/89/EEC) | OJ No. L83, 30.3.73, p. 21. OJ No. L344, 26.11.92, p. 35. |
| Free and total gossypol | Point 5 of Annex I to Directive 72/199/EEC | OJ No. L123, 29.5.72, p. 6 (OJ/SE 1966–72 supplement, p. 74). |
| Hydrocyanic acid | Point 2 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Iron, copper, manganese and zinc | Point 3 of the Annex to Directive 78/633/EEC | OJ No. L206, 29.7.78, p. 43. |
| Lactose | Point 9 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Magnesium | Point 2 of Annex I to Directive 73/46/EEC | OJ No. L83, 30.3.73, p. 21. |
| Menadione (vitamin K3) | Point 5 of Annex II to Directive 74/203/EEC | OJ No. L108, 22.4.74, p. 7. |
| Moisture | Part I of the Annex to Directive 71/393/EEC (as amended by Article 1 of Directive 73/47/EEC) | OJ No. L279, 20.12.71, p. 7 (OJ/SE 1971(III), p. 987). OJ No. L83, 30.3.73, p. 35. |
| Moisture in fats and oils | Point 1 of Annex I to Directive 73/46/EEC | OJ No. L83, 30.3.73, p. 21. |
| Oils and fats | Part IV of the Annex to Directive 71/393/EEC. (Part IV was replaced entirely by Annex I to Directive 84/4/EEC. That Annex was in turn replaced entirely by Part B of the Annex to Directive 98/64/EC) | OJ No. L279, 20.12.71, p. 7 (OJ/SE 1971(III), p. 987). OJ No. L15, 18.1.84, p. 28. OJ No. L257, 19.9.98, p. 14. |
| Pepsin activity | Point 4 of Annex I to Directive 72/199/EEC | OJ No. L123, 29.5.72, p. 6 (OJ/SE 1966–1972 supplement, p. 74). |
| Phosphorus | Part III of the Annex to Directive 71/393/EEC | OJ No. L279, 20.12.71, p. 7 (OJ/SE 1971(III), p. 987). |
| Potassium | Point 10 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Protein | Point 2 of Annex I to Directive 72/199/EEC (as replaced entirely by the Annex to Directive 93/28/EEC) | OJ No. L123, 29.5.72, p. 6 (OJ/SE 1966–1972 supplement, p. 74). OJ No. L179, 22.7.93, p. 8. |
| Proteins soluble in pepsin and hydrochloric acid | Point 3 of Annex I to Directive 72/199/EEC | OJ No. L123, 29.5.72, p. 6 (OJ/SE 1966–1972 supplement, p. 74). |
| Sodium | Point 11 of the Annex to Directive 71/250/EEC (as corrected by a corrigendum published in July 1975) | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). Consolidated edition of corrigenda to the first series of special editions of EC legislation (1952 to 1972). |
| Sugar | Point 12 of the Annex to Directive 71/250/EEC (as corrected by a corrigendum published in July 1975) | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). Consolidated edition of corrigenda to the first series of special editions of EC legislation (1952 to 1972). |
| Starch | Annex I to Directive 74/203/EEC(3)(3) | OJ No. L108, 22.4.74, p. 7. |
| Starch | Point 1 of Annex I to Directive 72/199/EEC (as corrected by a corrigendum published on 27 November 1980)(4)(4) | OJ No. L123, 29.5.72, p. 6 (OJ/SE 1966–1972 supplement, p. 74). OJ No. L320, 27.11.80, p. 43. |
| Theobromine | Point 13 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Urea | Point 14 of the Annex to Directive 71/250/EEC (as corrected by a corrigendum published in July 1975) | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). Consolidated edition of corrigenda to the first series of special editions of EC legislation (1952 to 1972). |
| Urease activity | Point 16 of the Annex of Directive 71/250/EEC (as corrected by a corrigendum published in July 1975) | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). Consolidated edition of corrigenda to the first series of special editions of EC legislation (1952 to 1972). |
| Vitamin A | Point 1 of Annex II to Directive 73/46/EEC | OJ No. L83, 30.3.73, p. 21. |
| Volatile mustard oil | Point 8 of the Annex to Directive 71/250/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
| Water soluble chlorides | Point 7 of the Annex to Directive 71/25/EEC | OJ No. L155, 12.7.71, p. 13 (OJ/SE 1971(II), p. 480). |
ANNEX IIMETHOD FOR DETERMINING URIC ACID
Scope and Field of Application
1. This method is for the determination of uric acid and its salts in dried poultry waste and in feeding stuffs containing dried poultry waste.
Principle
2. Uric acid is extracted with neutral ethanotic formaldehyde solution, precipitated as silver magnesium urate, redissolved in sodium thiosulphate solution and determined spectrophotometrically.
Reagents
3.—3.1 Sodium hydroxide solution: dissolve 50 g sodium hydroxide in 50 ml water, mix well and store in a suitable plastic container.
3.2 Formaldehyde solution: the strength of the commercially available solution should be checked as follows: mix 3 ml formaldehyde solution with 50 ml IN sodium hydroxide solution and 25 ml hydrogen peroxide solution (20 volumes). Heat on a steam bath until effervescence stops. Cool, and titrate with IN hydrochloric acid using phenolphthalein indicator. Carry out a blank titration using 3 ml water in place of the formaldehyde.
1 ml of 1N sodium hydroxide ≡ 0.0300 g formaldehyde
where B = blank titre; and
T = sample titre.

3.3 Neutral ethanolic formaldehyde solution: mix an appropriate volume of formaldehyde solution (3.2) containing 17.5 g of formaldehyde with 250 ml water and 500 ml ethanol. Adjust the pH of the solution to 7.0 with 0.1N sodium bhydroxide solution. Dilute to 1,000 ml with water, mix and gain adjust the pH to 7.0 if necessary.
3.4 Scucinate buffer solution: dissolve by heating, 29.5 g of succinic acid in 750 ml water and 20 ml sodium hydroxide solution (3.1). Cool, add an appropriate volume of formaldehyde solution (3.2) containing 17.5 g of formaldehyde, mix well and adjust the pH to 6.0 with sodium hydorxide solution (3.1). Dilute to 1,000 ml with water, mix and gain adjust the pH to 6.0 if necessary.
3.5 sodium thiosulphate solution: 25 g sodium thiosulphate (Na2S2O3.5H2O) per 1,000 ml.
3.6 Silver lactate solution: dissolve, by heating, 3 g silver lactate in 50 ml water and 1 ml lactic acid. Dilute to 100 ml with water, filter, and store in dark glassware. Do not expose to strong light.
3.7 Ammoniacal magnesium solution: dissolve 8.75 g magnesium sulphate (MgSO4.7H2O) and 17.5 g ammonium chloride in 50 ml water. Add 30 ml ammonia solution (d = 0.88 g/ml) mix well and dilute to 100 ml with water.
3.8 Benedict and Hitchcock reagent: mix 35 ml silver lactate solution (3.6) with 15 ml ammoniacal magnesium solution (3.7). Add 50 ml ammonia solution (d = 0.88 g /ml). Mix well, Prepare immediately before use.
3.9 Standard uric acid solution: weigh to the nearest 0.1 mg, 250 mg of uric acid and transfer to a 150 ml round-bottomed flask fitted with a reflux condenser. Add 100 ml ethanolic formaldehyde solution (3.3) and boil under reflux on a steam bath for 30 minutes, shaking frequently. Cool, transfer the solution to a 250 ml graduated flask, wash the round-bottomed flask with ethanolic formaldehyde solution (3.3) and combine the washings with the uric acid solution. Dilute to the mark with ethanolic formaldehyde solution (3.3) and mix. 1 ml contains 1 mg of uric acid.
3.10 Light petroleum, boiling range 40–60°C.
Apparatus
4.—4.1 Spectrophotometer, with 10 mm silica cells.
4.2 Percolation tubes, glass. Upper part: approximately 240 mm long, 18 mm internal diameter, lower part: approximately 120 mm long, 8 mm internal diameter.
Procedure
Extraction of Uric Acid
5.1.1 From dried poultry waste:
5.1.1Weigh to the nearest 0.001 g, about 0.4 g dried poultry waste and place in a 150 ml round-bottomed flask. Add 60 ml ethanolic formaldehyde solution (3.3), fit a reflux condenser onto the flask and heat on a steam bath for 1 hour. Cool and filter by suction through a sintered glass crucible (porosity 4) into a 100 ml graduated flask. Wash out the round-bottomed flask with 3 × 10 ml portions of ethanolic formaldehyde solution (3.3) passing each portion through the crucible into the graduated flask. Dilute to 100 ml with ethanolic formaldehyde solution and mix.
5.1.2 From feeding stuffs:
5.1.2Weigh to the nearest 0.001 g, between 4 g and 5 g of prepared sample. Transfer to a glass percolation tube (4.2) fitted with a small paper cup to retain the feed. Remove the fat from the feed by extraction with light petroleum (3.10). Transfer quantitatively the defatted sample to a 150 ml round-bottomed flask and remove the residual solvent with a slow current of air. Continue as in 5.1.1, second sentence “… Add 60 ml ethanolic formaldehyde solution (3.3) …”.
Determination
5.2 Transfer by pipette 20 ml of the sample extract prepared as in 5.1.1 or 5.1.2 to a 50 ml centrifuge tube. Add 10 ml of Benedict and Hitchcock reagent 93.8), mix well and allow to stand in the dark for 1 hour. Centrifuge at 2,000 rpm for 15 minutes, pour off the supernatant liquid and allow to drain for 10 minutes. Carefully wipe off any remaining liquid without disturbing the precipitate, and add 20 ml sodium thiosulphate solution (3.5) to each tube. Dissolve the precipitate by stirring with a thin glass rod. Transfer by pipette 5 ml of this solution into a 200 mg graduated flask containing 40 ml succinate buffer solution (3.4). Dilute to 200 ml with water and mix well. Measure the absorbance of the solution at 294 mm in 10 mm silica cells against a solution prepared by mixing 5 ml sodium thiosulphate solution (3.5) with 40 ml succinate buffer solution (3.4) and diluting to 200 ml with water. Determine the quantity of uric acid present by reference to the calibration curve (5.3).
Calibration Curve
5.3 Into a series of 50 ml centrifuge tubes, transfer by pipette 2, 4, 6, 8, 10 and 12 ml standard uric acid solution (3.9) (corresponding to 2, 4, 6, 8, 10 and 12 mg of uric acid) and make up to 20 ml with ethanolic formaldehyde solution (3.3). Add to each tube 10 ml Benedict and Hitchcock reagent (3.8), mix well and stand in the dark for 1 hour. Continue as in 5.2 from “… Centrifuge at 2,000 rpm. …”, Measure the absorbances of the solutions and plot the calibration curve using absorbances as the ordinates and the corresponding quantities of uric acid, in mg (as shown above) as the abseissae.
Expression of the Results
6. The uric acid nitrogen content per cent of the sample is given by the formula
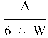
where:
=
mg uric acid (in the aliquot volume of the sample extract) as determined by photometric measurement; and
=
weight of sample in grams.
ANNEX IIIMETHOD FOR DETERMINING ISOBUTYLIDENEDIUREA
Scope and Field of Application
1. This method is for the determination of isobutylidenediurea in feeding staffs.
Principle
2. The sample is hydrolysed, liberating isobutyraldehyde, the concentration of which is determined by gas chromatography.
Reagents
3.—3.1 Toluene.
3.2 Sodium sulphate, anhydrous.
3.3 Buffer solution pH1: dissolve 27.2 g sodium acetate trihydrate in 300 ml IM hydrochloric acid and add 700 ml water.
3.4 Buffer solution pH 0.65: dissolve 27.2 g sodium acetate trihydrate in 400 ml IM hydrochloric acid and add 600 ml water.
3.5 Isobutylidenediurea.
3.6 internal standard solution: dilute 5 ml isopropyl acetate to 100 ml with toluene (3.1).
apparatus
4.—4.1 250 ml conical flasks with ground glass or PTFE stoppers.
4.2 Stoppered centrifuge tubes.
4.3 Gas chromatograph with flame ionisation detector.
4.4 Column:
either (i) 1.5 m glass column (4 mm internal diameter) packed with 5% QV17 on Gas Chrom Q, 80–100 mesh,
or (ii) 1.5 m glass column (4 mm internal diameter) packed with 5% Carbowax 20M-TPA on Diatomite C-AAW, 80–100 mesh.
4.5 Water bath: hotplate stirrer on which is placed a 2,000 ml beaker (or suitable vessel) containing water maintained at 40–50°C.
Procedure
Hydrolysis
5.—5.1 Weigh to the nearest 0.001 g, between 3 and 7 g of the prepared sample containing about 0.2 g of isobutylidenediurea into a concical flask (4.1). Add 100 ml buffer solution (3.4) and 20.0 ml toluene (3.1) to the sample and place in the flask a magnetic bar. Stopper firmly to ensure that the flask remains tightly closed during the hydrolysis.
Place the flask in the water bath (4.5) and stir vigorously for 20 minutes. Remove the flask and immerse in an ice-water bath for 5 minutes. Add 15 g sodium sulphate (3.2) and 5.0 ml internal standard solution (3.6) to the contents of the flask. Stopper the flask again, shake, return to the water bath (4.5) and warm for 3 minutes with stirring. Cool in the ice-water bath for 5 minutes. Transfer slowly between 15 and 25 ml of the mixture to the centrifuge tube (4.2), stopper, and centrifuge for 5 minutes to separate the layers. (Repeat the transfer if insufficient toluene is decanted). Transfer a portion of the upper (toluene) layer to a test tube with a pasteur pipette.
Determination
5.2 Inject between 0.5 and 1.0 μl of the toluene solution (5.1) into the gas chromatograph (4.3).
Suggested conditions:
Column 70iC Nitrogen 40 ml per minute Injection 150iC Hydrogen 30 ml per minute Detector 150iC Air 370 ml per minute Approximate retention times:
Isobutyraldehyde 1 min. Internal standard 1.5 min. Toluene 3 min. Measure the peak heights of the isobutyraldehyde and internal standard. Calculate the peak height ratio, isobutyraldehyde/internal standard, and from this value determine the quantity of isobutylidenediurea present by reference to the calibration curve (5.3).
Calibration curve
5.3 Weigh to the nearest mg, 100, 200 and 300 mg isobutylidenediurea (3.5) into three conical flasks (4.1). Add 100 ml buffer solution (3.3), 20.0 ml toluene (3.1) and a magnetic bar to each. Stopper the flasks firmly. Continue as in 5.1 from “… Place the flask in the water bath …”. Inject the toluene solutions into the gas chromatograph (4.3), and measure the peak heights. Calculate the peak height ratios, isobutyraldehyde/internal standard, and plot the calibration curve using peak height ratios as the ordinates and the corresponding weights or isobutylidenediurea as the abseissae.
Expression of the Results
6. The per cent content of isobutylidenediurea in the sample is given by the formula:

where:
=
weight of isobutyldenediurea (mg) read from the calibration curve; and
=
weight of sample in grams.