- Y Diweddaraf sydd Ar Gael (Diwygiedig)
- Gwreiddiol (Fel y’i mabwysiadwyd gan yr UE)
Regulation (EC) No 2003/2003 of the European Parliament and of the CouncilDangos y teitl llawn
Regulation (EC) No 2003/2003 of the European Parliament and of the Council of 13 October 2003 relating to fertilisers (Text with EEA relevance)
You are here:
- Rheoliadau yn deillio o’r UE
- 2003 No. 2003
- attachment 4

Pa Fersiwn
Nodweddion Uwch
- Dangos Graddfa Ddaearyddol(e.e. Lloegr, Cymru, Yr Alban aca Gogledd Iwerddon)
- Dangos Llinell Amser Newidiadau
Rhagor o Adnoddau
PDF o Fersiynau Diwygiedig
- ddiwygiedig 01/12/20201.61 MB
- ddiwygiedig 18/07/20191.65 MB
- ddiwygiedig 01/07/20171.58 MB
- ddiwygiedig 29/09/20161.58 MB
- ddiwygiedig 01/01/20161.50 MB
- ddiwygiedig 15/12/20141.51 MB
- ddiwygiedig 07/12/20141.40 MB
- ddiwygiedig 07/06/20141.55 MB
- ddiwygiedig 07/06/20131.30 MB
- ddiwygiedig 04/04/20131.38 MB
- ddiwygiedig 04/07/20121.91 MB
- ddiwygiedig 09/03/20112.04 MB
- ddiwygiedig 18/11/20090.71 MB
- ddiwygiedig 20/04/20091.20 MB
- ddiwygiedig 28/11/20081.20 MB
- ddiwygiedig 12/03/20071.18 MB
- ddiwygiedig 24/12/20041.17 MB
- ddiwygiedig 01/05/20041.17 MB
Deddfwriaeth yn deillio o’r UE
Pan adawodd y DU yr UE, cyhoeddodd legislation.gov.uk ddeddfwriaeth yr UE a gyhoeddwyd gan yr UE hyd at ddiwrnod cwblhau’r cyfnod gweithredu (31 Rhagfyr 2020 11.00 p.m.). Ar legislation.gov.uk, mae'r eitemau hyn o ddeddfwriaeth yn cael eu diweddaru'n gyson ag unrhyw ddiwygiadau a wnaed gan y DU ers hynny.


Mae'r eitem hon o ddeddfwriaeth yn tarddu o'r UE
Mae legislation.gov.uk yn cyhoeddi fersiwn y DU. Mae EUR-Lex yn cyhoeddi fersiwn yr UE. Mae Archif Gwe Ymadael â’r UE yn rhoi cipolwg ar fersiwn EUR-Lex o ddiwrnod cwblhau’r cyfnod gweithredu (31 Rhagfyr 2020 11.00 p.m.).
Changes over time for:
Alternative versions:
- 01/05/2004- Amendment
- 24/12/2004- Amendment
- 12/03/2007- Amendment
- 28/11/2008- Amendment
- 20/04/2009- Amendment
- 18/11/2009- Amendment
- 09/03/2011- Amendment
- 04/07/2012- Amendment
- 04/04/2013- Amendment
- 07/06/2013- Amendment
- 07/06/2014- Amendment
- 07/12/2014- Amendment
- 15/12/2014- Amendment
- 01/01/2016- Amendment
- 29/09/2016- Amendment
- 01/07/2017- Amendment
- 18/07/2019- Amendment
- Exit day: start of implementation period31/01/2020 11pm- Amendment
- 01/12/2020- Amendment
- End of implementation period31/12/2020- Amendment
- 11/01/2021- Amendment
- 27/02/2021- Amendment
Changes to legislation:
There are outstanding changes by UK legislation not yet made to Regulation (EC) No 2003/2003 of the European Parliament and of the Council. Any changes that have already been made to the legislation appear in the content and are referenced with annotations.
Changes to Legislation
Revised legislation carried on this site may not be fully up to date. Changes and effects are recorded by our editorial team in lists which can be found in the ‘Changes to Legislation’ area. Where those effects have yet to be applied to the text of the legislation by the editorial team they are also listed alongside the legislation in the affected provisions. Use the ‘more’ link to open the changes and effects relevant to the provision you are viewing.
Changes and effects yet to be applied to Attachment 4:
- Art. 2(1)(r) word substituted by S.I. 2025/82 Sch. 15 para. 67
Newidiadau ac effeithiau heb eu gweithredu eto ar yr eitem ddeddfwriaeth gyfan a’r darpariaethau cysylltiedig.
- Art. 2(1)(r) word substituted by S.I. 2025/82 Sch. 15 para. 67
ANNEX III
TECHNICAL PROVISIONS FOR AMMONIUM NITRATE FERTILISERS OF HIGH NITROGEN CONTENT
1.Characteristics of and limits for straight ammonium nitrate fertilisers of high nitrogen contentU.K.
1.1.Porosity (oil retention)U.K.
The oil retention of the fertiliser, which must first have undergone two thermal cycles of a temperature ranging from 25 to 50 °C and conforming with the provisions of part 2 of section 3. of this Annex, must not exceed 4 % by mass.
1.2.Combustible ingredientsU.K.
The percentage by mass of combustible material measured as carbon must not exceed 0,2 % for fertilisers having a nitrogen content of at least 31,5 % by mass and must not exceed 0,4 % for fertilisers having a nitrogen content of at least 28 % but less than 31,5 % by mass.
1.3.pHU.K.
A solution of 10 g of fertiliser in 100 ml of water must have a pH of at least 4.5.
1.4.Particle size analysisU.K.
Not more than 5 % by mass of the fertiliser must pass through a 1 mm mesh sieve and not more than 3 % by mass must pass through a 0,5 mm mesh sieve.
1.5.ChlorineU.K.
The maximum chlorine content is set at 0,02 % by mass.
1.6.Heavy metalsU.K.
Heavy metals should not be added deliberately, and any traces which are incidental to the production process should not exceed the limit fixed by [F1this Regulation].
Textual Amendments
F1Words in Annex 3 para. 1.6 substituted (31.12.2020) by The Fertilisers and Ammonium Nitrate Material (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/601), regs. 1(2), 5(22); 2020 c. 1, Sch. 5 para. 1(1)
The copper content shall not be higher than 10 mg/kg.
No limits are specified for other heavy metals.
2.Description of the test of resistance to detonation concerning ammonium nitrate fertilisers of high nitrogen contentU.K.
The test must be carried out on a representative sample of fertiliser. Before being tested for resistance to detonation, the whole mass of the sample is to be thermally cycled five times complying with the provisions of part 3 in section 3. of this Annex.
The fertiliser must be subjected to the test of resistance to detonation in a horizontal steel tube under the following conditions:
seamless steel tube,
Tube length: 1 000 mm at least,
Nominal external diameter: 114 mm at least,
Nominal wall thickness: 5 mm at least,
Booster: the type and mass of the booster chosen should be such as to maximise the detonation pressure applied to the sample in order to determine its susceptibility to the transmission of detonation,
Test temperature: 15-25 °C,
Witness lead cylinders for detecting detonation: 50 mm diameter and 100 mm high
placed at 150 mm intervals and supporting the tube horizontally. The test is to be carried out twice. The test is deemed conclusive if in both tests one or more of the supporting lead cylinders is crushed by less than 5 %.
3.Methods of checking compliance with the limits specified in Annexes III-1 and III-2U.K.
Method 1Methods for the application of thermal cyclesU.K.
1.Scope and field of applicationU.K.
This document defines the procedures for the application of thermal cycles prior to the execution of the oil retention test for straight ammonium nitrate fertilisers of high nitrogen content and of the test on the resistance to detonation for both, straight and compound ammonium nitrate fertiliser of high nitrogen content.
The methods of the closed thermal cycles as described in this section are regarded as simulating sufficiently the conditions to be taken into account within the scope of application of title II, chapter IV, however, these methods may not necessarily simulate all conditions arising during transport and storage;
2.Thermal cycles referred to in Annex III-1U.K.
2.1.Field of applicationU.K.
This procedure is for thermal cycling prior to determining the oil retention of the fertiliser.
2.2.Principle and definitionU.K.
In an Erlenmeyer flask, heat the sample from ambient temperature to 50 °C and maintain at this temperature for a period of two hours (phase at 50 °C). Thereupon cool the sample until a temperature of 25 °C is achieved and maintain at that temperature for two hours (phase at 25 °C). The combination of the successive phases at 50 °C and 25 °C forms one thermal cycle. After being subjected to two thermal cycles, the test sample is held at a temperature of 20 ± 3 °C for the determination of the oil retention value.
2.3.ApparatusU.K.
Normal laboratory apparatus, in particular:
water baths thermostated at 25 (± 1) and 50 (± 1) °C respectively,
Erlenmeyer flasks with an individual capacity of 150 ml.
2.4.ProcedureU.K.
Put each test sample of 70 (± 5) grams into an Erlenmeyer flask which is then sealed with a stopper.
Move each flask every two hours from the 50 °C bath to the 25 °C bath and vice versa.
Maintain the water in each bath at constant temperature and keep in motion by rapid stirring to ensure the water level comes above the level of the sample. Protect the stopper from condensation by a foam rubber cap.
3.Thermal cycles to be used for Annex III-2U.K.
3.1.Field of applicationU.K.
This procedure is for thermal cycling prior to the execution of the detonability test.
3.2.Principle and definitionU.K.
In a watertight box heat the sample from ambient temperature to 50 °C and maintain at this temperature for a period of one hour (phase at 50 °C). Thereupon cool the sample until a temperature of 25 °C is achieved and maintain at that temperature for one hour (phase at 25 °C). The combination of the successive phases at 50 °C and 25 °C forms one thermal cycle. After being subjected to the required number of thermal cycles, the test sample is held at a temperature of 20 ± 3 °C pending the execution of the detonability test.
3.3.ApparatusU.K.
A water bath, thermostated in a temperature range of 20 to 51 °C with a minimum heating and cooling rate of 10 °C/h, or two water baths, one thermostated at a temperature of 20 °C, the other at 51 °C. The water in the bath(s) is continuously stirred; the volume of the bath should be large enough to guarantee ample circulation of the water.
A stainless steel box, watertight all around and provided with a thermocouple in the centre. The outside width of the box is 45 (± 2) mm and the wall thickness is 1,5 mm (see Figure 1). The height and length of the box can be chosen to suit the dimensions of the water bath, e.g. length 600 mm, height 400 mm.
3.4.ProcedureU.K.
Place a quantity of fertilisers sufficient for a single detonation into the box and close the cover. Place the box in the water bath. Heat the water to 51 °C and measure the temperature in the centre of the fertiliser. One hour after the temperature at the centre has reached 50 °C cool the water. One hour after the temperature at the centre has reached 25 °C heat the water to start the second cycle. In the case of two water baths, transfer the box to the other bath after each heating/cooling period.
Figure 1

Method 2Determination of oil retentionU.K.
1.Scope and field of applicationU.K.
This document defines the procedure for the determination of oil retention of straight ammonium nitrate fertilisers of high nitrogen content.
The method is applicable to both prilled and granular fertilisers which do not contain oil-soluble materials.
2.DefinitionU.K.
Oil retention of a fertiliser: the quantity of oil retained by the fertiliser determined under the operating conditions specified, and expressed as a percentage by mass.
3.PrincipleU.K.
Total immersion of the test portion in gas oil for a specified period, followed by the draining away of surplus oil under specified conditions. Measurement of the increase in mass of the test portion.
4.ReagentU.K.
Gas oil
Viscosity max.
:
5 mPas at 40 °C
Density
:
0,8 to 0,85 g/ml at 20 °C
Sulphur content
:
≤ 1,0 % (m/m)
Ash
:
≤ 0,1 % (m/m)
5.ApparatusU.K.
Ordinary laboratory apparatus, and:
5.1.
Balance, capable of weighing to the nearest 0,01 gram.
5.2.
Beakers, of capacity 500 ml.
5.3.
Funnel, of plastic materials, preferably with a cylindrical wall at the upper end, diameter approximately 200 mm.
5.4.
Test sieve, aperture 0,5 mm, fitting into the funnel (5.3).
Note: The size of the funnel and sieve is such as to ensure that only a few granules lie one above another and the oil is able to drain easily.U.K.
5.5.
Filter paper, rapid filtering grade, creped, soft, mass 150 g/m2.
5.6.
Absorbent tissue (laboratory grade).
6.ProcedureU.K.
6.1.Two individual determinations are carried out in quick succession on separate portions of the same test sample.U.K.
[F26.2.Remove particles smaller than 0,5 mm using the test sieve (5.4). Weigh to the nearest 0,01 gram approximately 50 grams of the sample into the beaker (5.2). Add sufficient gas oil (section 4) to cover the prills or granules completely and stir carefully to ensure that the surfaces of all the prills or granules are fully wetted. Cover the beaker with a watch glass and leave to stand for one hour at 25 (± 2) °C.]U.K.
Textual Amendments
6.3.Filter the entire contents of the beaker through the funnel (5.3) containing the test sieve (5.4). Allow the portion retained by the sieve to remain there for one hour so that most of the excess oil can drain away.U.K.
6.4.Lay two sheets of filter paper (5.5) (about 500 × 500 mm) on top of each other on a smooth surface; fold the four edges of both filter papers upwards to a width of about 40 mm to prevent the prills from rolling away. Place two layers of absorbent tissue (5.6) in the centre of the filter papers. Pour the entire contents of the sieve (5.4) over the absorbent tissues and spread the prills evenly with a soft, flat brush. After two minutes lift one side of the tissues to transfer the prills to the filter papers beneath and spread them evenly over these with the brush. Lay another sheet of filter paper, similarly with its edges turned upward, on the sample and roll the prills between the filter papers with circular movements while exerting a little pressure. Pause after every eight circular movements to lift the opposite edges of the filter papers and return to the centre the prills that have rolled to the periphery. Keep to the following procedure: make four complete circular movements, first clockwise and then anticlockwise. Then roll the prills back to the centre as described above. This procedure to be carried out three times (24 circular movements, edges lifted twice). Carefully insert a new sheet of filter paper between the bottom sheet and the one above it and allow the prills to roll onto the new sheet by lifting the edges of the upper sheet. Cover the prills with a new sheet of filter paper and repeat the same procedure as described above. Immediately after rolling, pour the prills into a tared dish and reweigh to the nearest 0,01 gram to determine the mass of the quantity of gas oil retained.U.K.
6.5.Repeating the rolling procedure and reweighingU.K.
If the quantity of gas oil retained in the portion is found to be greater than 2 grams, place the portion on a fresh set of filter papers and repeat the rolling procedure, lifting the corners in accordance with section 6.4 (two times eight circular movements, lifting once). Then reweigh the portion.
7.Expression of the resultsU.K.
7.1.Method of calculation and formulaU.K.
The oil retention, from each determination (6.1) expressed as a percentage by mass of the sieved test portion, is given by the equation:

where:
m1 is the mass, in grams, of the sieved test portion (6.2),
m2 is the mass, in grams, of the test portion according to section 6.4 or 6.5 respectively as the result of the last weighing.
Take as the result the arithmetic mean of the two individual determinations.
Method 3Determination of the combustible ingredientsU.K.
1.Scope and field of applicationU.K.
This document defines the procedure for the determination of the combustible content of straight ammonium nitrate fertilisers of high nitrogen content.
2.PrincipleU.K.
The carbon dioxide produced by inorganic fillers is removed in advance with an acid. The organic compounds are oxidised by means of a chromic acid/sulphuric acid mixture. Carbon dioxide formed is absorbed in a barium hydroxide solution. The precipitate is dissolved in a solution of hydrochloric acid and measured by back-titration with sodium hydroxide solution.
3.ReagentsU.K.
3.1.Analytical-grade chromium (VI) trioxide Cr2O3;U.K.
3.2.Sulphuric acid, 60 % by volume: pour 360 ml of water into a one-litre beaker and carefully add 640 ml of sulphuric acid (density at 20 °C = 1.83 g/ml).U.K.
3.3.Silver nitrate: 0,1 mol/l solution.U.K.
3.4.Barium hydroxideU.K.
Weigh out 15 grams of barium hydroxide [Ba(OH)2. 8H2O], and dissolve completely in hot water. Allow to cool and transfer to a one-litre flask. Fill up to the mark and mix. Filter through a pleated filter paper.
3.5.Hydrochloric acid: 0,1 mol/l standard solution.U.K.
3.6.Sodium hydroxide: 0,1 mol/l standard solution.U.K.
3.7.Bromophenol blue: solution of 0,4 grams per litre in water.U.K.
3.8.Phenolphthalein: solution of 2 grams per litre in 60 % by volume ethanol.U.K.
3.9.Soda lime: particle dimensions, about 1,0 to 1,5 mm.U.K.
3.10.Demineralised water, freshly boiled to remove carbon dioxide.U.K.
4.ApparatusU.K.
4.1.Standard laboratory equipment, in particular:U.K.
filter crucible with a plate of sintered glass and a capacity of 15 ml; plate diameter: 20 mm; total height: 50 mm; porosity 4 (pore diameter from 5 to 15 μm),
600-ml beaker.
4.2.Compressed nitrogen supply.U.K.
4.3.Apparatus made up of the following parts and assembled, if possible, by means of spherical ground joints (see Figure 2).U.K.
4.3.1.
Absorption tube A about 200 mm long and 30 mm in diameter filled with soda lime (3.9) kept in place by fibreglass plugs.
4.3.2.
500-ml reaction flask B with side arm and a round bottom.
4.3.3.
Vigreux fractionating column about 150 mm long (C').
4.3.4.
Double-surface condenser C, 200 mm long.
4.3.5.
[F2Dreschel bottle D acting as a trap for any excess of acid which may distil over.]
4.3.6.
Ice bath E to cool the Drechsel bottle.
4.3.7.
Two absorption vessels F1 and F2, 32 to 35 mm in diameter, the gas distributor of which comprises a 10 mm disc of low-porosity sintered glass.
4.3.8.
Suction pump and suction regulating device G comprising a T-shaped glass piece inserted into the circuit, the free arm of which is connected to the fine capillary tube by a short rubber tube fitted with a screw clamp.
Caution: The use of boiling chromic acid solution in an apparatus under reduced pressure is a hazardous operation and requires appropriate precautions.U.K.
5.ProcedureU.K.
5.1.Sample for analysisU.K.
Weigh approximately 10 grams of ammonium nitrate to the nearest 0,001 grams.
5.2.Removal of carbonatesU.K.
[F2Place the sample for analysis in the reaction flask B. Add 100 ml of H 2 SO 4 (3.2). The prills or granules dissolve in about 10 minutes at ambient temperature. Assemble the apparatus as indicated in the diagram: connect one end of the absorption tube (A) to the nitrogen source (4.2) via a non-return flow device containing a pressure of 667 to 800 Pa and the other end to the feed tube which enters the reaction flask. Place the Vigreux fractionating column (C′) and the condenser (C) with cooling water supply in position. Adjust the nitrogen to provide a moderate flow through the solution, bring the solution to boiling point and heat for two minutes. At the end of this time there should be no more effervescence. If effervescence is seen, continue heating for 30 minutes. Allow solution to cool for at least 20 minutes with the nitrogen flowing through it.]
Complete assembly of the apparatus as indicated in the diagram by connecting the condenser tube to the Drechsel bottle (D) and the bottle to the absorption vessels F1 and F2. The nitrogen must continue to pass through the solution during the assembly operation. Rapidly introduce 50 ml of barium hydroxide solution (3.4) into each of the absorption vessels (F1 and F2).
Bubble a stream of nitrogen through for about 10 minutes. The solution must remain clear in the absorbers. If this does not happen, the carbonate removal process must be repeated.
5.3.Oxidation and absorptionU.K.
After withdrawing the nitrogen feed tube, rapidly introduce 20 grams of chromium trioxide (3.1) and 6 ml of silver nitrate solution (3.3) via the side arm of the reaction flask (B). Connect the apparatus to the suction pump and adjust the nitrogen flow so that a steady stream of gas bubbles passes through the sintered-glass absorbers F1 and F2.
Heat the reaction flask (B) until the liquid boils and keep it boiling for one and a half hours(1). It may be necessary to adjust the suction-regulating valve (G) to control the nitrogen flow since it is possible that the barium carbonate precipitated during the test may block the sintered-glass discs. The operation is satisfactory when the barium hydroxide solution in the absorber F2 remains clear. Otherwise repeat the test. Stop heating and dismantle the apparatus. Wash each of the distributors (3.10) both inside and outside to remove barium hydroxide and collect the washings in the corresponding absorber. Place the distributors one after the other in a 600-ml beaker which will subsequently be used for the determination.
Rapidly filter under vacuum firstly the contents of absorber F2 and then of absorber F1 using the sintered-glass crucible. Collect the precipitate by rinsing the absorbers with water (3.10) and wash the crucible with 50 ml of the same water. Place the crucible in the 600-ml beaker and add about 100 ml of boiled water (3.10). Introduce 50 ml of boiled water into each of the absorbers and pass nitrogen through the distributors for five minutes. Combine the water with that from the beaker. Repeat the operation once to ensure that the distributors are rinsed thoroughly.
5.4.Measurement of the carbonates originating from organic materialU.K.
Add five drops of phenolphthalein (3.8) to the contents of the beaker. The solution becomes red in colour. Add hydrochloric acid (3.5) drop by drop until the pink colour just disappears. Stir the solution well in the crucible to check that the pink colour does not reappear. Add five drops of bromphenol blue (3.7) and titrate with hydrochloric acid (3.5) until the solution turns yellow. Add a further 10 ml of hydrochloric acid.
Heat the solution to boiling point and continue boiling for a maximum of one minute. Check carefully that no precipitate remains in the liquid.
Allow to cool and back titrate with the sodium hydroxide solution (3.6).
6.Blank testU.K.
Carry out a blank test following the same procedure and using the same quantities of all reagents.
7.Expression of the resultsU.K.
The content of combustible ingredients (C), expressed as carbon, as a percentage by mass of the sample, is given by the formula:
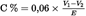
where:
E
=
the mass in grams of the test portion,
V1
=
the total volume in ml of 0,1 mol/l hydrochloric acid added after the change in colour of the phenolphthalein,
V2
=
the volume in ml of the 0,1 mol/l sodium hydroxide solution used for back titration.
Figure 2

Method 4Determination of the pH valueU.K.
1.Scope and field of applicationU.K.
This document defines the procedure for measuring the pH value of a solution of a straight ammonium nitrate fertiliser of high nitrogen content.
2.PrincipleU.K.
Measurement of the pH of an ammonium nitrate solution by means of a pH meter.
3.ReagentsU.K.
Distilled or demineralised water, free from carbon dioxide.
3.1.Buffer solution, pH 6,88 at 20 °CU.K.
Dissolve 3,40 ± 0,01 grams of potassium dihydrogen orthophosphate (KH2PO4) in approximately 400 ml of water. Then dissolve 3,55 ± 0,01 grams of disodium hydrogen orthophosphate (Na2HPO4) in approximately 400 ml of water. Transfer the two solutions without loss into a 1 000-ml graduated flask, make up to the mark and mix. Keep this solution in an airtight vessel.
3.2.Buffer solution, pH 4,00 at 20 °CU.K.
Dissolve 10,21 ± 0,01 grams of potassium hydrogen phthalate (KHC8O4H4) in water, transfer without loss into a 1 000-ml graduated flask, make up to the mark and mix.
Keep this solution in an airtight vessel.
3.3.Commercially available pH standard solutions may be used.U.K.
4.ApparatusU.K.
pH meter, equipped with glass and calomel electrodes or equivalent, sensitivity 0,05 pH unit.
5.ProcedureU.K.
5.1.Calibration of the pH meterU.K.
Calibrate the pH meter (4) at a temperature of 20 (± 1) °C, using the buffer solutions (3.1), (3.2) or (3.3). Pass a slow stream of nitrogen onto the surface of the solution and maintain this throughout the test.
5.2.DeterminationU.K.
Pour 100,0 ml of water onto 10 (± 0,01) grams of the sample in a 250 ml beaker. Remove the insolubles by filtering, decanting or centrifuging the liquid. Measure the pH value of the clear solution at a temperature of 20 (± 1) °C according to the same procedure as for the calibration of the meter.
6.Expression of the resultsU.K.
Express the result in pH units, to the nearest 0,1 unit, and state the temperature used.
Method 5Determination of the particle sizeU.K.
1.Scope and field of applicationU.K.
This document defines the procedure for the test sieving of straight ammonium nitrate fertilisers of high nitrogen content.
2.PrincipleU.K.
The test sample is sieved on a nest of three sieves, either by hand or by mechanical means. The mass retained on each sieve is recorded and the percentage of material passing the required sieves are calculated.
3.ApparatusU.K.
3.1.200-mm-diameter woven-wire test sieves with apertures of 2,0 mm, 1,0 mm and 0,5 mm respectively of standard ranges. One lid and one receiver for these sieves.U.K.
3.2.Balance to weigh to 0,1 gram.U.K.
3.3.Mechanical sieve shaker (if available) capable of imparting both vertical and horizontal motion to the test sample.U.K.
4.ProcedureU.K.
4.1.The sample is divided representatively into portions of approximately 100 grams.U.K.
4.2.Weigh one of these portions to the nearest 0,1 gram.U.K.
4.3.Arrange the nest of sieves in ascending order; receiver, 0,5 mm, 1 mm, 2 mm and place the weighed test portion on the top sieve. Fit the lid to the top of the nest of sieves.U.K.
4.4.Shake by hand or machine, imparting both a vertical and horizontal motion and if by hand, tapping occasionally. Continue this process for 10 minutes or until the quantity passing through each sieve in one minute is less than 0,1 gram.U.K.
4.5.Remove the sieves from the nest in turn and collect the material retained, brush gently from the reverse side with a soft brush, if necessary.U.K.
4.6.Weigh the material retained on each sieve and that collected in the receiver, to the nearest 0,1 gram.U.K.
5.Evaluation of the resultsU.K.
5.1.Convert the fraction masses to a percentage of the total of the fraction masses (not of the original charge).U.K.
Calculate the percentage in the receiver (i.e. < 0,5 mm): A %
Calculate the percentage retained on the 0,5 mm sieve: B %
Calculate the percentage passing 1,0 mm, i.e. (A + B) %
The sum of the fraction masses should be within 2 % of the initial mass taken.
5.2.At least two separate analyses should be carried out and the individual results for A should not differ by more than 1,0 % absolute and for B by more than 1,5 % absolute. Repeat the test if this is not the case.U.K.
6.Expression of the resultsU.K.
Report the mean of the two values obtained for A on the one hand and for A + B on the other.
Method 6Determination of the chlorine content (as chloride ion)U.K.
1.Scope and field of applicationU.K.
This document defines the procedure for the determination of the chlorine content (as chloride ion) of straight ammonium nitrate fertilisers with a high nitrogen content.
2.PrincipleU.K.
Chloride ions dissolved in water are determined by potentiometric titration with silver nitrate in an acidic medium.
3.ReagentsU.K.
Distilled or demineralised water, free from chloride ions.
3.1.Acetone AR.U.K.
3.2.Concentrated nitric acid (density at 20 °C = 1,40 g/ml)U.K.
3.3.Silver nitrate 0,1 mol/l standard solution. Store this solution in a brown glass bottle.U.K.
3.4.Silver nitrate 0,004 mol/l standard solution - prepare this solution at the time of use.U.K.
3.5.Potassium chloride 0,1 mol/l standard reference solution. Weigh, to the nearest 0,1 mg, 3,7276 grams of analytical-grade potassium chloride, previously dried for one hour in an oven at 130 °C and cooled in a desiccator to ambient temperature. Dissolve in a little water, transfer the solution without loss into a 500-ml standard flask, dilute to the mark and mix.U.K.
3.6.Potassium chloride, 0,004 mol/l standard reference solution — prepare this solution at the time of use.U.K.
4.ApparatusU.K.
4.1.Potentiometer with silver indicating electrode and calomel reference electrode, sensitivity 2 mV, covering the range - 500 to + 500 mV.U.K.
4.2.Bridge, containing a saturated potassium nitrate solution, connected to the calomel electrode (4.1), fitted at the ends with porous plugs.U.K.
4.3.Magnetic stirrer, with a Teflon-coated rod.U.K.
4.4.Microburette with fine-pointed tip, graduated in 0,01 ml divisions.U.K.
5.ProcedureU.K.
5.1.Standardisation of the silver nitrate solutionU.K.
Take 5,00 ml and 10,00 ml of the standard reference potassium chloride solution (3.6) and place in two low-form beakers of convenient capacity (for example 250 ml). Carry out the following titration of the contents of each beaker.
Add 5 ml of the nitric acid solution (3.2), 120 ml of the acetone (3.1) and sufficient water to bring the total volume to about 150 ml. Place the rod of the magnetic stirrer (4.3) in the beaker and set the stirrer in motion. Immerse the silver electrode (4.1) and the free end of the bridge (4.2) in the solution. Connect the electrodes to the potentiometer (4.1) and, after verifying the zero of the apparatus, note the value of the starting potential.
Titrate, using the microburette (4.4), adding initially 4 or 9 ml respectively of the silver nitrate solution corresponding to the standard reference potassium chloride solution used. Continue the addition in 0,1 ml portions for the 0,004 mol/l solutions and in 0,05 ml portions for the 0,1 mol/l solutions. After each addition, await the stabilisation of the potential.
Record the volumes added and the corresponding values of the potential in the first two columns of a table.
In a third column of the table, record the successive increments (Δ1E) of the potential E. In a fourth column, record the differences (Δ2E) positive or negative, between the potential increments (Δ1E). The end of the titration corresponds to the addition of the 0,1 or 0,05 ml portion (V1) of the silver nitrate solution which gives the maximum value of Δ1E.
In order to calculate the exact volume (Veq) of the silver nitrate solution corresponding to the end of the reaction, use the formula:

where:
V0 is the total volume, in ml, of the silver nitrate solution immediately lower than the volume which gives the maximum increment of Δ1E,
V1 is the volume, in ml, of the last portion of the silver nitrate solution added (0,1 or 0,05 ml),
b is the last positive value of Δ2E,
B is the sum of the absolute values of the last positive values of Δ2E and the first negative value of Δ2E (see example in Table 1).
5.2.Blank testU.K.
Carry out a blank test and take account thereof when calculating the final result.
The result V4 of the blank test on the reagents is given, in ml, by the formula:

where:
V2 is the value, in ml, of the exact volume (Veq) of the silver nitrate solution corresponding to the titration of 10 ml of the potassium chloride standard reference solution used,
V3 is the value, in ml, of the exact volume (Veq) of the silver nitrate solution corresponding to the titration of 5 ml of the potassium chloride standard reference solution used.
5.3.Check testU.K.
The blank test can at the same time serve as a check that the apparatus is functioning satisfactorily and that the test procedure is being implemented correctly.
5.4.DeterminationU.K.
Take a portion of sample in the range 10 to 20 grams and weigh to the nearest 0,01 gram. Transfer quantitatively to a 250-ml beaker. Add 20 ml of water, 5 ml of nitric acid solution (3.2), 120 ml of acetone (3.1) and sufficient water to bring the total volume to about 150 ml.
Place the rod of the magnetic stirrer (4.3) in the beaker, place the beaker on the stirrer and set the stirrer in motion. Immerse the silver electrode (4.1) and the free end of the bridge (4.2) in the solution, connect the electrodes to the potentiometer (4.1) and, after having verified the zero of the apparatus, note the value of the starting potential.
Titrate with the silver nitrate solution, by additions from the microburette (4.4) in increments of 0,1 ml. After each addition, await the stabilisation of the potential.
Continue the titration as specified in 5.1, starting from the fourth paragraph: ‘Record the volumes added and the corresponding values of the potential in the first two columns of a table …’.
6.Expression of the resultsU.K.
Express the result of the analysis as the percentage of chlorine contained in the sample as received for analysis. Calculate the percentage of chlorine (Cl) content from the formula:

where:
T is the concentration of silver nitrate solution used, in mol/l
V4 is the result, in ml, of the blank test (5.2),
V5 is the value, in ml, of Veq corresponding to the determination (5.4),
m is the mass, in grams, of the test portion.
Table 1: Example
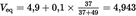
Method 7Determination of copperU.K.
1.Scope and field of applicationU.K.
This document defines the procedure for the determination of copper content of straight ammonium nitrate fertilisers of high nitrogen content.
2.PrincipleU.K.
The sample is dissolved in dilute hydrochloric acid and the copper is determined by atomic absorption spectrophotometry.
3.ReagentsU.K.
3.1.Hydrochloric acid (density at 20 °C = 1,18 g/ml).U.K.
3.2.Hydrochloric acid, 6 mol/l solution.U.K.
3.3.Hydrochloric acid 0,5 mol/l solution.U.K.
3.4.Ammonium nitrate.U.K.
3.5.Hydrogen peroxide, 30 % w/vU.K.
3.6.Copper solution(2) (stock): weigh, to the nearest 0,001 gram, 1 gram of pure copper, dissolve in 25 ml 6 mol/l hydrochloric acid solution (3.2), add 5 ml of hydrogen peroxide (3.5) in portions and dilute to 1 litre with water. 1 ml of this solution contains 1 000 μg of copper (Cu).U.K.
3.6.1.Copper solution (dilute): dilute 10 ml of stock solution (3.6) to 100 ml with water and then dilute 10 ml of the resulting solution, to 100 ml with water, 1 ml of the final dilution contains 10 μg of copper (Cu).U.K.
Prepare this solution at the time of use.
4.ApparatusU.K.
Atomic absorption spectrophotometer with a copper lamp (324,8 nm).
5.ProcedureU.K.
5.1.Preparation of the solution for analysisU.K.
Weigh, to the nearest 0,001 gram, 25 grams of the sample, place it in a 400-ml beaker, add carefully 20 ml of hydrochloric acid (3.1) (there may be a vigorous reaction due to carbon dioxide formation). Add more hydrochloric acid, if necessary. When effervescence has stopped, evaporate to dryness on a steam bath, stirring occasionally with a glass rod. Add 15 ml 6 mol/l hydrochloric acid solution (3.2) and 120 ml of water. Stir with the glass rod, which should be left in the beaker, and cover the beaker with a watch glass. Boil the solution gently until dissolution is complete and then cool.
Transfer the solution quantitatively into a 250-ml graduated flask, by washing the beaker with 5 ml 6 mol/l hydrochloric acid (3.2), and twice with 5 ml of boiling water, make up to the mark with 0,5 mol/l hydrochloric acid (3.3) and mix carefully.
Filter through a copper-free filter paper(3), discarding the first 50 ml.
5.2.Blank solutionU.K.
Prepare a blank solution from which only the sample has been omitted and allow for this in the calculation of the final results.
5.3.DeterminationU.K.
5.3.1.Preparation of sample and blank test solutionsU.K.
Dilute the sample solution (5.1) and the blank test solution (5.2) with 0,5 mol/l hydrochloric acid solution (3.3) to a concentration of copper within the optimal measuring range of the spectrophotometer. Normally no dilution is needed.
5.3.2.Preparation of the calibration solutionsU.K.
By diluting the standard solution (3.6.1) with 0,5 mol/l hydrochloric acid solution (3.3), prepare at least five standard solutions corresponding to the optimal measuring range of the spectrophotometer (0 to 5,0 mg/l Cu). Before making up to the mark, add to every solution ammonium nitrate (3.4) to give concentration of 100 mg per ml.
5.4.MeasurementU.K.
Set up the spectrophotometer (4) at a wavelength of 324,8 nm. Use an oxidising air-acetylene flame. Spray successively, in triplicate, the calibration solution (5.3.2), the sample solution and the blank solution (5.3.1), washing the instrument through with distilled water between each spraying. Plot the calibration curve using the mean absorbances of every standard used as the ordinates and the corresponding concentrations of copper in μg/ml as the abscissae.
Determine the concentration of copper in the final sample and blank solutions by reference to the calibration curve.
6.Expression of the resultsU.K.
Calculate the copper content of the sample taking into account the mass of the test sample, the dilutions carried out in the course of the analysis and the value of the blank. Express the result as mg Cu/kg.
4.Determination of resistance to detonationU.K.
4.1.Scope and field of applicationU.K.
This document defines the procedure for the determination or resistance to detonation of ammonium nitrate fertilisers of high nitrogen content.
4.2.PrincipleU.K.
The test sample is confined in a steel tube and subjected to detonation shock from an explosive booster charge. Propagation of the detonation is determined from the degree of crushing of lead cylinders on which the tube rests horizontally during the test.
4.3.MaterialsU.K.
4.3.1.Plastic explosive containing 83 to 86 % penthriteU.K.
Density
:
1 500 to 1 600 kg/m3
Detonation velocity
:
7 300 to 7 700 m/s
Mass
:
500 (± 1) gram.
4.3.2.Seven lengths of flexible detonating cord with non-metallic sleeveU.K.
Filling mass
:
11 to 13 g/m
Length of each cord
:
400 (± 2) mm.
4.3.3.Compressed pellet of secondary explosive, recessed to receive detonatorU.K.
Explosive
:
hexogen/wax 95/5 or tetryl or similar secondary explosive, with or without added graphite.
Density
:
1 500 to 1 600 kg/m3
Diameter
:
19 to 21 mm
Height
:
19 to 23 mm
Central recess to receive detonator
:
diameter 7 to 7,3 mm, depth 12 mm.
4.3.4.Seamless steel tube as specified in ISO 65 — 1981 — Heavy Series, with nominal dimensions DN 100 (4'')U.K.
Outside diameter
:
113,1 to 115,0 mm
Wall thickness
:
5,0 to 6,5 mm
Length
:
1 005 (± 2) mm.
4.3.5.Bottom placeU.K.
Material
:
steel of good weldable quality
Dimensions
:
160 × 160 mm
Thickness
:
5 to 6 mm
4.3.6.Six lead cylindersU.K.
Diameter
:
50 (± 1) mm
Height
:
100 to 101 mm
Materials
:
soft lead, at least 99,5 % purity.
4.3.7.Steel blockU.K.
Length
:
at least 1 000 mm
Width
:
at least 150 mm
Height
:
at least 150 mm
Mass
:
at least 300 kg if there is no firm base for the steel block.
4.3.8.Plastic or cardboard cylinder for booster chargeU.K.
Wall thickness
:
1,5 to 2,5 mm
Diameter
:
92 to 96 mm
Height
:
64 to 67 mm
4.3.9.Detonator (electric or non-electric) with initiation force 8 to 10U.K.
4.3.10.Wooden discU.K.
Diameter
:
92 to 96 mm. Diameter to be matched to the internal diameter of the plastic or cardboard cylinder (4.3.8)
Thickness
:
20 mm
4.3.11.Wooden rod of same dimensions as detonator (4.3.9)U.K.
4.3.12.Dressmaking pins (maximum length 20 mm)U.K.
4.4.ProcedureU.K.
4.4.1.Preparation of booster charge for insertion into steel tubeU.K.
There are two methods of initiation of the explosive in the booster charge, depending on the availability of equipment.
4.4.1.1.Seven-point simultaneous initiationU.K.
The booster charge prepared for use is shown in Figure 1.
4.4.1.1.1.Drill holes in the wooden disc (4.3.10) parallel to the axis of the disc through the centre and through six points symmetrically distributed around a concentric circle 55 mm in diameter. The diameter of the holes must be 6 to 7 mm (see Section A-B in Figure 1), depending on the diameter of the detonating cord used (4.3.2).U.K.
4.4.1.1.2.Cut seven lengths of flexible detonating cord (4.3.2) each 400 mm long, avoiding any loss of explosive at each end by making a clean cut and immediately sealing the end with adhesive. Push each of the seven lengths through the seven holes in the wooden disc (4.3.10) until their ends project a few centimetres on the other side of the disc. Then insert a small dressmaking pin (4.3.12) transversally into the textile sleeve of each length of cord 5 to 6 mm from the end and apply adhesive around the outside of the lengths of cord in a band 2 cm wide adjacent to the pin. Finally, pull the long piece of each cord to bring the pin into contact with the wooden disc.U.K.
4.4.1.1.3.Shape the plastic explosive (4.3.1) to form a cylinder 92 to 96 mm in diameter, depending on the diameter of the cylinder (4.3.8). Stand this cylinder upright on a level surface and insert the shaped explosive. Then insert the wooden disc(4) carrying the seven lengths of detonating cord into the top of the cylinder and press it down onto the explosive. Adjust the height of the cylinder (64 to 67 mm) so that its top edge does not extend beyond the level of the wood. Finally, fix the cylinder to the wooden disc for instance with staples or small nails, around its entire circumference.U.K.
4.4.1.1.4.Group the free ends of the seven lengths of detonating cord around the circumference of the wooden rod (4.3.11) so that their ends are all level in a plane perpendicular to the rod. Secure them in a bundle around the rod by means of adhesive tape(5).U.K.
4.4.1.2.Central initiation by a compressed pelletU.K.
The booster charge prepared for use is shown in Figure 2.
4.4.1.2.1.Preparing a compressed pelletU.K.
Taking the necessary safety precautions, place 10 grams of a secondary explosive (4.3.3) in a mould with an inside diameter of 19 to 21 mm and compress to the correct shape and density.
(The ratio of diameter: height should be roughly 1:1).
In the centre of the bottom of the mould there is a peg, 12 mm in height and 7,0 to 7,3 mm in diameter (depending on the diameter of the detonator used), which forms a cylindrical recess in the compressed cartridge for subsequent insertion of the detonator.
4.4.1.2.2.Preparing the booster chargeU.K.
Place the explosive (4.3.1) into the cylinder (4.3.8) standing upright on a level surface, then press it down with a wooden die to give the explosive a cylindrical shape with a central recess. Insert the compressed pellet into this recess. Cover the cylindrically shaped explosive containing the compressed pellet with a wooden disc (4.3.10) having a central hole 7,0 to 7,3 mm in diameter for insertion of a detonator. Fix the wooden disc and the cylinder together with a cross of adhesive tape. Ensure that the hole drilled in the disc and the recess in the compressed pellet are coaxial by inserting the wooden rod (4.3.11).
4.4.2.Preparing steel tubes for the detonation testsU.K.
At one end of the steel tube (4.3.4), drill two diametrically opposed holes 4 mm in diameter perpendicularly through the side wall at a distance of 4 mm from the edge.
Butt weld the bottom plate (4.3.5) to the opposite end of the tube, completely filling the right angle between the bottom place and the wall of the tube with weld metal around the entire circumference of the tube.
4.4.3.Filling and charging the steel tubeU.K.
See Figures 1 and 2.
4.4.3.1.The test sample, the steel tube and the booster charge must be conditioned to temperatures of 20 (± 5) °C. 16 to 18 kg of the test sample are needed for two detonation tests.U.K.
4.4.3.2.Place the tube upright with its square bottom place resting on a firm, flat surface, preferably concrete. Fill the tube to about one-third of its height with the test sample and drop it 10 cm vertically onto the floor five times to compact the prills or granules as densely as possible in the tube. To accelerate compaction, vibrate the tube by striking the side wall with a 750 to 1 000-gram hammer between drops for a total of 10 times.U.K.
Repeat this charging method with another portion of the test sample. Finally, a further addition shall be made such that, after compaction by raising and dropping the tube 10 times and a total of 20 intermittent hammer blows, the charge fills the tube to a distance of 70 mm from its orifice.
The filling height of the sample must be adjusted in the steel tube so that the booster charge (4.4.1.1 or 4.4.1.2) to be inserted later will be in close contact with the sample over its entire surface.
4.4.3.3.Insert the booster charge into the tube so that it is in contact with the sample; the top surface of the wooden disc must be 6 mm below the end of the tube. Ensure essential close contact between explosive and test sample by adding or removing small quantities of sample. As shown in Figures 1 and 2, split pins should be inserted through the holes near the open end of the tube and their legs opened flat against the tube.U.K.
4.4.4.Positioning of the steel tube and lead cylinders (see figure 3)U.K.
4.4.4.1.Number the bases of the lead cylinders (4.3.6) 1 to 6. Make six marks 150 mm apart on the centre line of a steel block (4.3.7) lying on a horizontal base, with the first mark at least 75 mm from the edge of the block. Place a lead cylinder upright on each of these marks, with the base of each cylinder centred on its mark.U.K.
4.4.4.2.Lay the steel tube prepared according to 4.4.3 horizontally on the lead cylinders so that the axis of the tube is parallel to the centre line of the steel block and the welded end of the tube extends 50 mm beyond lead cylinder No 6. To prevent the tube from rolling, insert small wooden wedges between the tops of the lead cylinders and the tube wall (one on each side) or place a cross of wood between the tube and the steel block.U.K.
Note: Make sure that the tube is in contact with all six lead cylinders; a slight curvature of the tube surface can be compensated for by rotating the tube about its longitudinal axis; if any of the lead cylinders is too tall, tap the cylinder in question carefully with a hammer until it is the required height.U.K.
4.4.5.Preparation for detonationU.K.
4.4.5.1.Set up the apparatus according to the 4.4.4 in a bunker or suitably prepared underground site (e.g. mine or tunnel). Ensure that the temperature of the steel tube is kept at 20 (± 5) °C before detonation.U.K.
Note: Should such firing sites not be available, the work can, if necessary, be done in a concrete-lined pit covered over with wooden beams. Detonation can cause steel fragments to be projected with high kinetic energy, therefore, firing must be carried out at a suitable distance from dwellings or thoroughfares.U.K.
4.4.5.2.If the booster charge with seven-point initiation is used, ensure that the detonation cords are stretched out as described in the footnote to 4.4.1.1.4 and arranged as horizontally as possible.U.K.
4.4.5.3.Finally, remove the wooden rod and replace with the detonator. Do not carry out firing until the danger zone has been evacuated and the test personnel have taken cover.U.K.
4.4.5.4.Detonate the explosive.U.K.
4.4.6.Allow sufficient time for the fumes (gaseous and sometimes toxic decomposition products such as nitrous gases) to disperse, then collect the lead cylinders and measure their heights with a Vernier caliperU.K.
Record for each of the marked lead cylinders, the degree of crushing expressed as a percentage of the original height of 100 mm. If the cylinders are crushed obliquely, record the highest and the lowest values and calculate the average.
4.4.7.A probe for continuous measurement of the detonation velocity can be used; the probe should be inserted longitudinally to the axis of the tube or along its side wallU.K.
4.4.8.Two detonation tests per sample are to be carried outU.K.
4.5.Test reportU.K.
Values for the following parameters are to be given in the test report for each of the detonation tests:
the values actually measures for the outside diameter of the steel tube and for the wall thickness,
the Brinell hardness of the steel tube,
the temperature of the tube and the sample shortly before firing,
the packing density (kg/m3) of the sample in the steel tube,
the height of each lead cylinder after firing, specifying the corresponding cylinder number,
method of initiation employed for the booster charge.
4.5.1.Evaluation of test resultsU.K.
If, in each firing, the crushing of at least one lead cylinder is less than 5 %, the test shall be considered conclusive and the sample in conformity with the requirements of Annex III.2.
Figure 1

Figure 2

Figure 3

Textual Amendments
F1Words in Annex 3 para. 1.6 substituted (31.12.2020) by The Fertilisers and Ammonium Nitrate Material (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/601), regs. 1(2), 5(22); 2020 c. 1, Sch. 5 para. 1(1)
(1)
A reaction time of one and a half hours, is sufficient in the case of most of the organic substances in the presence of silver nitrate catalyst.
(2)
Commercially available standard copper solution may be used.
(3)
Whatman 541 or equivalent.
(4)
The diameter of the disc must always correspond to the inside diameter of the cylinder.
(5)
NB: When the six peripheral lengths of cord are taut after assembly, the central cord must remain slightly slack.U.K.
Options/Cymorth
Print Options
PrintThe Whole Regulation
PrintThis Attachment only
Mae deddfwriaeth ar gael mewn fersiynau gwahanol:
Y Diweddaraf sydd Ar Gael (diwygiedig):Y fersiwn ddiweddaraf sydd ar gael o’r ddeddfwriaeth yn cynnwys newidiadau a wnaed gan ddeddfwriaeth ddilynol ac wedi eu gweithredu gan ein tîm golygyddol. Gellir gweld y newidiadau nad ydym wedi eu gweithredu i’r testun eto yn yr ardal ‘Newidiadau i Ddeddfwriaeth’.
Gwreiddiol (Fel y’i mabwysiadwyd gan yr UE): Mae'r wreiddiol version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Gweler y wybodaeth ychwanegol ochr yn ochr â’r cynnwys
Rhychwant ddaearyddol: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Dangos Llinell Amser Newidiadau: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Dewisiadau Agor
Dewisiadau gwahanol i agor deddfwriaeth er mwyn gweld rhagor o gynnwys ar y sgrin ar yr un pryd
Rhagor o Adnoddau
Gallwch wneud defnydd o ddogfennau atodol hanfodol a gwybodaeth ar gyfer yr eitem ddeddfwriaeth o’r tab hwn. Yn ddibynnol ar yr eitem ddeddfwriaeth sydd i’w gweld, gallai hyn gynnwys:
- y PDF print gwreiddiol y fel adopted version that was used for the EU Official Journal
- rhestr o newidiadau a wnaed gan a/neu yn effeithio ar yr eitem hon o ddeddfwriaeth
- pob fformat o’r holl ddogfennau cysylltiedig
- slipiau cywiro
- dolenni i ddeddfwriaeth gysylltiedig ac adnoddau gwybodaeth eraill
Llinell Amser Newidiadau
Mae’r llinell amser yma yn dangos y fersiynau gwahanol a gymerwyd o EUR-Lex yn ogystal ag unrhyw fersiynau dilynol a grëwyd ar ôl y diwrnod ymadael o ganlyniad i newidiadau a wnaed gan ddeddfwriaeth y Deyrnas Unedig.
Cymerir dyddiadau fersiynau’r UE o ddyddiadau’r dogfennau ar EUR-Lex ac efallai na fyddant yn cyfateb â’r adeg pan ddaeth y newidiadau i rym ar gyfer y ddogfen.
Ar gyfer unrhyw fersiynau a grëwyd ar ôl y diwrnod ymadael o ganlyniad i newidiadau a wnaed gan ddeddfwriaeth y Deyrnas Unedig, bydd y dyddiad yn cyd-fynd â’r dyddiad cynharaf y daeth y newid (e.e. ychwanegiad, diddymiad neu gyfnewidiad) a weithredwyd i rym. Am ragor o wybodaeth gweler ein canllaw i ddeddfwriaeth ddiwygiedig ar Ddeall Deddfwriaeth.
Rhagor o Adnoddau
Defnyddiwch y ddewislen hon i agor dogfennau hanfodol sy’n cyd-fynd â’r ddeddfwriaeth a gwybodaeth am yr eitem hon o ddeddfwriaeth. Gan ddibynnu ar yr eitem o ddeddfwriaeth sy’n cael ei gweld gall hyn gynnwys:
- y PDF print gwreiddiol y fel adopted fersiwn a ddefnyddiwyd am y copi print
- slipiau cywiro
liciwch ‘Gweld Mwy’ neu ddewis ‘Rhagor o Adnoddau’ am wybodaeth ychwanegol gan gynnwys
- rhestr o newidiadau a wnaed gan a/neu yn effeithio ar yr eitem hon o ddeddfwriaeth
- manylion rhoi grym a newid cyffredinol
- pob fformat o’r holl ddogfennau cysylltiedig
- dolenni i ddeddfwriaeth gysylltiedig ac adnoddau gwybodaeth eraill
 Mae’r holl gynnwys ar gael dan Drwydded Llywodraeth Agored v3.0 ac eithrio ble nodir yn wahanol. Yn ychwanegol mae’r safle hwn â chynnwys sy’n deillio o EUR-Lex, a ailddefnyddiwyd dan delerau Penderfyniad y Comisiwn 2011/833/EU ar ailddefnyddio dogfennau o sefydliadau’r UE. Am ragor o wybodaeth gweler ddatganiad cyhoeddus Swyddfa Gyhoeddiadau’r UE ar ailddefnyddio.
Mae’r holl gynnwys ar gael dan Drwydded Llywodraeth Agored v3.0 ac eithrio ble nodir yn wahanol. Yn ychwanegol mae’r safle hwn â chynnwys sy’n deillio o EUR-Lex, a ailddefnyddiwyd dan delerau Penderfyniad y Comisiwn 2011/833/EU ar ailddefnyddio dogfennau o sefydliadau’r UE. Am ragor o wybodaeth gweler ddatganiad cyhoeddus Swyddfa Gyhoeddiadau’r UE ar ailddefnyddio.