- Y Diweddaraf sydd Ar Gael (Diwygiedig)
- Pwynt Penodol mewn Amser (31/12/2020)
- Gwreiddiol (Fel y’i mabwysiadwyd gan yr UE)
Commission Regulation (EC) No 152/2009Dangos y teitl llawn
Commission Regulation (EC) No 152/2009 of 27 January 2009 laying down the methods of sampling and analysis for the official control of feed (Text with EEA relevance)
You are here:
- Rheoliadau yn deillio o’r UE
- 2009 No. 152
- Whole Regulation
- Blaenorol
- Nesaf

Pa Fersiwn
Nodweddion Uwch
- Dangos Graddfa Ddaearyddol(e.e. Lloegr, Cymru, Yr Alban aca Gogledd Iwerddon)
- Dangos Llinell Amser Newidiadau
Rhagor o Adnoddau
PDF o Fersiynau Diwygiedig
- ddiwygiedig 16/11/20202.10 MB
- ddiwygiedig 24/05/20171.85 MB
- ddiwygiedig 26/04/20171.62 MB
- ddiwygiedig 17/07/20142.16 MB
- ddiwygiedig 01/01/20142.40 MB
- ddiwygiedig 12/02/20132.46 MB
- ddiwygiedig 18/04/20122.15 MB
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


Mae hon yn eitem o ddeddfwriaeth sy’n deillio o’r UE
Mae unrhyw newidiadau sydd wedi cael eu gwneud yn barod gan y tîm yn ymddangos yn y cynnwys a chyfeirir atynt gydag anodiadau.Ar ôl y diwrnod ymadael bydd tair fersiwn o’r ddeddfwriaeth yma i’w gwirio at ddibenion gwahanol. Y fersiwn legislation.gov.uk yw’r fersiwn sy’n weithredol yn y Deyrnas Unedig. Y Fersiwn UE sydd ar EUR-lex ar hyn o bryd yw’r fersiwn sy’n weithredol yn yr UE h.y. efallai y bydd arnoch angen y fersiwn hon os byddwch yn gweithredu busnes yn yr UE. EUR-Lex Y fersiwn yn yr archif ar y we yw’r fersiwn swyddogol o’r ddeddfwriaeth fel yr oedd ar y diwrnod ymadael cyn cael ei chyhoeddi ar legislation.gov.uk ac unrhyw newidiadau ac effeithiau a weithredwyd yn y Deyrnas Unedig wedyn. Mae’r archif ar y we hefyd yn cynnwys cyfraith achos a ffurfiau mewn ieithoedd eraill o EUR-Lex. The EU Exit Web Archive legislation_originated_from_EU_p3
Changes over time for: Commission Regulation (EC) No 152/2009
Version Superseded: 31/12/2022
Alternative versions:
- 27/01/2009- Amendment
- 18/04/2012- Amendment
- 12/02/2013- Amendment
- 01/01/2014- Amendment
- 17/07/2014- Amendment
- 26/04/2017- Amendment
- 24/05/2017- Amendment
- Exit day: start of implementation period31/01/2020 11pm- Amendment
- 16/11/2020- Amendment
- End of implementation period31/12/2020- Amendment
- 31/12/2020
Point in time - 31/12/2022- Amendment
Status:
Point in time view as at 31/12/2020.
Changes to legislation:
There are currently no known outstanding effects for the Commission Regulation (EC) No 152/2009.
Changes to Legislation
Revised legislation carried on this site may not be fully up to date. At the current time any known changes or effects made by subsequent legislation have been applied to the text of the legislation you are viewing by the editorial team. Please see ‘Frequently Asked Questions’ for details regarding the timescales for which new effects are identified and recorded on this site.
Commission Regulation (EC) No 152/2009
of 27 January 2009
laying down the methods of sampling and analysis for the official control of feed
(Text with EEA relevance)
THE COMMISSION OF THE EUROPEAN COMMUNITIES,
Having regard to the Treaty establishing the European Community,
Having regard to Regulation (EC) No 882/2004 of the European Parliament and of the Council of 29 April 2004 on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules(1), and in particular Article 11(4)(a), (b) and (c) thereof,
Whereas:
(1) The following acts were adopted for the implementation of Directive 70/373/EEC and remain in force in accordance with Article 61(2) of Regulation (EC) No 882/2004:
First Commission Directive 71/250/EEC of 15 June 1971 establishing Community methods of analysis for the official control of feedingstuffs(2),
Second Commission Directive 71/393/EEC of 18 November 1971 establishing Community methods of analysis for the official control of feedingstuffs(3),
Third Commission Directive 72/199/EEC of 27 April 1972 establishing Community methods of analysis for the official control of feedingstuffs(4),
Fourth Commission Directive 73/46/EEC of 5 December 1972 establishing Community methods of analysis for the official control of feedingstuffs(5),
First Commission Directive 76/371/EEC of 1 March 1976 establishing Community methods of sampling for the official control of feedingstuffs(6),
Seventh Commission Directive 76/372/EEC of 1 March 1976 establishing Community methods of analysis for the official control of feedingstuffs(7),
Eight Commission Directive 78/633/EEC of 15 June 1978 establishing Community methods of analysis for the official control of feedingstuffs(8),
Ninth Commission Directive 81/715/EEC of 31 July 1981 establishing Community methods of analysis for the official control of feedingstuffs(9),
Tenth Commission Directive 84/425/EEC of 25 July 1984 establishing Community methods of analysis for the official control of feedingstuffs(10),
Commission Directive 86/174/EEC of 9 April 1986 fixing the method of calculation for the energy value of compound poultry-feed(11),
Eleventh Commission Directive 93/70/EEC of 28 July 1993 establishing Community methods of analysis for official control of feedingstuffs(12),
Twelfth Commission Directive 93/117/EC of 17 December 1993 establishing Community methods of analysis for official control of feedingstuffs(13),
Commission Directive 98/64/EC of 3 September 1998 establishing Community methods of analysis for the determination of amino-acids, crude oils and fats, and olaquindox in feedingstuffs and amending Directive 71/393/EEC(14),
Commission Directive 1999/27/EC of 20 April 1999 establishing Community methods of analysis for the determination of amprolium, diclazuril and carbadox in feedingstuffs and amending Directives 71/250/EEC, 73/46/EEC and repealing Directive 74/203/EEC(15),
Commission Directive 1999/76/EC of 23 July 1999 establishing a Community method of analysis for the determination of lasalocid sodium in feedingstuffs(16),
Commission Directive 2000/45/EC of 6 July 2000 establishing Community methods of analysis for the determination of vitamin A, vitamin E and tryptophan in feedingstuffs(17),
Commission Directive 2002/70/EC of 26 July 2002 establishing requirements for the determination of levels of dioxins and dioxin-like PCBs in feedingstuffs(18),
Commission Directive 2003/126/EC of 23 December 2003 on the analytical method for the determination of constituents of animal origin for the official control of feedingstuffs(19).
(2) Since Directive 70/373/EEC was replaced by Regulation (EC) No 882/2004 it is appropriate to replace the implementing acts to that Directive by a single Regulation. At the same time the methods should be adapted in the light of developments in scientific and technological knowledge. Methods which are no longer valid for their intended purpose should be deleted. It is foreseen to update the sampling provisions in due time to take into account the recent developments in the way feed are produced, stored, transported and marketed, nevertheless it is appropriate to maintain for the time being the existing provisions on sampling.
(3) Directives 71/250/EEC, 71/393/EEC, 72/199/EEC, 73/46/EEC, 76/371/EEC, 76/372/EEC, 78/633/EEC, 81/715/EEC, 84/425/EEC, 86/174/EEC, 93/70/EEC, 93/117/EC, 98/64/EC, 1999/27/EC, 1999/76/EC, 2000/45/EC, 2002/70/EC and 2003/126/EC should therefore be repealed.
(4) The measures provided for in this Regulation are in accordance with the opinion of the Standing Committee on the Food Chain and Animal Health,
HAS ADOPTED THIS REGULATION:
[F1Article 1U.K.
Sampling for the official control of feed, in particular as regards the determination of constituents, including material which contains or consists of or is produced from genetically modified organisms (GMOs), feed additives as defined by Regulation (EC) No 1831/2003 of the European Parliament and of the Council(20), undesirable substances as defined by Directive 2002/32/EC of the European Parliament and of the Council(21) shall be carried out in accordance with the methods set out in Annex I.
The method of sampling set out in Annex I is applicable for the control of feed as regards the determination of pesticide residues as defined in Regulation (EC) No 396/2005 of the European Parliament and of the Council(22) and control of compliance with Regulation (EU) No 619/2011.
[F2In this Regulation, “reference laboratory” means a reference laboratory prescribed by the appropriate authority under Regulation 2017/625.]]
Textual Amendments
F1Substituted by Commission Regulation (EU) No 691/2013 of 19 July 2013 amending Regulation (EC) No 152/2009 as regards methods of sampling and analysis (Text with EEA relevance).
F2Words in Art. 1 inserted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 86; 2020 c. 1, Sch. 5 para. 1(1)
Article 2U.K.
Preparation of samples for analysis and expression of results shall be carried out in accordance with the methods set out in Annex II.
Article 3U.K.
Analysis for the official control of feed shall be carried out using the methods set out in Annex III (Methods of analysis to control the composition of feed materials and compound feed, Annex IV (Methods of analysis to control the level of authorised additives in feed), Annex V (Methods of analysis to control undesirable substances in feed) and Annex VI (Methods of analysis for the determination of constituents of animal origin for the official control of feed).
Article 4U.K.
The energy value of compound poultry feed shall be calculated in accordance with Annex VII.
Article 5U.K.
The methods of analysis to control illegal presence of no longer authorised additives in feed set out in Annex VIII shall be used for confirmatory purposes.
Article 6U.K.
Directives 71/250/EEC, 71/393/EEC, 72/199/EEC, 73/46/EEC, 76/371/EEC, 76/372/EEC, 78/633/EEC, 81/715/EEC, 84/425/EEC, 86/174/EEC, 93/70/EEC, 93/117/EC, 98/64/EC, 1999/27/EC, 1999/76/EC, 2000/45/EC, 2002/70/EC and 2003/126/EC are repealed.
References to the repealed Directives shall be construed as references to this Regulation and shall be read in accordance with the correlation tables in Annex IX.
Article 7U.K.
This Regulation shall enter into force on the twentieth day following that of 20th day following its publication in the Official Journal of the European Union.
It shall apply from 26 August 2009.
F3...
Textual Amendments
F3Words in Signature omitted (31.12.2020) by virtue of The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 87; 2020 c. 1, Sch. 5 para. 1(1)
[F1ANNEX IU.K. METHODS OF SAMPLING
1. PURPOSE AND SCOPE U.K.
Samples intended for the official control of feed shall be taken according to the methods described below. Samples thus obtained shall be considered as representative of the sampled portions.
The purpose of representative sampling is to obtain a small fraction from a lot in such a way that a determination of any particular characteristic of this fraction will represent the mean value of the characteristic of the lot. The lot shall be sampled by repeatedly taking incremental samples at various single positions in the lot. These incremental samples shall be combined by mixing to form an aggregate sample from which representative final samples shall be prepared by representative dividing.
If by a visual inspection, portions of the feed to be sampled show a difference in quality from the rest of the feed from the same lot, such portions shall be separated from the rest of the feed and treated as a separate sublot. If it is not possible to divide the feed into separate sublots, the feed shall be sampled as one lot. In such cases, mention shall be made of this fact in the sampling report.
Where a feed sampled in accordance with the provisions of this Regulation is identified as not satisfying [F4retained EU law], is part of a lot of feed of the same class or description, it shall be presumed that all of the feed in that lot is so affected, unless following a detailed assessment there is no evidence that the rest of the lot fails to satisfy [F5retained EU law].
Textual Amendments
F4Words in Annex 1 point 1 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(a)(i); 2020 c. 1, Sch. 5 para. 1(1)
F5Words in Annex 1 point 1 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(a)(ii); 2020 c. 1, Sch. 5 para. 1(1)
2. DEFINITIONS U.K.
— Lot (or batch)
:
an identified quantity of feed determined to have common characteristics, such as origin, variety, type of packaging, packer, consignor or labelling, and in case of a production process, a unit of production from a single plant using uniform production parameters or a number of such units, when produced in continuous order and stored together.
— Sampled portion
:
A lot or an identified part of the lot or sublot.
— Sealed sample
:
a sample sealed in such a manner as to prevent any access to the sample without breaking or removing the seal.
— Incremental sample
:
A quantity taken from one point in the sampled portion.
— Aggregate sample
:
An aggregate of incremental samples taken from the same sampled portion.
— Reduced sample
:
A part of the aggregate sample, obtained from the latter by a process of representative reduction.
— Final sample
:
A part of the reduced sample or of the homogenised aggregate sample.
— Laboratory sample
:
a sample intended for the laboratory (as received by the laboratory) and can be the final, reduced or aggregate sample.
3. GENERAL PROVISIONS U.K.
Sampling personnel: the samples shall be taken by persons authorised for that purpose by the competent authority.
The sample has to be sealed in such a manner as to prevent any access to the sample without breaking or removing the seal. The seal’s mark should be clearly identifiable and clearly visible. Alternatively, the sample can be put in a recipient which can be closed in such a manner that it cannot be opened without irreversibly damaging the receptacle or container, avoiding the re-use of the receptacle or container.
Identification of the sample: the sample has to be indelibly marked and must be identified in such a way that there is an unambiguous link to the sampling report.
From each aggregate sample at least two final samples are taken: at least one for control (enforcement) and one for the feed business operator (defence). Eventually, one final sample may be taken for reference. In case the complete aggregate sample is homogenized, the final samples are taken from the homogenized aggregate sample, unless such procedure conflicts with [F6the right of the feed business operator to challenge the method of sampling].
Textual Amendments
F6Words in Annex 1 point 3 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(b); 2020 c. 1, Sch. 5 para. 1(1)
4. APPARATUS U.K.
4.1.The sampling apparatus must be made of materials which cannot contaminate the products to be sampled. Apparatus which is intended to be used multiple times must be easy to clean to avoid any cross-contamination.U.K.
4.2. Apparatus recommended for the sampling of solid feed U.K.
4.2.1. Manual sampling U.K.
4.2.1.1. Flat-bottomed shovel with vertical sides U.K.
4.2.1.2.Sampling spear with a long split or compartments. The dimensions of the sampling spear must be appropriate to the characteristics of the sampled portion (depth of container, dimensions of sack, etc.) and to the particle size of the feed.U.K.
In case the sampling spear has several apertures, in order to ensure that the sample is taken at the different locations alongside the spear, the apertures should be separated by compartments or sequentially staggered apertures.
4.2.2. Mechanical sampling U.K.
Appropriate mechanical apparatus may be used for the sampling of moving feed. Appropriate means that at least the whole section of the flow is sampled.
Sampling of feed in motion (at high flow rates) can be performed by automatic samplers.
4.2.3. Divider U.K.
If possible and appropriate, apparatus designed to divide the sample into approximately equal parts should be used for the preparation of reduced samples in a representative way.
5. QUANTITATIVE REQUIREMENTS AS REGARDS NUMBER OF INCREMENTAL SAMPLES U.K.
The quantitative requirements in points 5.1 and 5.2 as regards the number of incremental samples are applicable for sampled portion sizes up to a maximum of 500 tonnes and which can be sampled in a representative way. The sampling procedure described is equally valid for quantities larger than prescribed maximum sampled portion size provided that the maximum number of incremental samples given in the tables below is ignored, the number of incremental samples being determined by the square-root formula given in the appropriate part of the procedure (see point 5.3) and the minimum aggregate sample size increased proportionally. This does not prevent a large lot being divided into smaller sublots and each sublot sampled in accordance with the procedure described in points 5.1 and 5.2.
The size of the sampled portion must be such that each of its constituent parts can be sampled.
For very large lots or sublots (> 500 tonnes) and for lots which are transported or stored in such a way that sampling cannot be done in accordance with the sampling procedure provided for in points 5.1 and 5.2 of this chapter, the sampling procedure as provided for in point 5.3 is to be applied.
In case the feed business operator is required by legislation to comply with this Regulation within the frame of a mandatory monitoring system, the feed business operator may deviate from the quantitative requirements as provided for in this chapter to take into account operational characteristics on the condition that the feed business operator has demonstrated to the satisfaction of the competent authority the equivalence of the sampling procedure as regards representativeness and after authorisation from the competent authority.
In exceptional cases, if it is not possible to carry out the method of sampling set out as regards the quantitative requirements because of the unacceptable commercial damage to the lot (because of packaging forms, means of transport, way of storage etc.) an alternative method of sampling may be applied provided that it is as representative as possible and is fully described and documented.
5.1. Quantitative requirements as regards incremental samples in relation to the control of substances or products uniformly distributed throughout the feed U.K.
5.1.1. Loose solid feed U.K.
a Where the number obtained is a fraction, it shall be rounded up to the next whole number. | |
| Size of sampled portion | Minimum number of incremental samples |
|---|---|
| ≤ 2,5 tonnes | 7 |
| > 2,5 tonnes | √ 20 times the number of tonnes making up the sampled portion a , up to 40 incremental samples |
5.1.2. Loose liquid feed U.K.
5.1.3. Packaged feed U.K.
Feed (solid and liquid) can be packaged in bags, sacks, cans, barrels etc. which are referred to in the table as units. Large units (≥ 500 kg or litres) have to be sampled in accordance with the provisions foreseen for loose feed (see points 5.1.1 and 5.1.2).
a In the case where opening of an unit might affect the analysis (e.g. perishable wet feeds) an incremental sample shall be the unopened unit. | |
b For units whose contents do not exceed 1 kg or one litre, an incremental sample shall be the contents of one original unit. | |
c Where the number obtained is a fraction, it shall be rounded up to the next whole number. | |
| Size of sampled portion | Minimum number of units from which (at least) one incremental sample has to be taken a |
|---|---|
| 1 to 20 units | 1 unit b |
| 21 to 150 units | 3 units b |
| 151 to 400 units | 5 units b |
| > 400 units | ¼ of the √ number of units making up the sampled portion c , up to 40 units |
5.1.4. Feed blocks and mineral licks U.K.
Minimum one block or lick to be sampled per sampled portion of 25 units, up to a maximum of four blocks or licks.
For blocks or licks weighing not more than 1 kg each, an incremental sample shall be the contents of one block or one lick.
5.1.5. Roughages/forage U.K.
a It is acknowledged that in certain situations (e.g. silages) it is not possible to take the required incremental samples, without causing unacceptable damage to the lot. An alternative method of sampling may be applied in such situations and a guidance for sampling such lots will be elaborated before the entry into application of this Regulation. | |
b Where the number obtained is a fraction, it shall be rounded up to the next whole number. | |
| Size of sampled portion | Minimum number of incremental samples a |
|---|---|
| ≤ 5 tonnes | 5 |
| > 5 tonnes | √ 5 times the number of tonnes making up the sampled portion b , up to 40 incremental samples |
5.2. Quantitative requirements as regards incremental samples in relation to the control of constituents or substances likely to be distributed non-uniformly in feed U.K.
These quantitative requirements as regards incremental samples are to be used in the following situations:
control of aflatoxins, rye ergot, other mycotoxins and harmful botanical impurities in feed materials;
control of cross contamination by a constituent, including GM material, or substance for which non-uniform distribution is expected in feed materials.
In case the control authority has strong suspicion that such a non-uniform distribution occurs also in case of cross contamination by a constituent or substance in a compound feed, the quantitative requirements as provided for in the table below can be applied.
| Size of sampled portion | Minimum number of incremental samples |
|---|---|
| < 80 tonnes | See quantitative requirements under point 5.1. The number of incremental samples to be taken has to be multiplied by 2,5. |
| ≥ 80 tonnes | 100 |
5.3. Quantitative requirements as regards the incremental samples in the case of very large lots U.K.
In the case of large sampled portions (sampled portions > 500 tonnes), the number of incremental samples to be taken = 40 incremental samples + √ tonnes in relation to the control of substances or products uniformly distributed throughout the feed or 100 incremental samples + √ tonnes in relation to the control of constituents or substances likely to be distributed non-uniformly in feed materials.
6. QUANTITATIVE REQUIREMENTS AS REGARDS AGGREGATE SAMPLE U.K.
a In case the sampled feed is of high value, a smaller quantity of aggregate sample can be taken on the condition this is described and documented in the sampling report. | ||
b In accordance with the provisions of Commission Regulation (EU) No 619/2011 of 24 June 2011 laying down the methods of sampling and analysis for the official control of feed as regards presence of genetically modified material for which an authorisation procedure is pending or the authorisation of which has expired (OJ L 166, 25.6.2011, p. 9), the aggregate sample for the control of the presence of genetically modified material must contain at least 35 000 seeds/grains. This means that for maize the size of the aggregate sample must be at least 10,5 kg and for soybean 7 kg. For other seeds and grains such as barley, millet, oat, rice, rye, wheat and rapeseed, the aggregate sample size of 4 kg corresponds to more than 35 000 seeds. | ||
c In case of packaged feed, it may also not be possible to achieve the size of 4 kg for the aggregate sample depending of the size of the individual units. | ||
d In case it concerns roughage or forage with a low specific gravity (e.g. hay, straw), the aggregate sample should have a minimum size of 1 kg. | ||
| A single aggregate sample per sampled portion is required. | ||
|---|---|---|
| Nature of feed | Minimum size of aggregate sample a b | |
| 6.1. | Loose feed | 4 kg |
| 6.2. | Packaged feed: | 4 kg c |
| 6.3. | Liquid or semi-liquid feed: | 4 litres |
| 6.4. | Feed blocks or mineral licks: | |
| 6.4.1. | each weighing more than 1 kg | 4 kg |
| 6.4.2. | each weighing not more than 1 kg | weight of four original blocks or licks |
| 6.5. | Roughage/forage | 4 kg d |
7. QUANTITATIVE REQUIREMENTS AS REGARDS FINAL SAMPLES U.K.
Final samples U.K.
Analysis of at least one final sample is required. The amount in the final sample for analysis shall be not less than the following:
a In accordance with the provisions of Regulation (EU) No 619/2011, the final sample for the control of the presence of genetically modified material must contain at least 10 000 seeds/grains. This means that for maize the size of the final sample must be at least 3 000 g and for soybean 2 000 g. For other seeds and grains such as barley, millet, oat, rice, rye, wheat and rapeseed, the final sample size of 500 g corresponds to more for 10 000. | |
b In case the size of the aggregate sample is significantly less than 4 kg or litre (see footnotes point (6), also a smaller quantity of final sample can be taken on the condition this is described and documented in the sampling report. | |
c In case of sampling pulses, cereal grains and tree nuts for the determination of pesticide residues, the minimum size of the final sample shall be 1 kg in accordance with the provisions of Commission Directive 2002/63/EC (OJ L 187, 16.7.2002, p. 30). | |
| Solid feed | 500 g a b c |
| Liquid or semi-liquid feed | 500 ml a |
8. METHOD OF SAMPLING FOR VERY LARGE LOTS OR LOTS STORED OR TRANSPORTED IN A WAY WHEREBY SAMPLING THROUGHOUT THE LOT IS NOT FEASIBLE U.K.
8.1. General principles U.K.
In case the way of transport or storage of a lot does not enable to take incremental samples throughout the whole lot, sampling of such lots should preferably be done when the lot is in flow.
In the case of large warehouses destined to store feed, operators should be encouraged to install equipment in the warehouse enabling (automatic) sampling across the whole stored lot.
In case of applying the sampling procedures as provided for in this chapter 8, the feed business operator or his representative is informed of the sampling procedure. In case this sampling procedure is questioned by the feed business operator or his representative, the feed business operator or his representative shall enable the competent authority to sample throughout the whole lot at his/her cost.
8.2. Large lots transported by ship U.K.
8.2.1. Dynamic sampling of large lots transported by ship U.K.
The sampling of large lots in ships is preferably carried out while the product is in flow (dynamic sampling).
The sampling is to be done per hold (entity that can physically be separated). Holds are however emptied partly one after the other so that the initial physical separation does no longer exist after transfer into storage facilities. Sampling can therefore be performed in function of the initial physical separation or in function of the separation after transfer into the storage facilities.
The unloading of a ship can last for several days. Normally, sampling has to be performed at regular intervals during the whole duration of unloading. It is however not always feasible or appropriate for an official inspector to be present for sampling during the whole operation of unloading. Therefore sampling is allowed to be undertaken of part (sampled portion) of the whole lot. The number of incremental samples is determined by taking into account the size of the sampled portion.
In the case of sampling a part of a lot of feed of the same class or description and that part of the lot has been identified as not satisfying [F7retained EU law], it shall be presumed that all of the feed in that lot is so affected, unless following a detailed assessment there is no evidence that the rest of the lot fails to satisfy [F8retained EU Law].
Textual Amendments
F7Words in Annex 1 point 8.2.1 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(c)(i); 2020 c. 1, Sch. 5 para. 1(1)
F8Words in Annex 1 point 8.2.1 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(c)(ii); 2020 c. 1, Sch. 5 para. 1(1)
Even if the official sample is taken automatically, the presence of an inspector is necessary. However in case the automatic sampling is done with preset parameters which cannot be changed during the sampling and the incremental samples are collected in a sealed receptacle, preventing any possible fraud, then the presence of an inspector is only required at the beginning of the sampling, every time the receptacle of the samples needs to be changed and at the end of the sampling.
8.2.2. Sampling of lots transported by ship by static sampling U.K.
In case the sampling is done in a static way the same procedure as foreseen for storage facilities (silos) accessible from above has to be applied (see point 8.4.1).
The sampling has to be performed on the accessible part (from above) of the lot/hold. The number of incremental samples is determined by taking into account the size of the sampled portion. In the case of sampling a part of a lot of feed of the same class or description and that part of the lot has been identified as not satisfying [F9retained EU law], it shall be presumed that all of the feed in that lot is so affected, unless following a detailed assessment there is no evidence that the rest of the lot fails to satisfy [F10retained EU Law].
Textual Amendments
F9Words in Annex 1 point 8.2.2 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(d)(i); 2020 c. 1, Sch. 5 para. 1(1)
F10Words in Annex 1 point 8.2.2 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(d)(ii); 2020 c. 1, Sch. 5 para. 1(1)
8.3. Sampling of large lots stored in warehouses U.K.
The sampling has to be performed on the accessible part of the lot. The number of incremental samples is determined by taking into account the size of the sampled portion. In the case of sampling a part of a lot of feed of the same class or description and that part of the lot has been identified as not satisfying [F11retained EU law], it shall be presumed that all of the feed in that lot is so affected, unless following a detailed assessment there is no evidence that the rest of the lot fails to satisfy [F12retained EU Law].
Textual Amendments
F11Words in Annex 1 point 8.3 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(e)(i); 2020 c. 1, Sch. 5 para. 1(1)
F12Words in Annex 1 point 8.3 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(e)(ii); 2020 c. 1, Sch. 5 para. 1(1)
8.4. Sampling of storage facilities (silos) U.K.
8.4.1. Sampling of silos (easily) accessible from above U.K.
The sampling has to be performed on the accessible part of the lot. The number of incremental samples is determined by taking into account the size of the sampled portion. In the case of sampling a part of a lot of feed of the same class or description and that part of the lot has been identified as not satisfying [F13retained EU law], it shall be presumed that all of the feed in that lot is so affected, unless following a detailed assessment there is no evidence that the rest of the lot fails to satisfy [F14retained EU Law].
Textual Amendments
F13Words in Annex 1 point 8.4.1 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(f)(i); 2020 c. 1, Sch. 5 para. 1(1)
F14Words in Annex 1 point 8.4.1 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(f)(ii); 2020 c. 1, Sch. 5 para. 1(1)
8.4.2. Sampling of silos not accessible from above (closed silos) U.K.
8.4.2.1. Silos not accessible from above (closed silos) with size > 100 tonnes U.K.
Feed stored in such silos cannot be sampled in a static way. Therefore in case the feed in the silo has to be sampled and there is no possibility to move the consignment, the agreement has to be made with the operator that he or she has to inform the inspector about when the silo will be unloaded in order to enable sampling when the feed is in flow.
8.4.2.2. Silos not accessible from above (closed silos) with size < 100 tonnes U.K.
Sampling procedure involves the release into a receptacle of a quantity of 50 to 100 kg and taking the sample from it. The size of the aggregate sample corresponds to the whole lot and the number of incremental samples relate to the quantity of the silo released in a receptacle for sampling. In the case of sampling a part of a lot of feed of the same class or description and that part of the lot has been identified as not satisfying [F15retained EU law], it shall be presumed that all of the feed in that lot is so affected, unless following a detailed assessment there is no evidence that the rest of the lot fails to satisfy [F16retained EU Law].
Textual Amendments
F15Words in Annex 1 point 8.4.2.2 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(g)(i); 2020 c. 1, Sch. 5 para. 1(1)
F16Words in Annex 1 point 8.4.2.2 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 88(g)(ii); 2020 c. 1, Sch. 5 para. 1(1)
8.5. Sampling of loose feed in large closed containers U.K.
Such lots can often only be sampled when unloaded. It is in certain cases not possible to unload at the point of import or control and therefore the sampling should take place when such containers are unloaded.
9. INSTRUCTIONS FOR TAKING, PREPARING AND PACKAGING THE SAMPLES U.K.
9.1. General U.K.
The samples must be taken and prepared without unnecessary delay bearing in mind the precautions necessary to ensure that the product is neither changed nor contaminated. Instruments and also surfaces and containers intended to receive samples must be clean and dry.
9.2. Incremental samples U.K.
Incremental samples must be taken at random throughout the whole sampled portion and they must be of approximately equal sizes.
The incremental sample size is at least 100 grams or 25 grams in case of roughage or forage with low specific gravity.
In case that in accordance with the rules for the sampling procedure established in point 8 less than 40 incremental samples have to be taken, the size of the incremental samples shall be determined in function of the required size of the aggregate sample to be achieved (see point (6).
In case of sampling of small lots of packaged feed where according to the quantitative requirements a limited number of incremental samples have to be taken, an incremental sample shall be the contents of one original unit whose contents do not exceed 1 kg or one litre.
In case of sampling of packaged feed composed of small units (e.g. < 250 g), the size of the incremental sample depends on the size of the unit.
9.2.1. Loose feed U.K.
Where appropriate, sampling may be carried out when the sampled portion is being moved (loading or unloading).
9.2.2. Packaged feed U.K.
Having selected the required number of units for sampling as indicated in chapter 5, part of the contents of each unit shall be removed using a spear or shovel. Where necessary, the samples shall be taken after emptying the units separately.
9.2.3. Homogeneous or homogenisable liquid or semi-liquid feed U.K.
Having selected the required number of units for sampling as indicated in chapter 5, the contents shall be homogenised if necessary and an amount taken from each unit.
The incremental samples may be taken when the contents are being discharged.
9.2.4. Non-homogenisable, liquid or semi-liquid feed U.K.
Having selected the required number of units for sampling as indicated in chapter 5, samples shall be taken from different levels.
Samples may also be taken when the contents are being discharged but the first fractions shall be discarded.
In either case the total volume taken must not be less than 10 litres.
9.2.5. Feed blocks and mineral licks U.K.
Having selected the required number of blocks or licks for sampling as indicated in chapter 5, a part of each block or lick can be taken. In case of suspicion of a non-homogeneous block or lick, the whole block or lick can be taken as sample.
For blocks or licks weighing not more than 1 kg each, an incremental sample shall be the contents of one block or one lick.
9.3. Preparation of aggregate samples U.K.
The incremental samples shall be mixed to form a single aggregate sample.
9.4. Preparation of final samples U.K.
The material in the aggregate sample shall be carefully mixed(23).
Each sample shall be put into an appropriate container/receptacle. All necessary precautions shall be taken to avoid any change of composition of the sample, contamination or adulteration which might arise during transportation or storage.
In case of the control of constituents or substances uniformly distributed throughout the feed, the aggregate sample can be representatively reduced to at least 2,0 kg or 2,0 litres (reduced sample)(24) preferably either by using a mechanical or automatic divider. For the control of the presence of pesticide residues in pulses, cereal grains and tree nuts, the minimum size of the reduced sample shall be 3 kg. In case the nature of the feed does not allow using a divider or the divider is not available, then the sample can be reduced by the quartering method. From the reduced samples the final samples (for control, defence and reference) shall then be prepared of approximately the same amount and conforming to the quantitative requirements of chapter 7. In case of the control of constituents, including genetically modified material, or substances likely to be distributed non-uniformly in feed materials, the aggregate sample shall be:
completely homogenized and divided afterwards into final samples or
reduced to at least 2 kg or 2 litres(25) by using a mechanical or automatic divider. Only in the case that the nature of the feed does not allow for using a divider, the sample can, if necessary, be reduced by quartering method. For the control of the presence of genetically modified material in the frame of Regulation (EU) No 619/2011, the reduced sample must contain at least 35 000 seeds/grains to enable to obtain the final samples for enforcement, defence and reference of at least 10 000 seeds grain (see footnote (**) in chapter 6 and footnote (*) in chapter 7).
9.5. Packaging of samples U.K.
The containers or packages shall be sealed and labelled in such a manner that they cannot be opened without damaging the seal. The total label must be incorporated in the seal.
9.6. Sending of samples to the laboratory U.K.
The sample shall be sent without unnecessary delay to the designated analytical laboratory, together with the information necessary for the analyst.
10. SAMPLING RECORD U.K.
A record must be kept of each sample, permitting each sampled portion and its size to be identified unambiguously.
The record shall also mention any deviation of the sampling procedure as provided for in this Regulation.
Besides making the record available to the official control laboratory, the record shall be made available to the feed business operator and/or the laboratory designated by the feed business operator.]
[F1ANNEX IIU.K. GENERAL PROVISIONS ON METHODS OF ANALYSIS FOR FEED
A. PREPARATION OF SAMPLES FOR ANALYSIS U.K.
1. Purpose U.K.
The procedures described below concern the preparation for analysis of samples, sent to the control laboratories after sampling in accordance with the provisions laid down in Annex I.
The laboratory samples must be prepared in such a way that the amounts weighed out, as provided for in the methods of analysis, are homogeneous and representative of the final samples.
2. Precautions to be taken U.K.
The sample preparation procedure to be followed is dependent on the methods of analysis to be used and the constituents or substances to be controlled. It is therefore of major importance that it is ensured that the followed sample preparation procedure is appropriate for the used method of analysis and for constituents or substances to be controlled.
All the necessary operations must be performed in such a way as to avoid as far as possible contamination of the sample and changes of its composition.
Grinding, mixing and sieving shall be carried out without delay with minimal exposure of the sample to the air and light. Mills and grinders likely to appreciably heat the sample shall not be used.
Manual grinding is recommended for feed which are particularly sensitive to heat. Care shall also be taken to ensure that the apparatus itself is not a source of contamination.
If the preparation cannot be carried out without significant changes in the moisture content of the sample, determine the moisture content before and after preparation according to the method laid down in Part A of Annex III.
3. Procedure U.K.
3.1. General procedure U.K.
The test aliquot is taken from the final sample. Coning and quartering is not recommended because this might provide test aliquots with high splitting error.
3.1.1. Feed which can be ground as such U.K.
Mix the sieved final sample and collect it in a suitable clean, dry container fitted with an air-tight stopper. Mix again in order to ensure full homogenisation, immediately before weighing out the amount for analysis (test aliquot).
3.1.2. Feed which can be ground after drying U.K.
Unless otherwise specified in the methods of analysis, dry the final sample to bring its moisture content down to a level of 8 to 12 %, according to the preliminary drying procedure described under point 4.3 of the method of determination of moisture mentioned in Part A of Annex III). Then proceed as indicated in section 3.1.1.
3.1.3. Liquid or semi-liquid feed U.K.
Collect the final sample in a suitable clean, dry container, fitted with an air-tight stopper. Mix thoroughly in order to ensure full homogenisation immediately before weighing out the amount for analysis (test aliquot).
3.1.4. Other feed U.K.
Final samples which cannot be prepared according to one of the above procedures shall be treated by any other procedure which ensures that the amounts weighed out for the analysis (test aliquot) are homogeneous and representative of the final samples.
3.2. Specific procedure in case of examination by visual inspection or by microscopy or in cases where the whole aggregate sample is homogenised U.K.
In case of an examination by visual inspection (without making use of microscope), the whole laboratory sample is used for examination.
In case of a microscopic examination, the laboratory may reduce the aggregate sample, or further reduce the reduced sample. The final samples for defence and eventually reference purposes are taken following a procedure equivalent to the procedure followed for the final sample for enforcement.
In case the whole aggregate sample is homogenized, the final samples are taken from the homogenized aggregate sample.
4. Storage of samples U.K.
Samples must be stored at a temperature that will not alter their composition. Samples intended for the analysis of vitamins or substances which are particularly sensitive to light shall be stored in such conditions that the sample is not adversely affected by light.
B. PROVISIONS RELATING TO REAGENTS AND APPARATUS USED IN METHODS OF ANALYSIS U.K.
1.Unless otherwise specified in the methods of analysis, all analytical reagents must be analytically pure (a.p.). When trace analysis is carried out, the purity of the reagents must be checked by a blank test. Depending upon the results obtained, further purification of the reagents may be required.U.K.
2.Any operation involving preparation of solutions, dilution, rinsing or washing, mentioned in the methods of analysis without indication as to the nature of the solvent or diluent employed, implies that water must be used. As a general rule, water shall be demineralised or distilled. In particular cases, which are indicated in the methods of analysis, it must be submitted to special procedures of purification.U.K.
3.In view of the equipment normally found in control laboratories, only those instruments and apparatus which are special or require specific usage are referred to in the methods of analysis. They must be clean, especially when very small amounts of substances have to be determined.U.K.
C. APPLICATION OF METHODS OF ANALYSIS AND EXPRESSION OF THE RESULTS U.K.
1. Extraction procedure U.K.
Several methods determine a specific extraction procedure. As a general rule, other extraction procedures than the procedure referred to in the method can be applied on the condition that the used extraction procedure has been proven to have the equivalent extraction efficiency for the matrix analysed as the procedure mentioned in the method.
2. Clean-up procedure U.K.
Several methods determine a specific clean-up procedure. As a general rule, other clean-up procedures than the procedure referred to in the method can be applied on the condition that the used clean-up procedure has been proven to result in equivalent analytical results for the matrix analysed as the procedure mentioned in the method.
3. Number of determinations U.K.
In case of the analysis of undesirable substances, if the result of the first determination is significantly (> 50 %) lower than the specification to be controlled, no additional determinations are necessary, on the condition that the appropriate quality procedures are applied. In other cases a duplicate analysis (second determination) is necessary to exclude the possibility of internal cross-contamination or an accidental mix-up of samples. The mean of the two determinations, taking into account the measurement uncertainty is used for verification of compliance.
In case of the control of the declared content of a substance or ingredient, if the result of the first determination confirms the declared content, i.e. the analytical result falls within the acceptable range of variation of the declared content, no additional determinations are necessary, on the condition that the appropriate quality procedures are applied. In other cases a duplicate analysis (second determination) is necessary to exclude the possibility of internal cross-contamination or an accidental mix-up of samples. The mean of the two determinations, taking into account the measurement uncertainty is used for verification of compliance.
In some cases this acceptable range of variation is defined in legislation such as in Regulation (EC) No 767/2009 of the European Parliament and of the Council of 13 July 2009 on the placing on the market and use of feed, amending European Parliament and Council Regulation (EC) No 1831/2003 and repealing Council Directive 79/373/EEC, Commission Directive 80/511/EEC, Council Directives 82/471/EEC, 83/228/EEC, 93/74/EEC, 93/113/EC and 96/25/EC and Commission Decision 2004/217/EC(26).
4. Reporting of the method of analysis used U.K.
The analysis report shall mention the method of analysis used.
5. Reporting of the analytical result U.K.
The analytical result shall be expressed in the manner laid down in the method of analysis to an appropriate number of significant figures and shall be corrected, if necessary, to the moisture content of the final sample prior to preparation.
6. Measurement uncertainty and recovery rate in case of analysis of undesirable substances U.K.
As regards undesirable substances within the meaning of Directive 2002/32/EC, a product intended for animal feed shall be considered as non-compliant with the established maximum content, if the analytical result, relative to a feed with a moisture content of 12 %, is deemed to exceed the maximum content taking into account expanded measurement uncertainty and correction for recovery. In order to assess compliance, the analysed concentration is used after being corrected for recovery and after deduction of the expanded measurement uncertainty. This procedure is only applicable in cases where the method of analysis enables the estimation of measurement uncertainty and correction for recovery (e.g. not possible in case of microscopic analysis).
The analytical result shall be reported as follows (in so far the used method of analysis enables to estimate the measurement uncertainty and recovery rate):
(a)
corrected for recovery, the level of recovery being indicated. The correction for recovery is not necessary in case the recovery rate is between 90-110 %.
(b)
as ‘x +/– U’, whereby x is the analytical result and U is the expanded measurement uncertainty, using a coverage factor of 2 which gives a level of confidence of approximately 95 %.
However, if the result of the analysis is significantly (> 50 %) lower than the specification to be controlled, and on the condition that the appropriate quality procedures are applied and the analysis serves only the purpose of checking compliance with legal provisions, the analytical result might be reported without correction for recovery and the reporting of the recovery rate and measurement uncertainty might be omitted in these cases.]
ANNEX IIIU.K.METHODS OF ANALYSIS TO CONTROL THE COMPOSITION OF FEED MATERIALS AND COMPOUND FEED
A.DETERMINATION OF MOISTUREU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the moisture content of feed. In case of feed containing volatile substances, such as organic acids, it is to be observed that also significant amount of volatile substances are determined together with the moisture content.
It does not cover the analysis of milk products as feed materials, the analysis of mineral substances and mixtures composed predominantly of mineral substances, the analysis of animal and vegetable fats and oils or the analysis of the oil seeds and oleaginous fruit.
2.PrincipleU.K.
The sample is desiccated under specified conditions which vary according to the nature of the feed. The loss in weight is determined by weighing. It is necessary to carry out preliminary drying when dealing with solid feed which has high moisture content.
3.ApparatusU.K.
3.1.Crusher of non-moisture-absorbing material which is easy to clean, allows rapid, even crushing without producing any appreciable heating, prevents contact with the outside air as far as possible and meets the requirements laid down in 4.1.1 and 4.1.2 (e.g. hammer or water cooled micro-crushers, collapsible cone mills, slow motion or cog wheeled crushers).U.K.
3.2.Analytical balance, accurate to 1 mg.U.K.
3.3.Dry containers of non-corrodible metal or of glass with lids ensuring airtight closure; working surface allowing the test sample to be spread at about 0,3 g/cm2.U.K.
3.4.Electrically heated isothermal oven (± 2 oC) properly ventilated and ensuring rapid temperature regulation(27).U.K.
3.5.Adjustable electrically heated vacuum oven fitted with an oil pump and either a mechanism for introducing hot dried air or a drying agent (e.g. calcium oxide).U.K.
3.6.Desiccator with a thick perforated metal or porcelain plate, containing an efficient drying agent.U.K.
4.ProcedureU.K.
N.B.
The operations described in this section must be carried out immediately after opening the packages of samples. Analysis must be carried out at least in duplicate.
4.1.PreparationU.K.
4.1.1.Feed other than those coming under 4.1.2 and 4.1.3U.K.
Take at least 50 g of the sample. If necessary, crush or divide in such a way as to avoid any variation in moisture content (see 6).
4.1.2.Cereals and groatsU.K.
Take at least 50 g of the sample. Grind into particles of which at least 50 % will pass through a 0,5 mm mesh sieve and will leave no more than 10 % reject on a 1 mm round-meshed sieve.
4.1.3.Feed in liquid or paste form, feed predominantly composed of oils and fatsU.K.
Take about 25 g of the sample, weigh to the nearest 10 mg, add an appropriate quantity of anhydrous sand weighed to the nearest 10 mg and mix until a homogeneous product is obtained.
4.2.DryingU.K.
4.2.1.Feed other than those coming under 4.2.2 and 4.2.3U.K.
Weigh a container (3.3) with its lid to the nearest 1 mg. Weigh into the weighed container, to the nearest 1 mg, about 5 g of the sample and spread evenly. Place the container, without its lid, in the oven preheated to 103 oC. To prevent the oven temperature from falling unduly, introduce the container as rapidly as possible. Leave to dry for four hours reckoned from the time when the oven temperature returns to 103 oC. Replace the lid on the container, remove the latter from the oven, leave to cool for 30 to 45 minutes in the desiccator (3.6) and weigh to the nearest 1 mg.
For feed composed predominantly of oils and fats, dry in the oven for an additional 30 minutes at 130 oC. The difference between the two weighings must not exceed 0,1 % of moisture.
4.2.2.Cereals, flour, groats and mealU.K.
Weigh a container (3.3) with its lid to the nearest 0,5 mg. Weigh into the weighed container, to the nearest 1 mg, about 5 g of the crushed sample and spread evenly. Place the container, without its lid, in the oven preheated to 130 oC. To prevent the oven temperature from falling unduly, introduce the container as rapidly as possible. Leave to dry for two hours reckoned from the time when the oven temperature returns to 130 oC. Replace the lid on the container, remove the latter from the oven, leave to cool for 30 to 45 minutes in the desiccator (3.6) and weigh to the nearest 1 mg.
4.2.3.Compound feed containing more than 4 % of sucrose or lactose: feed materials such as locust beans, hydrolysed cereal products, malt seeds, dried beet chips, fish and sugar solubles; compound feed containing more than 25 % of mineral salts including water of crystallisation.U.K.
Weigh a container (3.3) with its lid to the nearest 0,5 mg. Weigh into the weighed container, to the nearest 1 mg, about 5 g of the sample and spread evenly. Place the container, without its lid, in the vacuum oven (3.5) preheated to between 80 oC and 85 oC. To prevent the oven temperature from falling unduly, introduce the container as rapidly as possible.
Bring the pressure up to 100 Torr and leave to dry for four hours at this pressure, either in a current of hot, dry air or using a drying agent (about 300 g for 20 samples). In the latter instance, disconnect the vacuum pump when the prescribed pressure has been reached. Reckon drying time from the moment when the oven temperature returns to 80 oC to 85 oC. Carefully bring the oven back to atmospheric pressure. Open the oven, place the lid on the container immediately, remove the container from the oven, leave to cool for 30 to 45 minutes in the desiccator (3.6) and weigh to the nearest 1 mg. Dry for an additional 30 minutes in the vacuum oven at 80 oC to 85 oC and reweigh. The difference between the two weighings must not exceed 0,1 % of moisture.
4.3.Preliminary dryingU.K.
4.3.1.Feed other than those coming under 4.3.2U.K.
Solid feed with a high moisture content which makes crushing difficult must be subjected to preliminary drying as follows:
Weigh 50 g of uncrushed sample to the nearest 10 mg (compressed or agglomerated feed may be roughly divided if necessary) in a suitable container (e.g. a 20 × 12 cm aluminium plate with a 0,5 cm rim). Leave to dry in an oven from 60 oC to 70 oC until the moisture content has been reduced to between 8 % and 12 %. Remove from the oven, leave to cool uncovered in the laboratory for one hour and weigh to the nearest 10 mg. Crush immediately as indicated in 4.1.1 and dry as indicated in 4.2.1 or 4.2.3 according to the nature of the feed.
4.3.2.CerealsU.K.
Grain with a moisture content of over 17 % must be subjected to preliminary drying as follows:
Weigh 50 g of unground grain to the nearest 10 mg in a suitable container (e.g. a 20 × 12 cm aluminium plate with a 0,5 cm rim). Leave to dry for 5 to 7 minutes in an oven at 130 oC. Remove from the oven, leave to cool uncovered in the laboratory for two hours and weigh to the nearest 10 mg. Grind immediately as indicated in 4.1.2 and dry as indicated in 4.2.2.
5.Calculation of resultsU.K.
The moisture content (X), as a percentage of the sample, is calculated by using the following formulae:
5.1.Drying without preliminary dryingU.K.

where:
m
=
initial weight, in grammes, of the test sample,
m0
=
weight, in grammes, of the dry test sample.
5.2.Drying with preliminary dryingU.K.

where:
m
=
initial weight, in grammes, of the test sample,
m1
=
weight, in grammes, of the test sample after preliminary drying,
m2
=
weight, in grammes, of the test sample after crushing or grinding,
m0
=
weight, in grammes, of the dry test sample.
5.3.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample shall not exceed 0,2 % of the absolute value of moisture.
6.ObservationU.K.
If crushing proves necessary and if this is seen to alter the moisture content of the product, the results of the analysis of the components of the feed must be corrected on the basis of the moisture content of the sample in its initial state.
B.DETERMINATION OF MOISTURE IN ANIMAL AND VEGETABLE FATS AND OILSU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine the water and volatile substances content of animal and vegetable fats and oils.
2.PrincipleU.K.
The sample is dried to constant weight (loss in weight between two successive weighings must be less than or equal to 1 mg) at 103 oC. The loss in weight is determined by weighing.
3.ApparatusU.K.
3.1.Flat-bottomed dish, of a corrosion-resistant material, 8 to 9 cm in diameter and approximately 3 cm high.U.K.
3.2.Thermometer with a strengthened bulb and expansion tube at the top end, graduated from approximately 80 oC to at least 110 oC, and approximately 10 cm in length.U.K.
3.3.Sand bath or electric hot-plate.U.K.
3.4.Desiccator, containing an efficient drying agent.U.K.
3.5.Analytical balance.U.K.
4.ProcedureU.K.
Weigh out to the nearest mg approximately 20 g of the homogenised sample into the dry, weighed dish (3.1) containing the thermometer (3.2). Heat on the sand bath or hot-plate (3.3), stirring continuously with the thermometer, so that the temperature reaches 90 oC in about 7 minutes.
Reduce the heat, watching the frequency with which bubbles rise from the bottom of the dish. The temperature must not exceed 105 oC. Continue to stir, scraping the bottom of the dish, until bubbles stop forming.
In order to ensure complete elimination of moisture, reheat several times to 103 oC ± 2 oC, cooling to 93 oC between successive heatings. Then leave to cool to room temperature in the desiccator (3.4) and weigh. Repeat this operation until the loss in weight between two successive weighings no longer exceeds 2 mg.
N.B
:
An increase in the weight of the sample after repeated heating indicates an oxidation of the fat, in which case calculate the result from the weighing carried out immediately before the weight began to increase.
5.Calculation of resultsU.K.
The moisture content (X), as a percentage of the sample, is given by the following formula:

where:
m
=
weight, in grammes, of the test sample,
m1
=
weight, in grammes, of the dish with its contents before heating,
m2
=
weight, in grammes, of the dish with its contents after heating.
Results lower than 0,05 % must be recorded as ‘lower than 0,05 %’.
RepeatabilityU.K.
The difference in moisture between the results of two parallel determinations carried out on the same sample must not exceed 0,05 %, in absolute value.
C.DETERMINATION OF THE CONTENT OF CRUDE PROTEINU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine the crude protein content of feed on the basis of the nitrogen content, determined according to the Kjeldahl method.
2.PrincipleU.K.
The sample is digested by sulphuric acid in the presence of a catalyst. The acid solution is made alkaline with sodium hydroxide solution. The ammonia is distilled and collected in a measured quantity of sulphuric acid, the excess of which is titrated with a standard solution of sodium hydroxide.
Alternatively, the liberated ammonia is distilled into an excess of boric acid solution, followed by titration with hydrochloric acid or sulphuric acid solution.
3.ReagentsU.K.
3.1.Potassium sulphate.U.K.
3.2.Catalyst: copper (II) oxide CuO or copper (II) sulphate pentahydrate, CuSO4 5H2O.U.K.
3.3.Granulated zinc.U.K.
3.4.Sulphuric acid, ρ20 = 1,84 g/ml.U.K.
3.5.Sulphuric acid, standard volumetric solution, c(H2SO4) = 0,25 mol/l.U.K.
3.6.Sulphuric acid, standard volumetric solution, c(H2SO4) = 0,1 mol/l.U.K.
3.7.Sulphuric acid, standard volumetric solution, c(H2SO4) = 0,05 mol/l.U.K.
3.8.Methyl red indicator; dissolve 300 mg of methyl red in 100 ml of ethanol, σ = 95 %-96 % (v/v).U.K.
3.9.Sodium hydroxide solution (Technical grade may be used) β = 40 g/100 ml (m/v: 40 %).U.K.
3.10.Sodium hydroxide, standard volumetric solution c(NaOH) = 0,25 mol/l.U.K.
3.11.Sodium hydroxide, standard volumetric solution c(NaOH) = 0,1 mol/l.U.K.
3.12.Granulated pumice stone, washed in hydrochloric acid and ignited.U.K.
3.13.Acetanilide (m.p. = 114 oC, N-content = 10,36 %).U.K.
3.14.Sucrose (nitrogen free).U.K.
3.15.Boric acid (H3BO3).U.K.
3.16.Methyl red indicator solution: dissolve 100 mg methyl red in 100 ml ethanol or methanol.U.K.
3.17.Bromocresol green solution: dissolve 100 mg bromocresol green in 100 ml ethanol or methanol.U.K.
3.18.Boric acid solution (10 g/l to 40 g/l depending on the apparatus used).U.K.
When colorimetric end-point detection is applied, methyl red and bromocresol indicators must be added to the boric acid solutions. If 1 litre of the boric acid solution is prepared, before adjusting to volume, 7 ml methyl red indicator solution (3.16) and 10 ml bromocresol green solution (3.17) shall be added.
Dependent on the water used, the pH of the boric acid solution might differ from batch to batch. Often an adjustment with a small volume of alkali is necessary to obtain a positive blank.
Note
:
The addition of about 3 ml to 4 ml of NaOH (3.11) into 1 litre of 10 g/l boric acid usually gives good adjustments. Store the solution at room temperature and protect the solution from light and sources of ammonia fumes during storage.
3.19.Hydrochloric acid standard volumetric solution c(HCl) = 0,1 mol/l.U.K.
Note:
Other concentrations of volumetric solutions (3.5, 3.6, 3.7, 3.10, 3.11, and 3.19) can be used, if this is corrected for in the calculations. The concentrations shall always be expressed to four decimal places.
4.ApparatusU.K.
Apparatus suitable for performing digestion, distillation and titration according to the Kjeldahl procedure.
5.ProcedureU.K.
5.1.DigestionU.K.
Weigh 1 g of the sample to the nearest 0,001 g and transfer the sample to the flask of the digestion apparatus. Add 15 g of potassium sulphate (3.1), an appropriate quantity of catalyst (3.2) (0,3 to 0,4 g of copper (II) oxide or 0,9 to 1,2 g of copper (II) sulphate pentahydrate), 25 ml of sulphuric acid (3.4) and if required, a few granules of pumice stone (3.12) and mix.
Heat the flask moderately at first, swirling from time to time if necessary until the mass has carbonised and the foam has disappeared; then heat more intensively until the liquid is boiling steadily. Heating is adequate if the boiling acid condenses on the wall of the flask. Prevent the sides from becoming overheated and organic particles from sticking to them.
When the solution becomes clear and light green continue to boil for another two hours, then leave to cool.
5.2.DistillationU.K.
Add carefully enough water to ensure complete dissolution of the sulphates. Allow to cool and then add a few granules of zinc (3.3), if required. Proceed according to 5.2.1 or 5.2.2.
5.2.1.Distillation into sulphuric acidU.K.
Place in the collecting flask of the distillation apparatus an exactly measured quantity of 25 ml of sulphuric acid (3.5) or (3.7) depending on the presumed nitrogen content. Add a few drops of methyl red indicator (3.8).
Connect the digestion flask to the condenser of the distillation apparatus and immerse the end of the condenser in the liquid contained in the collecting flask to a depth of at least 1 cm (see observation 8.3). Slowly pour 100 ml of sodium hydroxide solution (3.9) into the digestion flask without loss of ammonia (see observation 8.1). Heat the flask until the ammonia has distilled over.
5.2.2.Distillation into boric acidU.K.
Where titration of the ammonia content of the distillate is performed manually, the procedure mentioned below applies. Where the distillation unit is fully automated to include titration of the ammonia content of the distillate, follow the manufacturer's instructions for operation of the distillation unit.
Place a collecting flask containing 25 ml to 30 ml of the boric acid solution (3.18) under the outlet of the condenser in such a way that the delivery tube is below the surface of the excess boric acid solution. Adjust the distillation unit to dispense 50 ml of sodium hydroxide solution (3.9). Operate the distillation unit in accordance with the manufacturer's instructions and distil off the ammonia liberated by the addition of the sodium hydroxide solution. Collect distillate in the boric acid receiving solution. The amount of distillate (time of steam distillation) depends on the amount of nitrogen in the sample. Follow the instructions of the manufacturer.
Note:
In a semi-automatic distillation unit, the addition of excess sodium hydroxide and the steam distillation are performed automatically.
5.3.TitrationU.K.
Proceed according to 5.3.1 or 5.3.2.
5.3.1.Sulphuric acidU.K.
Titrate the excess sulphuric acid in the collecting flask with sodium hydroxide solution (3.10 or 3.11) depending on the concentration of the sulphuric acid used, until the end-point is reached.
5.3.2.Boric acidU.K.
Titrate the contents of the collecting flask with the hydrochloric acid standard volumetric solution (3.19) or with the sulphuric acid standard volumetric solution (3.6) using a burette and read the amount of titrant used.
When colorimetric end-point detection is applied, the end-point is reached at the first trace of pink colour in the contents. Estimate the burette reading to the nearest 0,05 ml. An illuminated magnetic stirrer plate or a photometric detector may aid visualisation of the end-point.
This can be done automatically using a steam distiller with automatic titration.
Follow the manufacturers' instructions for operation of the specific distiller or distiller/titrator.
Note
:
When an automatic titration system is used, titration begins immediately after distillation starts and the 1 % boric acid solution (3.18) is used.
Where a fully automatic distillation unit is employed, the automatic titration of the ammonia can also be carried out with end-point detection using a potentiometric pH system.
In this case an automatic titrator, with a pH-meter is used. The pH-meter shall be calibrated properly in the range of pH 4 to pH 7 following normal laboratory pH-calibration procedures.
The pH end-point of the titration is reached at pH 4,6, being the steepest point in the titration curve (inflection point).
5.4.Blank testU.K.
To confirm that the reagents are free from nitrogen, carry out a blank test (digestion, distillation and titration) using 1 g of sucrose (3.14) in place of the sample.
6.Calculation of resultsU.K.
Calculations are performed according to 6.1 or 6.2.
6.1.Calculation for titration according to 5.3.1U.K.
The content of crude protein, expressed as a percentage by weight, is calculated according to the following formula:

where:
Vo
=
is the volume (ml) of NaOH (3.10 or 3.11) used in the blank test,
V1
=
is the volume (ml) of NaOH (3.10 or 3.11) used in the sample titration,
c
=
is the concentration (mol/l) of sodium hydroxide (3.10 or 3.11),
m
=
is the weight (g) of sample.
6.2.Calculation for titration according to 5.3.2U.K.
6.2.1.Titration with hydrochloric acidU.K.
The content of crude protein, expressed as a percentage by weight, is calculated according to the following formula:

where:
m
=
is the weight (g) of the test portion,
c
=
is the concentration (mol/l) of the standard volumetric solution of the hydrochloric acid (3.19),
V0
=
is the volume (in ml) of hydrochloric acid used for the blank test,
V1
=
is the volume (in ml) of hydrochloric acid used for the test portion.
6.2.2.Titration with sulphuric acidU.K.
The content of crude protein, expressed as a percentage by weight, is calculated according to the following formula:

where:
m
=
is the weight (g) of the test portion,
c
=
is the concentration (mol/l) of the standard volumetric solution of sulphuric acid (3.6),
V0
=
is the volume (in ml) of sulphuric acid (3.6) used for the blank test,
V1
=
is the volume (in ml) of sulphuric acid (3.6) used for test portion.
7.Verification of the methodU.K.
7.1.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed:
0,2 % in absolute value, for crude protein contents of less than 20 %,
1,0 % relative to the higher value, for crude protein contents from 20 % to 40 %,
0,4 % in absolute value, for crude protein contents of more than 40 %.
7.2.AccuracyU.K.
Carry out the analysis (digestion, distillation and titration) on 1,5 to 2,0 g of acetanilide (3.13) in the presence of 1 g of sucrose (3.14); 1 g acetanilide consumes 14,8 ml of sulphuric acid (3.5). Recovery must be at least 99 %.
8.ObservationsU.K.
8.1.Apparatus may be of the manual, semi-automatic or automatic type. If the apparatus requires transference between the digestion and distillation steps, this transfer must be carried out without loss. If the flask of the distillation apparatus is not fitted with a dropping funnel, add the sodium hydroxide immediately before connecting the flask to the condenser, pouring the liquid slowly down the side.U.K.
8.2.If the digest solidifies, recommence the determination using a larger amount of sulphuric acid (3.4) than that specified above.U.K.
8.3.For products with a low nitrogen content, the volume of sulphuric acid (3.7) to be placed in the collecting flask may be reduced, if necessary, to 10 or 15 ml and made up to 25 ml with water.U.K.
8.4.For routine analysis, alternative methods of analysis can be applied for the determination of crude protein but the Kjeldahl method described in this Part C is the reference method. The equivalence of the results obtained with the alternative method (e.g. DUMAS) compared to the reference method must be demonstrated for each matrix individually. As the results obtained with an alternative method, even after having verified the equivalency, might deviate slightly from the results obtained with the reference method, it is necessary to mention in the analytical report the method of analysis used for the determination of crude protein.U.K.
D.DETERMINATION OF UREAU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine the level of urea in feed.
2.PrincipleU.K.
The sample is suspended in water with a clarifying agent. The suspension is filtered. The urea content of the filtrate is determined after the addition of 4-dimethylaminobenzaldehyde (4-DMAB) by measuring the optical density at a wavelength of 420 nm.
3.ReagentsU.K.
3.1.Solution of 4-dimethylaminobenzaldehyde: dissolve 1,6 g of 4-DMAB in 100 ml of 96 % ethanol and add 10 ml of hydrochloric acid (ρ201,19 g/ml). This reagent keeps for a maximum period of two weeks.U.K.
3.2.Carrez solution I: dissolve in water 21,9 g of zinc acetate, Zn(CH3COO)2 2H2O and 3 g of glacial acetic acid. Make up to 100 ml with water.U.K.
3.3.Carrez solution II: dissolve in water 10,6 g of potassium ferrocyanide, K4 Fe (CN)6 3H2O. Make up to 100 ml with water.U.K.
3.4.Active carbon which does not absorb urea (to be checked).U.K.
3.5.Urea, 0,1 % solution (w/v).U.K.
4.ApparatusU.K.
4.1.Mixer (tumbler): approximately 35 to 40 r.p.m.U.K.
4.2.Test tubes: 160 × 16 mm with ground-glass stoppers.U.K.
4.3.Spectrophotometer.U.K.
5.ProcedureU.K.
5.1.Analysis of sampleU.K.
Weigh out 2 g of the sample to the nearest mg and place with 1 g of active carbon (3.4) in a 500 ml volumetric flask. Add 400 ml of water and 5 ml of Carrez solution I (3.2), mix for approximately 30 seconds and add 5 ml of Carrez solution II (3.3). Mix for 30 minutes in the tumbler. Make up to volume with water, shake and filter.
Remove 5 ml of the transparent colourless filtrates, place in test tubes with ground-glass stoppers, add 5 ml of 4-DMAB solution (3.1) and mix. Place the tubes in a water bath at 20 oC (+/- 4 oC). After 15 minutes measure the optical density of the sample solution with the spectrophotometer at 420 nm. Compare with the blank test solution of the reagents.
5.2.Calibration curveU.K.
Remove volumes of 1, 2, 4, 5 and 10 ml of the urea solution (3.5), place in 100 ml volumetric flasks and make up the volume with water. Remove 5 ml from each solution, add 5 ml of 4-DMAB solution (3.1) to each of them, homogenise and measure the optical density as shown above in comparison with a control solution containing 5 ml of 4-DMAB and 5 ml of water free from urea. Plot the calibration curve.
6.Calculation of resultsU.K.
Determine the amount of urea in the sample using the calibration curve.
Express the result as a percentage of the sample.
7.ObservationsU.K.
7.1.In the case of contents of urea exceeding 3 %, reduce the sample to 1 g or dilute the original solution so that there are not more than 50 mg of urea in 500 ml.U.K.
7.2.In the case of low contents of urea, increase the sample as long as the filtrate remains transparent and colourless.U.K.
7.3.If the sample contains simple nitrogenous compounds such as amino acids, the optical density shall be measured at 435 nm.U.K.
E.DETERMINATION OF VOLATILE NITROGENOUS BASESU.K.
I.BY MICRODIFFUSIONU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine the content of volatile nitrogenous bases, expressed as ammonia, in feed.
2.PrincipleU.K.
The sample is extracted with water and the solution clarified and filtered. The volatile nitrogenous bases are displaced by microdiffusion using a solution of potassium carbonate, collected in a solution of boric acid and titrated with sulphuric acid.
3.ReagentsU.K.
3.1.Trichloroacetic acid, solution 20 % (w/v).U.K.
3.2.Indicator: dissolve 33 mg of bromocresol green and 65 mg of methyl red in 100 ml of 95 %-96 % (v/v) of ethanol.U.K.
3.3.Boric acid solution: in a 1 litre graduated flask dissolve 10 g of boric acid in 200 ml of 95 %-96 % (v/v) ethanol and 700 ml of water. Add 10 ml of indicator (3.2). Mix and, if necessary, adjust the colour of the solution to light red by adding a solution of sodium hydroxide. 1 ml of this solution will fix a maximum of 300 μg of NH3.U.K.
3.4.Saturated potassium carbonate solution: dissolve 100 g of potassium carbonate in 100 ml of boiling water. Leave to cool, filter.U.K.
3.5.Sulphuric acid 0,01 mol/litre.U.K.
4.ApparatusU.K.
4.1.Mixer (tumbler): approximately 35 to 40 r.p.m.U.K.
4.2.Glass or plastic Conway cells (see diagram).U.K.
4.3.Microburettes graduated in 1/100 ml.U.K.
5.ProcedureU.K.
Weigh 10 g of sample to the nearest 1 mg and place with 100 ml of water in a 200 ml graduated flask. Mix or stir in the tumbler for 30 minutes. Add 50 ml of trichloroacetic acid solution (3.1), make up to volume with water, shake vigorously and filter through a pleated filter.
Using a pipette, introduce 1 ml of boric acid solution (3.3) into the central part of the Conway cell and 1 ml of the sample filtrate into the crown of the cell. Cover partially with the greased lid. Drop 1 ml of saturated potassium carbonate solution (3.4) quickly into the crown and close the lid so that the cell is airtight. Turn the cell carefully rotating it in a horizontal plane so that the two reagents are mixed. Leave to incubate either for at least four hours at room temperature or for one hour at 40 oC.
Using a microburette (4.3), titrate the volatile bases in the boric acid solution with sulphuric acid (3.5).
Carry out a blank test using the same procedure but without a sample to be analysed.
6.Calculation of resultsU.K.
1 ml of H2SO40,01 mol/litre corresponds to 0,34 mg of ammonia.
Express the result as a percentage of the sample.
RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample shall not exceed:
10 %, in relative value, for ammonia contents of less than 1,0 %,
0,1 %, in absolute value, for ammonia contents of 1,0 % or more.
7.ObservationU.K.
If the ammonia content of the sample exceeds 0,6 %, dilute the initial filtrate.
CONWAY CELLScale 1/1U.K.

II.BY DISTILLATIONU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the content of volatile nitrogenous bases, expressed as ammonia, in fish-meal containing practically no urea. It is applicable only to ammonia contents of less than 0,25 %.
2.PrincipleU.K.
The sample is extracted with water and the solution clarified and filtered. The volatile nitrogenous bases are displaced at boiling point by adding magnesium oxide and collected in a specific quantity of sulphuric acid, the excess of which is back-titrated with a solution of sodium hydroxide.
3.ReagentsU.K.
3.1.Trichloroacetic acid, solution 20 % (w/v).U.K.
3.2.Magnesium oxide.U.K.
3.3.Anti-foaming emulsion (e.g. silicone).U.K.
3.4.Sulphuric acid 0,05 mol/litre.U.K.
3.5.Sodium hydroxide solution 0,1 mol/litre.U.K.
3.6.Methyl red solution 0,3 % in 95 %-96 % (v/v) ethanol.U.K.
4.ApparatusU.K.
4.1.Mixer (tumbler): approximately 35 to 40 r.p.m.U.K.
4.2.Distilling apparatus of the Kjeldahl type.U.K.
5.ProcedureU.K.
Weigh 10 g of the sample to the nearest 1 mg and place with 100 ml of water in a 200 ml graduated flask. Mix or stir in the tumbler for 30 minutes. Add 50 ml of trichloroacetic acid solution (3.1), make up to volume with water, shake vigorously and filter through a pleated filter.
Take a quantity of clear filtrate appropriate for the presumed content of volatile nitrogenous bases (100 ml is usually suitable). Dilute to 200 ml and add 2 g of magnesium oxide (3.2) and a few drops of anti-foaming emulsion (3.3). The solution must be alkaline to litmus paper; otherwise add some magnesium oxide (3.2). Proceed according to 5.2 and 5.3 of the method of analysis for the determination of the crude protein content (Part C of this Annex).
Carry out a blank test using the same procedure but without a sample to be analysed.
6.Calculation of resultsU.K.
1 ml of H2SO40,05 mol/litre corresponds to 1,7 mg of ammonia.
Express the result as a percentage of the sample.
RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample shall not exceed, in relative value, 10 % of ammonia.
F.DETERMINATION OF AMINO ACIDS (EXCEPT TRYPTOPHANE)U.K.
1.Purpose and scopeU.K.
This method makes the determination possible of free (synthetic and natural) and total (peptide bound and free) amino acids in feed, using an amino acid analyser. It is applicable to the following amino acids: cyst(e)ine, methionine, lysine, threonine, alanine, arginine, aspartic acid, glutamic acid, glycine, histidine, isoleucine, leucine, phenylalanine, proline, serine, tyrosine and valine.
The method does not distinguish between the salts of amino acids and it cannot differentiate between D and L forms of amino acids. It is not valid for the determination of tryptophan or hydroxy analogues of amino acids.
2.PrincipleU.K.
2.1.Free amino acidsU.K.
The free amino acids are extracted with diluted hydrochloric acid. Co-extracted nitrogenous macromolecules are precipitated with sulfosalicylic acid and removed by filtration. The filtered solution is adjusted to pH 2,2. The amino acids are separated by ion exchange chromatography and determined by reaction with ninhydrin with photometric detection at 570 nm.
2.2.Total amino acidsU.K.
The procedure chosen depends on the amino acids under investigation. Cyst(e)ine and methionine must be oxidised to cysteic acid and methionine sulphone respectively prior to hydrolysis. Tyrosine must be determined in hydrolysates of unoxidised samples. All the other amino acids listed in paragraph 1 can be determined in either the oxidised or unoxidised sample.
Oxidation is performed at 0 oC with a performic acid/phenol mixture. Excess oxidation reagent is decomposed with sodium disulphite. The oxidised or unoxidised sample is hydrolysed with hydrochloric acid (3.20) for 23 hours. The hydrolysate is adjusted to pH 2,2. The amino acids are separated by ion exchange chromatography and determined by reaction with ninhydrin using photometric detection at 570 nm (440 nm for proline).
3.ReagentsU.K.
Double distilled water or water of equivalent quality must be used (conductivity < 10 μS).
3.1.Hydrogen peroxide, w (w/w) = 30 %.U.K.
3.2.Formic acid, w (w/w) = 98 %-100 %.U.K.
3.3.Phenol.U.K.
3.4.Sodium disulphite.U.K.
3.5.Sodium hydroxide.U.K.
3.6.5-Sulfosalicylic acid dihydrate.U.K.
3.7.Hydrochloric acid, density approximately 1,18 g/ml.U.K.
3.8.tri-Sodium citrate dihydrate.U.K.
3.9.2,2'-Thiodiethanol (thiodiglycol).U.K.
3.10.Sodium chloride.U.K.
3.11.Ninhydrin.U.K.
3.12.Light petroleum, boiling range 40-60 oC.U.K.
3.13.Norleucine, or other compound suitable for use as internal standard.U.K.
3.14.Nitrogen gas (< 10 ppm oxygen).U.K.
3.15.1-Octanol.U.K.
3.16.Amino acids.U.K.
3.16.1.Standard substances listed under paragraph 1. Pure compounds containing no water of crystallisation. Dry under vacuum over P2O5 or H2SO4 for 1 week prior to use.U.K.
3.16.2.Cysteic acid.U.K.
3.16.3.Methionine sulphone.U.K.
3.17.Sodium hydroxide solution, c = 7,5 mol/l:U.K.
Dissolve 300 g NaOH (3.5) in water and make up to 1 litre.
3.18.Sodium hydroxide solution, c = 1 mol/l:U.K.
Dissolve 40 g NaOH (3.5) in water and make up to 1 litre.
3.19.Formic acid — phenol solution:U.K.
Mix 889 g formic acid (3.2) with 111 g water and add 4,73 g phenol (3.3).
3.20.Hydrolysis mixture, c = 6 mol HCl/l containing 1 g phenol/l:U.K.
Add 1 g phenol (3.3) to 492 ml HCl (3.7) and make up to 1 litre with water.
3.21.Extraction mixture, c = 0,1 mol HCl/l containing 2 % thiodiglycol: Take 8,2 ml HCl (3.7), dilute with approximately 900 ml water, add 20 ml thiodiglycol (3.9) and make up to 1 litre with water, (do not mix 3.7 and 3.9 directly).U.K.
3.22.5-Sulfosalicylic acid, ß = 6 %:U.K.
Dissolve 60 g 5-sulfosalicylic acid (3.6) in water and make up to 1 l with water.
3.23.Oxidation mixture (Performic acid — phenol):U.K.
Mix 0,5 ml hydrogen peroxide (3.1) with 4,5 ml formic acid-phenol solution (3.19) in a small beaker. Incubate at 20-30 oC for 1 hour in order to form performic acid, then cool on an ice-water bath (15 min.) before adding to the sample.
Caution: Avoid contact with skin and wear protective clothing.
3.24.Citrate buffer, c = 0,2 mol Na+/l, pH 2,2:U.K.
Dissolve 19,61 g sodium citrate (3.8), 5 ml thiodiglycol (3.9), 1 g phenol (3.3) and 16,5 ml HCl (3.7) in approximately 800 ml water. Adjust pH to 2,2. Make up to 1 litre with water.
3.25.Elution buffers, prepared according to conditions for the analyser used (4.9).U.K.
3.26.Ninhydrin reagent, prepared according to conditions for the analyser used (4.9).U.K.
3.27.Standard solutions of amino acids. These solutions shall be stored below 5 oC.U.K.
3.27.1.Stock standard solution of amino acids (3.16.1).U.K.
c = 2,5 μmol/ml of each in hydrochloric acid.
May be obtained commercially.
3.27.2.Stock standard solution of cysteic acid and methionine sulphone, c = 1,25 μmol/ml.U.K.
Dissolve 0,2115 g cysteic acid (3.16.2) and 0,2265 g methionine sulphone (3.16.3) in citrate buffer (3.24) in a 1 litre graduated flask and make up to mark with citrate buffer. Store below 5 oC for not more than 12 months. This solution is not used if the stock standard solution (3.27.1) contains cysteic acid and methionine sulphone.
3.27.3.Stock standard solution of the internal standard e.g. norleucine, c = 20 μmol/ml.U.K.
Dissolve 0,656 g norleucine (3.13) in citrate buffer (3.24) in a graduated flask and make up to 250 ml with citrate buffer. Store below 5 oC for no more than 6 months.
3.27.4.Calibration solution of standard amino acids for use with hydrolysates, c = 5 nmol/50 μl of cysteic acid and methionine sulphone and c = 10 nmol/50 μl of the other amino acids. Dissolve 2,2 g sodium chloride (3.10) in 100 ml beaker with 30 ml citrate buffer (3.24). Add 4,0 ml stock standard solution of amino acids (3.27.1), 4,0 ml stock standard solution of cysteic acid and methionine sulphone (3.27.2) and 0,5 ml stock standard solution of internal standard (3.27.3) if used. Adjust pH to 2,2 with sodium hydroxide (3.18).U.K.
Transfer quantitatively to a 50 ml graduated flask and make up to the mark with citrate buffer (3.24) and mix.
Store below 5 oC for not more than 3 months.
See also observation 9.1.
3.27.5.Calibration solution of standard amino acids for use with hydrolysates prepared according to paragraph 5.3.3.1 and for use with extracts (5.2). The calibration solution is prepared according to 3.27.4 but omitting sodium chloride.U.K.
Store below 5 oC for not more than 3 months.
4.ApparatusU.K.
4.1.100 or 250 ml round bottomed flask fitted with a reflux condenser.U.K.
4.2.100 ml borosilicate glass bottle with screw cap with rubber/teflon liner (e.g. Duran, Schott) for use in the oven.U.K.
4.3.Oven with forced ventilation and a temperature regulator with an accuracy better than ± 2 oC.U.K.
4.4.pH-meter (three decimal places).U.K.
4.5.Membrane filter (0,22 μm).U.K.
4.6.Centrifuge.U.K.
4.7.Rotary vacuum evaporator.U.K.
4.8.Mechanical shaker or magnetic stirrer.U.K.
4.9.Amino acid analyser or HPLC equipment with ion exchange column, device for ninhydrin, post column derivatisation and photometric detector.U.K.
The column is filled with sulfonated polystyrene resins capable of separating the amino acids from each other and from other ninhydrin-positive materials. The flow in the buffer and ninhydrin lines is provided by pumps having a flow stability of ±0,5 % in the period covering both the standard calibration run and the analysis of the sample.
With some amino acid analysers hydrolysis procedures can be used in which the hydrolysate has a sodium concentration of c = 0,8 mol/l and contains all the residual formic acid from the oxidation step. Others do not give a satisfactory separation of certain amino acids if the hydrolysate contains excess formic acid and/or high sodium ion concentrations. In this case the volume of acid is reduced by evaporation to approx. 5 ml after the hydrolysis and prior to pH adjustment. The evaporation shall be performed under vacuum at 40 o C maximum.
5.ProcedureU.K.
5.1.Preparation of the sampleU.K.
The sample is ground to pass through a 0,5 mm sieve. Samples high in moisture must be either air-dried at a temperature not exceeding 50 oC or freeze dried prior to grinding. Samples with a high fat content shall be extracted with light petroleum (3.12) prior to grinding.
5.2.Determination of free amino acids in feed and premixturesU.K.
Weigh to the nearest 0,2 mg an appropriate amount (1-5 g) of the prepared sample (5.1), into a conical flask and add 100,0 ml of extraction mixture (3.21). Shake the mixture for 60 min. using a mechanical shaker or a magnetic stirrer (4.8). Allow the sediment to settle and pipette 10,0 ml of the supernatant solution into a 100 ml beaker.
Add 5,0 ml of sulfosalicylic acid solution (3.22), with stirring and continue to stir with the aid of magnetic stirrer for 5 min. Filter or centrifuge the supernatant in order to remove any precipitate. Place 10,0 ml of the resulting solution into a 100 ml beaker and adjust the pH to 2,2 using sodium hydroxide solution (3.18), transfer to a volumetric flask of appropriate volume using citrate buffer (3.24), and make up to the mark with the buffer solution (3.24).
If an internal standard is being used add 1,0 ml of internal standard (3.27.3) for each 100 ml final solution and make up to the mark with the buffer solution (3.24).
Proceed to the chromatography step according to paragraph 5.4.
If the extracts are not being examined the same day, they must be stored below 5 oC.
5.3.Determination of total amino acidsU.K.
5.3.1.OxidationU.K.
Weigh to the nearest 0,2 mg from 0,1 to 1 g of the prepared sample (5.1) into:
a 100 ml round-bottomed flask (4.1) for open hydrolysis (5.3.2.3) or,
a 250 ml round-bottomed flask (4.1) if a low sodium concentration is required (5.3.3.1) or,
a 100 ml bottle fitted with a screw cap (4.2), for closed hydrolysis (5.3.2.4).
The weighed sample portion must have a nitrogen content of about 10 mg and a moisture content not exceeding 100 mg.
Place the flask/bottle in an ice-water bath and cool to 0 oC, add 5 ml of oxidation mixture (3.23) and mix using a glass spatula with a bent tip. Seal the flask/bottle containing the spatula with an air-tight film, place the ice-water bath containing the sealed container in a refrigerator at 0 oC and leave for 16 hours. After 16 hours remove from the refrigerator and decompose the excess oxidation reagent by the addition of 0,84 g of sodium disulphite (3.4).
Proceed to 5.3.2.1.
5.3.2.HydrolysisU.K.
5.3.2.1.Hydrolysis of oxidised samplesU.K.
To the oxidised sample prepared according to 5.3.1 add 25 ml of hydrolysis mixture (3.20) taking care to wash down any sample residue adhering to the sides of the vessel and the spatula.
Depending on the hydrolysis procedure being used, proceed according to 5.3.2.3 or 5.3.2.4.
5.3.2.2.Hydrolysis of unoxidised samplesU.K.
Weigh into either a 100 ml or a 250 ml round-bottom flask (4.1) or a 100 ml bottle fitted with a screw cap (4.2), to the nearest 0,2 mg, from 0,1 to 1 g of the prepared sample (5.1). The weighed sample portion must have a nitrogen content of about 10 mg. Add carefully 25 ml of hydrolysis mixture (3.20) and mix with the sample. Proceed according to either 5.3.2.3 or 5.3.2.4.
5.3.2.3.Open hydrolysisU.K.
Add 3 glass beads to the mixture in the flask (prepared in accordance with 5.3.2.1 or 5.3.2.2) and boil with continuous bubbling under reflux for 23 hours. On completion of hydrolysis, wash the condenser down with 5 ml of citrate buffer (3.24). Disconnect the flask and cool it in an ice bath.
Proceed according to 5.3.3.
5.3.2.4.Closed HydrolysisU.K.
Place the bottle containing the mixture prepared in accordance with 5.3.2.1 or 5.3.2.2 in an oven (4.3) at 110 oC. During the first hour in order to prevent a build up of pressure (due to the evolution of gaseous substances) and to avoid explosion, place the screw cap over the top of the vessel. Do not close the vessel with the cap. After one hour close the vessel with the cap and leave in the oven (4.3) for 23 hours. On completion of hydrolysis, remove the bottle from the oven, carefully open the cap of the bottle and place the bottle in an ice-water bath. Leave to cool.
Depending on the procedure for pH adjustment (5.3.3), quantitatively transfer the contents of the bottle to a 250 ml beaker or a 250 ml round-bottom flask, using citrate buffer (3.24).
Proceed according to 5.3.3.
5.3.3.Adjustment of pHU.K.
Depending on the sodium tolerance of the amino acid analyser (4.9) proceed according to 5.3.3.1 or 5.3.3.2 for the pH adjustment.
5.3.3.1.For Chromatographic Systems (4.9) requiring a low sodium concentrationU.K.
It is advisable to use an internal stock standard solution (3.27.3) when amino acid analysers requiring a low sodium concentration are employed (when the acid volume has to be reduced).
In this case add 2,0 ml of the internal stock standard solution (3.27.3) to the hydrolysate before the evaporation.
Add 2 drops of 1-octanol (3.15) to the hydrolysate obtained in accordance with paragraph 5.3.2.3 or 5.3.2.4.
Using a rotary evaporator (4.7) reduce the volume to 5-10 ml under vacuum at 40 oC. If the volume is accidentally reduced to less than 5 ml the hydrolysate must be discarded and the analysis recommenced.
Adjust the pH to 2,2 with sodium hydroxide solution (3.18) and proceed to paragraph 5.3.4.
5.3.3.2.For all other Amino Acid Analysers (4.9)U.K.
Take the hydrolysates obtained in accordance with 5.3.2.3 or 5.3.2.4 and partly neutralise them by carefully adding with stirring, 17 ml of sodium hydroxide solution (3.17), ensuring that the temperature is kept below 40 oC.
Adjust the pH to 2,2 at room temperature using sodium hydroxide solution (3.17) and finally sodium hydroxide solution (3.18). Proceed to 5.3.4.
5.3.4.Sample solution for chromatographyU.K.
Quantitatively transfer the pH adjusted hydrolysate (5.3.3.1 or 5.3.3.2) with citrate buffer (3.24) to a 200 ml graduated flask, and make up to the mark with buffer (3.24).
If an internal standard has not already been used, add 2,0 ml of internal standard (3.27.3) and make up to the mark with citrate buffer (3.24). Mix thoroughly.
Proceed to the chromatography step (5.4).
If the sample solutions are not being examined the same day they must be stored below 5 oC.
5.4.ChromatographyU.K.
Before chromatography bring the extract (5.2) or hydrolysate (5.3.4) to room temperature. Shake the mixture and filter a suitable amount through a 0,22 μm membrane filter (4.5). The resulting clear solution is subjected to ion exchange chromatography, using an amino acid analyser (4.9).
The injection may be performed manually or automatically. It is important that the same quantity of solution ± 0,5 % is added to the column for the analysis of standards and samples except when an internal standard is used, and that the sodium:amino acid ratios in the standard and sample solutions are as similar as is practicable.
In general the frequency of calibration runs depends on the stability of the ninhydrin reagent and the analytical system. The standard or sample is diluted with citrate buffer (3.24) to give a peak area of the standard of 30 %-200 % of the sample amino acid peak area.
The chromatography of amino acids will vary slightly according to the type of analyser employed and resin used. The chosen system must be capable of separating the amino acids from each other and from the ninhydrin-positive materials. In the range of operation the chromatographic system must give a linear response to changes in the amounts of amino acids added to the column.
During the chromatography step the valley:peak height ratios mentioned below apply, when an equimolar solution (of the amino acids being determined) is analysed. This equimolar solution must contain at least 30 % of the maximum load of each amino acid which can be accurately measured with the amino acid analyser system (4.9).
For separation of threonine-serine the valley:peak height ratio of the lower of the two overlapping amino acids on the chromatogram must not exceed 2:10. (if only cyst(e)ine, methionine, threonine and lysine are determined, insufficient separation from adjoining peaks will adversely influence the determination). For all other amino acids the separation must be better than 1:10.
The system must ensure that lysine is separated from ‘lysine artifacts’ and ornithine.
6.Calculation of resultsU.K.
The area of the sample and standard peaks is measured for each individual amino acid and the amount (X), in g amino acid per kg sample, is calculated as follows:

If an internal standard is used multiply by: 
A
=
peak area, hydrolysate or extract
B
=
peak area, calibration standard solution
C
=
peak area, internal standard in hydrolysate or extract
D
=
peak area, internal standard, calibration standard solution
M
=
molar weight of the amino acid being determined
c
=
concentration of standard in μmol/ml
m
=
sample weight (g) (corrected to original weight if dried or defatted)
V
=
ml total hydrolysate (5.3.4) or ml calculated total dilution volume of extract (6.1)
Cystine and cysteine are both determined as cysteic acid in hydrolysates of oxidised sample, but calculated as cystine (C6H12N2O4S2, M 240,3 g/mol) by using M 120,15 g/mol (= 0,5 x 240,3 g/mol).
Methionine is determined as methionine sulphone in hydrolysates of oxidised sample, but calculated as methionine by using M of methionine: 149,21 g/mol.
Added free methionine is determined after extraction as methionine, for the calculation the same M is used.
6.1.The total dilution volume of extracts (F) for determination of free amino acids (5.2) is calculated as follows:U.K.
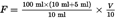
V
=
Volume of final extract
7.Evaluation of the methodU.K.
The method has been tested in an intercomparison made at international level in 1990 using four different feed (mixed pig feed, broiler compound, protein concentrate, premixture). The results, after elimination of outliers, of mean and standard deviation are given in the tables in this point:
Means in g/kg
n = Number of participating laboratories. | ||||
| Reference material | Amino Acid | |||
|---|---|---|---|---|
| Threonine | Cyst(e)ine | Methionine | Lysine | |
| Mixed Pig Feed | 6,94 n = 15 | 3,01 n = 17 | 3,27 n = 17 | 9,55 n = 13 |
| Broiler Compound | 9,31 n = 16 | 3,92 n = 18 | 5,08 n = 18 | 13,93 n = 16 |
| Protein Concentrate | 22,32 n = 16 | 5,06 n = 17 | 12,01 n = 17 | 47,74 n = 15 |
| Premixture | 58,42 N = 16 | — | 90,21 n = 16 | 98,03 n = 16 |
7.1.RepeatabilityU.K.
The repeatability expressed as ‘within laboratory standard deviation’ of the abovementioned intercomparison is given in the tables below:
Within Laboratory Standard Deviation (Sr) in g/kg
n = Number of participating laboratories. | ||||
| Reference material | Amino Acid | |||
|---|---|---|---|---|
| Threonine | Cyst(e)ine | Methionine | Lysine | |
| Mixed Pig Feed | 0,13 n = 15 | 0,1 n = 17 | 0,11 n = 17 | 0,26 n = 13 |
| Broiler Compound | 0,2 n = 16 | 0,11 n = 18 | 0,16 n = 18 | 0,28 n = 16 |
| Protein Concentrate | 0,48 n = 16 | 0,13 n = 17 | 0,27 n = 17 | 0,99 n = 15 |
| Premixture | 1,3 N = 16 | — | 2,19 n = 16 | 2,06 n = 16 |
Coefficient of Variation (%) for Within Laboratory Standard Deviation (Sr)
n = Number of participating laboratories. | ||||
| Reference material | Amino Acid | |||
|---|---|---|---|---|
| Threonine | Cyst(e)ine | Methionine | Lysine | |
| Mixed Pig Feed | 1,9 n = 15 | 3,3 n = 17 | 3,4 n = 17 | 2,8 n = 13 |
| Broiler Compound | 2,1 n = 16 | 2,8 n = 18 | 3,1 n = 18 | 2,1 n = 16 |
| Protein Concentrate | 2,7 n = 16 | 2,6 n = 17 | 2,2 n = 17 | 2,4 n = 15 |
| Premixture | 2,2 n = 16 | — | 2,4 n = 16 | 2,1 n = 16 |
7.2ReproducibilityU.K.
The results for between laboratory standard deviation by the abovementioned intercomparison are given in the table below:
Between Laboratory Standard Deviation (SR) in g/kg
n = Number of participating laboratories. | ||||
| Reference material | Amino Acid | |||
|---|---|---|---|---|
| Threonine | Cyst(e)ine | Methionine | Lysine | |
| Mixed Pig Feed | 0,28 n = 15 | 0,3 n = 17 | 0,23 n = 17 | 0,3 n = 13 |
| Broiler Compound | 0,48 n = 16 | 0,34 n = 18 | 0,55 n = 18 | 0,75 n = 16 |
| Protein Concentrate | 0,85 n = 16 | 0,62 n = 17 | 1,57 n = 17 | 1,24 n = 15 |
| Premixture | 2,49 n = 16 | — | 6,2 n = 16 | 6,62 n = 16 |
Coefficient of Variation (%) for Between Laboratory Standard Deviation (SR)
n = Number of participating laboratories. | ||||
| Reference material | Amino Acid | |||
|---|---|---|---|---|
| Threonine | Cyst(e)ine | Methionine | Lysine | |
| Mixed Pig Feed | 4,1 n = 15 | 9,9 n = 17 | 7,0 n = 17 | 3,2 n = 13 |
| Broiler Compound | 5,2 n = 16 | 8,8 n = 18 | 10,9 n = 18 | 5,4 n = 16 |
| Protein Concentrate | 3,8 n = 16 | 12,3 n = 17 | 13,0 n = 17 | 3,0 n = 15 |
| Premixture | 4,3 n = 16 | — | 6,9 n = 16 | 6,7 n = 16 |
8.Use of reference materialsU.K.
The correct application of the method shall be verified by making replicate measurements of certified reference materials when available. Calibration with certified amino acid calibration solution is recommended.
9.ObservationsU.K.
9.1.Because of differences between amino acid analysers the final concentrations of the calibration solutions of standard amino acids (see 3.27.4 and 3.27.5) and of the hydrolysate (see 5.3.4) shall be taken as a guideline.U.K.
The range of linear response of the apparatus has to be checked for all amino acids.
The standard solution is diluted with citrate buffer to give peak areas in the middle of the range.
9.2.Where high performance liquid chromatographic equipment is used to analyse the hydrolysates, the experimental conditions must be optimised in accordance with the manufacturer's recommendations.U.K.
9.3.By applying the method to feed containing more than 1 % chloride (concentrate, mineral feeds, supplementary feeds) underestimation of methionine could occur and special treatment has to be done.U.K.
G.DETERMINATION OF TRYPTOPHANU.K.
1.Purpose and scopeU.K.
The method makes the determination possible of the total and free tryptophan in feed. It does not distinguish between D- and L- forms.
2.PrincipleU.K.
For the determination of the total tryptophan, the sample is hydrolysed under alkaline conditions with saturated barium hydroxide solution and heated to 110 oC for 20 hours. After hydrolysis internal standard is added.
For the determination of free tryptophan, the sample is extracted under mild acidic conditions in the presence of internal standard.
The tryptophan and the internal standard in the hydrolysate or in the extract are determined by HPLC with fluorescence detection.
3.ReagentsU.K.
3.1.Double distilled water or water of equivalent quality must be used (conductivity < 10 μS/cm).U.K.
3.2.Standard substance: tryptophan (purity/content ≥ 99 %) dried under vacuum over phosphorous pentoxide.U.K.
3.3.Internal standard substance: α-methyl-tryptophan (purity/content ≥ 99 %), dried under vacuum over phosphorous pentoxide.U.K.
3.4.Barium hydroxide octa-hydrate (care shall be taken not to expose the Ba(OH)2 .8 H2O excessively to air in order to avoid formation of BaCO3, which could disturb the determination) (see observation 9.3).U.K.
3.5.Sodium hydroxide.U.K.
3.6.Ortho-phosphoric acid, w (w/w) = 85 %.U.K.
3.7.Hydrochloric acid, ρ201,19 g/ml.U.K.
3.8.Methanol, equivalent to HPLC grade.U.K.
3.9.Light petroleum, boiling range 40-60 oC.U.K.
3.10.Sodium hydroxide solution, c = 1 mol/l:U.K.
Dissolve 40,0 g NaOH (3.5) in water and make up to 1 litre with water (3.1).
3.11.Hydrochloric acid, c = 6 mol/l:U.K.
Take 492 ml HCl (3.7) and make up to 1 litre with water.
3.12.Hydrochloric acid, c = 1 mol/l:U.K.
Take 82 ml HCl (3.7) and make up to 1 litre with water.
3.13.Hydrochloric acid, c = 0,1 mol/l:U.K.
Take 8,2 ml HCl (3.7) and make up to 1 litre with water.
3.14.Ortho-phosphoric acid, c = 0,5 mol/l:U.K.
Take 34 ml ortho-phosphoric acid (3.6) and make up to 1 litre with water (3.1).
3.15.Concentrated solution of tryptophan (3.2), c = 2,5 μmol/ml:U.K.
In a 500 ml volumetric flask dissolve 0,2553 g tryptophan (3.2) in hydrochloric acid (3.13) and make up to the mark with hydrochloric acid (3.13). Store at - 18 oC for a maximum of 4 weeks.
3.16.Concentrated internal standard solution, c = 2,5 μmol/ml:U.K.
In a 500 ml volumetric flask dissolve 0,2728 g α-methyl-tryptophan (3.3) in hydrochloric acid (3.13) and make up to the mark with hydrochloric acid (3.13). Store at - 18 oC for a maximum of 4 weeks.
3.17.Calibration standard solution of tryptophan and internal standard:U.K.
Take 2,0 ml concentrated solution of tryptophan (3.15), and 2,0 ml of concentrated internal standard (α-methyl-tryptophan) solution (3.16). Dilute with water (3.1) and methanol (3.8) to approximately the same volume and to approximately the same concentration of methanol (10 %-30 %) as the finished hydrolysate.
This solution must be prepared freshly before use.
Protect from direct sunlight during preparation.
3.18.Acetic acidU.K.
3.19.1,1,1-trichloro-2-methyl-2-propanol.U.K.
3.20.Ethanolamine w (w/w) > 98 %.U.K.
3.21.Solution of 1 g 1,1,1-trichloro-2-methyl-2-propanol (3.19) in 100 ml methanol (3.8).U.K.
3.22.Mobile phase for HPLC: 3,0 g acetic acid (3.18) + 900 ml water (3.1) +50,0 ml solution (3.21) of 1,1,1-trichloro-2-methyl-2-propanol (3.19) in methanol (3.8) (1g/100ml). Adjust pH to 5,0 using ethanolamine (3.20). Make up to 1 000 ml with water (3.1).U.K.
4.ApparatusU.K.
4.1.HPLC equipment with a spectrofluorometric detector.U.K.
4.2.Liquid chromatographic column, 125 mm x 4 mm, C18, 3 μm packing, or equivalent.U.K.
4.3.pH-meter.U.K.
4.4.Polypropylene flask, capacity 125 ml, with wide neck and screw cap.U.K.
4.5.Membrane filter, 0,45 μm.U.K.
4.6.Autoclave, 110 (± 2) oC, 1,4 (±0,1) bar.U.K.
4.7.Mechanical shaker or magnetic stirrer.U.K.
4.8.Vortex mixer.U.K.
5.ProcedureU.K.
5.1.Preparation of samplesU.K.
The sample is ground to pass through a 0,5 mm sieve. Samples high in moisture must be either air-dried at a temperature not exceeding 50 oC or freeze dried prior to grinding. Samples with high fat content shall be extracted with light petroleum (3.9) prior to grinding.
5.2.Determination of free tryptophan (extract)U.K.
Weigh to the nearest 1 mg an appropriate amount (1-5 g) of the prepared sample (5.1), into a conical flask. Add 100,0 ml hydrochloric acid, (3.13) and 5,0 ml concentrated internal standard solution (3.16). Shake or mix for 60 min. using a mechanical shaker or a magnetic stirrer (4.7). Allow the sediment to settle and pipette 10,0 ml of the supernatant solution into a beaker. Add 5 ml ortho-phosphoric acid (3.14). Adjust the pH to 3 using sodium hydroxide (3.10). Add sufficient methanol (3.8) to give a concentration of between 10 % and 30 % of methanol in the final volume. Transfer to a volumetric flask of appropriate volume and dilute with water to a volume necessary for the chromatography (approx. the same volume as the calibration standard solution (3.17)).
Filter a few ml of the solution through a 0,45 μm membrane filter (4.5) before injection on the HPLC column. Proceed to the chromatography step according to paragraph 5.4.
Protect standard solution and extracts against direct sunlight. If it is not possible to analyse the extracts the same day, the extracts may be stored at 5 oC for a maximum of 3 days.
5.3.Determination of total tryptophan (hydrolysate)U.K.
Weigh to the nearest 0,2 mg from 0,1 to 1 g of the prepared sample (5.1) into the polypropylene flask (4.4). The weighed sample portion shall have a nitrogen content of about 10 mg. Add 8,4 g barium hydroxide octa-hydrate (3.4) and 10 ml water. Mix on a vortex mixer (4.8) or magnetic stirrer (4.7). Leave the teflon coated magnet in the mixture. Wash down the walls of the vessel with 4 ml water. Put on the screw cap and close the flask loosely. Transfer to an autoclave (4.6) with boiling water and steam for 30-60 minutes. Close the autoclave and autoclave at 110 (± 2) oC for 20 hours.
Before opening the autoclave reduce the temperature to just under 100 oC. In order to avoid crystallisation of Ba(OH)2 · 8 H2O, add to the warm mixture 30 ml water which is at room temperature. Shake or stir gently. Add 2,0 ml concentrated internal standard (α-methyl-tryptophan) solution (3.16). Cool the vessels on water/ice bath for 15 minutes.
Then, add 5 ml ortho-phosphoric acid (3.14). Keep the vessel in the cooling bath and neutralise with HCl (3.11) whilst stirring and adjust the pH to 3,0 using HCl (3.12). Add sufficient methanol to give a concentration of between 10 % and 30 % of methanol in the final volume. Transfer to a volumetric flask of appropriate volume and dilute with water to the defined volume necessary for the chromatography (for example 100 ml). The addition of methanol shall not cause precipitation.
Filter a few ml of the solution through a 0,45 μm membrane filter (4.5) before injection on the HPLC column. Proceed to the chromatography step according to paragraph 5.4.
Protect standard solution and hydrolysates against direct sunlight. If it is not possible to analyse the hydrolysates the same day, they may be stored at 5 oC for a maximum of 3 days.
5.4.HPLC determinationU.K.
The following conditions for isocratic elution are offered for guidance; other conditions may be used, provided they yield equivalent results (see also observations 9.1 and 9.2):
| Liquid chromatographic column (4.2): | 125 mm x 4 mm, C18, 3 μm packing or equivalent |
| Column temperature: | Room temperature |
| Mobile phase (3.22): | 3,0 g acetic acid (3.18) + 900 ml water (3.1) +50,0 ml solution (3.21) of 1,1,1-trichloro-2- methyl-2-propanol (3.19) in methanol (3.8) (1 g/100 ml). Adjust pH to 5,0 using ethanolamine (3.20). Make up to 1 000 ml with water (3.1) |
| Flow rate: | 1 ml/min. |
| Total run time: | approx. 34 min. |
| Detection wavelength: | excitation: 280 nm, emission: 356 nm. |
| Injection volume | 20 μl |
6.Calculation of resultsU.K.
The amount of tryptophane (X), in g per 100g sample, is calculated as follows:

A
=
peak area of internal standard, calibration standard solution (3.17)
B
=
peak area of tryptophan, extract (5.2) or hydrolysate (5.3)
V1
=
volume in ml (2 ml) of concentrated tryptophan solution (3.15) added to the calibration solution (3.17)
c
=
concentration in μmol/ml (= 2,5) of concentrated tryptophan solution (3.15) added to calibration solution (3.17)
V2
=
volume in ml of concentrated internal standard solution (3.16) added at the extraction (5.2) (= 5,0 ml) or to the hydrolysate (5.3) (= 2,0 ml)
C
=
peak area of internal standard, extract (5.2) or hydrolysate (5.3)
D
=
peak area of tryptophan, calibration standard solution (3.17)
V3
=
volume in ml (= 2,0 ml) of concentrated internal standard solution (3.16) added to calibration standard solution (3.17)
m
=
sample weight in g (corrected to original weight if dried and/or defatted)
M
=
molar weight of tryptophan (= 204,23 g/mol)
7.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed 10 % relative to the highest result.
8.Results of a collaborative studyU.K.
An EC collaborative study (4th intercomparison) was arranged in which three samples were analysed by up to 12 laboratories to certify the method for hydrolysis. Replicate (5) analyses were performed on each sample. The results are given in the following table:
| Sample 1Pig feed | Sample 2Pig feed supplemented with L-tryptophan | Sample 3Feed concentrate for pigs | |
|---|---|---|---|
| L | 12 | 12 | 12 |
| n | 50 | 55 | 50 |
| Mean [g/kg] | 2,42 | 3,4 | 4,22 |
| sr [g/kg] | 0,05 | 0,05 | 0,08 |
| r [g/kg] | 0,14 | 0,14 | 0,22 |
| CVr [%] | 1,9 | 1,6 | 1,9 |
| SR [g/kg] | 0,15 | 0,2 | 0,09 |
| R [g/kg] | 0,42 | 0,56 | 0,25 |
| CVR [%] | 6,3 | 6,0 | 2,2 |
L
=
number of laboratories submitting results
n
=
number of single results retained eliminating outliers (identified by Cochran, Dixon outlier test)
sr
=
standard deviation of repeatability
SR
=
standard deviation of reproducibility
r
=
repeatability
R
=
reproducibility
CVr
=
coefficient of variation of repeatability, %
CVR
=
coefficient of variation of reproducibility, %
Another EC collaborative study (3rd intercomparison) was arranged in which two samples were analysed by up to 13 laboratories to certify the method for extraction of free tryptophan. Replicate (5) analyses were performed on each sample. The results are given in the following table:
| Sample 4Wheat and soya mixture | Sample 5Wheat and soya mixture (= sample 4) with added tryptophan (0,457g/kg1) | |
|---|---|---|
| L | 12 | 12 |
| n | 55 | 60 |
| Mean [g/kg] | 0,391 | 0,931 |
| sr [g/kg] | 0,005 | 0,012 |
| r [g/kg] | 0,014 | 0,034 |
| CVr [%] | 1,34 | 1,34 |
| SR [g/kg] | 0,018 | 0,048 |
| R [g/kg] | 0,05 | 0,134 |
| CVR [%] | 4,71 | 5,11 |
L
=
number of laboratories submitting results
n
=
number of single results retained after eliminating outliers (identified by Cochran, Dixon outlier test)
sr
=
standard deviation of repeatability
SR
=
standard deviation of reproducibility
r
=
repeatability
R
=
reproducibility
CVr
=
coefficient of variation of repeatability, %
CVR
=
coefficient of variation of reproducibility, %
Another EC intercomparison study was arranged in which four samples were analysed by up to 7 laboratories with the aim of a tryptophan certification for hydrolysis. The results are given below. Replicate (5) analyses were performed on each sample.
| Sample 1Mixed pig feed(CRM 117) | Sample 2Low fat fish meal(CRM 118) | Sample 3Soybean meal(CRM 119) | Sample 4Skimmed milk powder(CRM 120) | |
|---|---|---|---|---|
| L | 7 | 7 | 7 | 7 |
| n | 25 | 30 | 30 | 30 |
| Mean [g/kg] | 2,064 | 8,801 | 6,882 | 5,236 |
| sr [g/kg] | 0,021 | 0,101 | 0,089 | 0,04 |
| r [g/kg] | 0,059 | 0,283 | 0,249 | 0,112 |
| CVr [%] | 1,04 | 1,15 | 1,3 | 0,76 |
| SR [g/kg] | 0,031 | 0,413 | 0,283 | 0,221 |
| R [g/kg] | 0,087 | 1,156 | 0,792 | 0,619 |
| CVR [%] | 1,48 | 4,69 | 4,11 | 4,22 |
L
=
number of laboratories submitting results
n
=
number of single results retained after eliminating outliers (identified by Cochran, Dixon outlier test)
sr
=
standard deviation of repeatability
SR
=
standard deviation of reproducibility
r
=
repeatability
R
=
reproducibility
CVr
=
coefficient of variation of repeatability, %
CVR
=
coefficient of variation of reproducibility, %
9.ObservationsU.K.
9.1.Following special chromatographic conditions may give better separation between tryptophan and α-methyl-tryptophan.U.K.
Isocratic elution followed by gradient column cleaning:
| Liquid chromatographic column: | 125 mm x 4 mm, C18, 5 μm packing or equivalent | ||
| Column temperature: | 32 oC | ||
| Mobile phase: | A: 0,01 mol/l KH2PO4/méthanol, 95+5 (V+V). B: methanol | ||
| Gradient program: | 0 min. | 100 % A | 0 % B |
| 15 min. | 100 % A | 0 % B | |
| 17 min. | 60 % A | 40 % B | |
| 19 min. | 60 % A | 40 % B | |
| 21 min. | 100 % A | 0 % B | |
| 33 min. | 100 % A | 0 % B | |
| Flow rate: | 1,2 ml/min. | ||
| Total run time: | approx. 33 min. | ||
9.2.The chromatography will vary according to the type of HPLC and column packing material used. The chosen system must be capable of giving baseline separation between the tryptophan and the internal standard. Moreover it is important that degradation products are well separated from the tryptophan and the internal standard. Hydrolysates without internal standard shall be run in order to check the base line under the internal standard for impurities. It is important that the run time is sufficiently long for the elution of all the degradation products, otherwise late eluting peaks may interfere with subsequent chromatographic runs.U.K.
In the range of operation, the chromatographic system shall give linear response. The linear response shall be measured with a constant (the normal) concentration of the internal standard and varying concentrations of tryptophan. It is of importance that the size of both the tryptophan and internal standard peaks are within the linear range of the HPLC/fluorescence system. If either the tryptophan and/or the internal standard peak(s) is (are) too small or too high the analysis shall be repeated with another sample size and/or a changed final volume.
9.3.Barium hydroxideU.K.
With age barium hydroxide becomes more difficult to dissolve. This results in an unclear solution for the HPLC determination, which may produce low results for tryptophan.
H.DETERMINATION OF CRUDE OILS AND FATSU.K.
1.Purpose and scopeU.K.
This method is for the determination of crude oils and fats in feed. It does not cover the analysis of oil seeds and oleaginous fruit.
The use of the two procedures described below depends on the nature and composition of the feed and the reason for carrying out the analysis.
1.1.Procedure A — Directly extractable crude oils and fatsU.K.
This method is applicable to feed materials of plant origin, except those included within the scope of Procedure B.
1.2.Procedure B — Total crude oils and fatsU.K.
This method is applicable to feed materials of animal origin and to all compound feeds. It is to be used for all materials from which the oils and fats cannot be completely extracted without prior hydrolysis (e.g. glutens, yeast, potato proteins and products subjected to processes such as extrusion, flaking and heating).
1.3.Interpretation of resultsU.K.
In all cases where a higher result is obtained by using Procedure B than by Procedure A, the result obtained by Procedure B shall be accepted as the true value.
2.PrincipleU.K.
2.1.Procedure AU.K.
The sample is extracted with light petroleum. The solvent is distilled off and the residue dried and weighed.
2.2.Procedure BU.K.
The sample is treated under heating with hydrochloric acid. The mixture is cooled and filtered. The residue is washed and dried and submitted to the determination according to Procedure A.
3.ReagentsU.K.
3.1.Light petroleum, boiling range: 40 to 60 oC. The bromine value must be less than 1 and the residue on evaporation less than 2 mg/100 ml.U.K.
3.2.Sodium sulfate, anhydrous.U.K.
3.3.Hydrochloric acid, c = 3 mol/lU.K.
3.4.Filtration aid, e.g. Kieselguhr, Hyflo-supercel.U.K.
4.ApparatusU.K.
4.1.Extraction apparatus. If fitted with a siphon (Soxhlet apparatus), the reflux rate shall be such as to produce about 10 cycles per hour; if of the non-siphoning type, the reflux rate shall be about 10 ml per minute.U.K.
4.2.Extraction thimbles, free of matter soluble in light petroleum and having a porosity consistent with the requirements of point 4.1.U.K.
4.3.Drying oven, either a vacuum oven set at 75 ± 3 oC or an air-oven set at 100 ± 3 oC.U.K.
5.ProcedureU.K.
5.1.Procedure A (see point 8.1)U.K.
Weigh 5 g of the sample to the nearest 1 mg, transfer it to an extraction thimble (4.2) and cover with a fat-free wad of cotton wool.
Place the thimble in an extractor (4.1) and extract for six hours with light petroleum (3.1). Collect the light petroleum extract in a dry, weighed flask containing fragments of pumice stone(28).
Distil off the solvent. Dry the residue maintaining the flask for one and a half hours in the drying oven (4.3). Leave to cool in a desiccator and weigh. Dry again for 30 minutes to ensure that the weight of the oils and fats remains constant (loss in weight between two successive weighings must be less than or equal to 1 mg).
5.2.Procedure BU.K.
Weigh 2,5 g of the sample to the nearest 1 mg (see point 8.2), place in a 400 ml beaker or a 300 ml conical flask and add 100 ml of hydrochloric acid (3.3) and fragments of pumice stone. Cover the beaker with a watch glass or fit the conical flask with a reflux condenser. Bring the mixture to a gentle boil over a low flame or a hot-plate and keep it there for one hour. Do not allow the product to stick to the sides of the container.
Cool and add a quantity of filtration aid (3.4) sufficient to prevent any loss of oil and fat during filtration. Filter through a moistened, fat-free, double filter paper. Wash the residue in cold water until a neutral filtrate is obtained. Check that the filtrate does not contain any oil or fats. Their presence indicates that the sample must be extracted with light petroleum, using Procedure A, before hydrolysis.
Place the double filter paper containing the residue on a watch glass and dry for one and a half hours in the air oven (4.3) at 100 ± 3 oC.
Place the double filter paper containing the dry residue in an extraction thimble (4.2) and cover with a fat-free wad of cotton wool. Place the thimble in an extractor (4.1) and proceed as indicated in the second and third paragraphs of point 5.1.
6.Expression of resultU.K.
Express the weight of the residue as a percentage of the sample.
7.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample by the same analyst shall not exceed:
0,2 %, in absolute value, for contents of crude oils and fats lower than 5 %,
4,0 % relative to the highest result for contents of 5 % to 10 %,
0,4 %, in absolute value, for contents above 10 %.
8.ObservationsU.K.
8.1.For products with a high content of oils and fats, which are difficult to crush or unsuitable for drawing a homogeneous reduced test sample, proceed as follows.U.K.
Weigh 20 g of the sample to the nearest 1 mg and mix with 10 g or more of anhydrous sodium sulfate (3.2). Extract with light petroleum (3.1) as indicated in point 5.1. Make up the extract obtained to 500 ml with light petroleum (3.1) and mix. Take 50 ml of the solution and place in a small, dry, weighed flask containing fragments of pumice stone. Distil off the solvent, dry and proceed as indicated in the last paragraph of point 5.1.
Eliminate the solvent from the extraction residue left in the thimble, crush the residue to a fineness of 1 mm, return it to the extraction thimble (do not add sodium sulfate) and proceed as indicated in the second and third paragraphs of point 5.1.
Calculate the content of oils and fats as a percentage of the sample by using the following formula:
(10m1 + m2) × 5
where:
m1
=
weight in grams of the residue after the first extraction (aliquot part of the extract),
m2
=
weight in grams of the residue after the second extraction.
8.2.For products low in oils and fats the test sample may be increased to 5 g.U.K.
8.3.Pet foods containing a high content of water may need to be mixed with anhydrous sodium sulfate prior to hydrolysis and extraction as per Procedure B.U.K.
8.4.In paragraph 5.2 it may be more effective to use hot water in place of cold water to wash the residue after filtration.U.K.
8.5.The drying time of 1,5 h may need to be extended for some feed. Excessive drying shall be avoided as this can lead to low results. A microwave oven can also be used.U.K.
8.6.Pre-extraction by Procedure A prior to hydrolysis and re-extraction by Procedure B is recommended if the crude oil/fat content is greater than 15 %. To some extent this depends on the nature of the feed and the nature of the oil/fat in the feed.U.K.
I.DETERMINATION OF CRUDE FIBREU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine fat-free organic substances in feed which are insoluble in acid and alkaline media and are conventionally described as crude fibre.
2.PrincipleU.K.
The sample, defatted where necessary, is treated successively with boiling solutions of sulphuric acid and potassium hydroxide of specified concentrations. The residue is separated by filtration on a sintered-glass filter washed, dried, weighed and ashed within a range of 475 to 500 oC. The loss of weight resulting from ashing corresponds to the crude fibre present in the test sample.
3.ReagentsU.K.
3.1.Sulphuric acid, c = 0,13 mol/l.U.K.
3.2.Anti-foaming agent (e.g. n-octanol).U.K.
3.3.Filter aid (Celite 545 or equivalent), heated at 500 oC for four hours (8.6).U.K.
3.4.Acetone.U.K.
3.5.Light petroleum boiling-range 40 to 60 oC.U.K.
3.6.Hydrochloric acid, c = 0,5 mol/l.U.K.
3.7.Potassium hydroxide solution, c = 0,23 mol/l.U.K.
4.ApparatusU.K.
4.1.Heating unit for digestion with sulphuric acid and potassium hydroxide solution, equipped with a support for the filter crucible (4.2) and provided with an outlet tube with a tap to the liquid outlet and vacuum, possibly with compressed air. Before use each day preheat the unit with boiling water for five minutes.U.K.
4.2.Glass filter crucible with fused sintered glass filter plate pore size 40-90 μm. Before first use, heat to 500 oC for a few minutes and cool (8.6).U.K.
4.3.Cylinder of at least 270 ml with a reflux condenser, suitable for boiling.U.K.
4.4.Drying oven with thermostat.U.K.
4.5.Muffle furnace with thermostat.U.K.
4.6.Extraction unit consisting of a support plate for the filter crucible (4.2) and with a discharge pipe with a tap to the vacuum and liquid outlet.U.K.
4.7.Connecting rings to assemble the heating unit (4.1), crucible (4.2) and cylinder (4.3) and to connect the cold extraction unit (4.6) and crucible.U.K.
5.ProcedureU.K.
Weigh out 1 g of the prepared sample to the nearest 1 mg and place it in the crucible (4.2), (see observations 8.1, 8.2 and 8.3) and add 1 g of filter aid (3.3).
Assemble the heating unit (4.1) and the filter crucible (4.2), then attach the cylinder (4.3) to the crucible. Pour 150 ml of boiling sulphuric acid (3.1) into the assembled cylinder and crucible and if necessary add a few drops of anti-foaming agent (3.2).
Bring the liquid to the boil within 5 ± 2 minutes and boil vigorously for exactly 30 minutes.
Open the tap to the discharge pipe (4.1) and, under vacuum, filter the sulphuric acid through the filter crucible and wash the residue with three consecutive 30 ml portions of boiling water, ensuring that the residue is filtered dry after each washing.
Close the outlet tap and pour 150 ml boiling potassium hydroxide solution (3.7) to the assembled cylinder and crucible and add a few drops of anti-foaming agent (3.2). Bring the liquid to boiling point within 5 ± 2 minutes and boil vigorously for exactly 30 minutes. Filter and repeat the washing procedure used for the sulphuric acid step.
After the final washing and drying, disconnect the crucible and its contents and reconnect it to the cold extraction unit (4.6). Apply the vacuum and wash the residue in the crucible with three consecutive 25 ml portions of acetone (3.4) ensuring that the residue is filtered dry after each washing.
Dry the crucible to constant weight in the oven at 130 oC. After each drying cool in the desiccator and weigh rapidly. Place the crucible in a muffle furnace and ash to constant weight (loss in weight between two successive weightings must be less than or equal to 2 mg) at 475 oC to 500 oC for at least 30 minutes.
After each heating cool first in the furnace and then in the desiccator before weighing.
Carry out a blank test without the sample. Loss of weight resulting from ashing must not exceed 4 mg.
6.Calculation of resultsU.K.
The crude fibre content as a percentage of the sample is given by the expression:

where:
m
=
weight of sample in g,
m0
=
loss of weight after ashing during the determination, in g,
m1
=
loss of weight after ashing during the blank test, in g.
7.RepeatabilityU.K.
The difference between two parallel determinations carried out on the same sample must not exceed:
0,6 % in absolute value for crude fibre contents lower than 10 %,
6 % relative to the higher result, for crude fibre contents equal to or greater than 10 %.
8.ObservationsU.K.
8.1.Feed containing more than 10 % crude fat must be defatted prior to analysis with light petroleum (3.5). Connect the filter crucible (4.2) and its contents to the cold extraction unit (4.6) and apply vacuum and wash the residue with three consecutive 30 ml portions of light petroleum, ensuring that the residue is dry. Connect the crucible and its contents to the heating unit (4.1) and continue as described under 5.U.K.
8.2.Feed containing fats which cannot be extracted directly with light petroleum (3.5) must be defatted as shown in 8.1 and defatted once more after boiling with acid. After boiling with acid and the subsequent washing connect the crucible and its contents to the cold extraction unit (4.6) and wash three times with 30 ml acetone followed by three further washings with 30 ml portions of light petroleum. Filter under vacuum until dry and continue the analysis as described under 5, beginning with potassium hydroxide treatment.U.K.
8.3.If the feed contains over 5 % of carbonates, expressed as calcium carbonate, connect the crucible (4.2) with the weighed sample to the heating unit (4.1). Wash the sample three times with 30 ml hydrochloric acid (3.6). After each addition let the sample stand for about one minute before filtering. Wash once with 30 ml water and then continue as described under 5.U.K.
8.4.If an apparatus in the form of a stand is used (several crucibles attached to the same heating unit) no two individual determinations on the same sample for analysis may be carried out in the same series.U.K.
8.5.If after boiling it is difficult to filter the acidic and basic solutions, use compressed air through the discharge pipe of the heating unit and then continue filtering.U.K.
8.6.The temperature for ashing shall not be higher than 500 oC in order to extend the lifetime of the glass filter crucibles. Care must be taken to avoid excessive thermal shock during heating and cooling cycles.U.K.
J.DETERMINATION OF SUGARU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine the amount of reducing sugars and total sugars after inversion, expressed as glucose or where appropriate as sucrose, converting by the factor 0,95. It is applicable to compound feed. Special methods are provided for other feed. Where necessary, lactose shall be measured separately and taken into account when calculating the results.
2.PrincipleU.K.
The sugars are extracted in dilute ethanol; the solution is clarified with Carrez solutions I and II. After eliminating the ethanol, the quantities before and after inversion are determined by the Luff-Schoorl method.
3.ReagentsU.K.
3.1.Ethanol solution 40 % (v/v) density: 0,948 g/ml at 20 oC, neutralised to phenolphthalein.U.K.
3.2.Carrez solution I: dissolve in water 21,9 g of zinc acetate Zn (CH3COO)2 2H2O and 3 g of glacial acetic acid. Make up to 100 ml with water.U.K.
3.3.Carrez solution II: dissolve in water 10,6 g of potassium ferrocyanide K4Fe (CN)6 3H2O. Make up to 100 ml with water.U.K.
3.4.Methyl orange, solution 0,1 % (w/v).U.K.
3.5.Hydrochloric acid 4 mol/litre.U.K.
3.6.Hydrochloric acid 0,1 mol/litre.U.K.
3.7.Sodium hydroxide solution 0,1 mol/litre.U.K.
3.8.Luff-Schoorl reagent:U.K.
Stirring carefully, pour the citric acid solution (3.8.2) into the sodium carbonate solution (3.8.3). Add the copper sulphate solution (3.8.1) and make up to 1 litre with water. Leave to settle overnight and filter.
Check the concentration of the reagent thus obtained (Cu 0,05 mol/litre; Na2 CO3 1 mol/litre), see (5.4) last paragraph. The solution's pH shall be approximately 9,4.
3.8.1.Copper sulphate solution: dissolve 25 g of copper sulphate, Cu SO4 5H2O, free from iron, in 100 ml of water.U.K.
3.8.2.Citric acid solution: dissolve 50 g of citric acid, C6H8O7·H2O in 50 ml of water.U.K.
3.8.3.Sodium carbonate solution: dissolve 143,8 g of anhydrous sodium carbonate in approximately 300 ml of warm water. Leave to cool.U.K.
3.9.Sodium thiosulphate solution 0,1 mol/litre.U.K.
3.10.Starch solution: add a mixture of 5 g of soluble starch in 30 ml of water to 1 litre of boiling water. Boil for three minutes, leave to cool and if necessary add 10 mg of mercuric iodide as a preservative.U.K.
3.11.Sulphuric acid 3 mol/litre.U.K.
3.12.Potassium iodide, solution 30 % (w/v).U.K.
3.13.Granulated pumice stone boiled in hydrochloric acid, washed in water and dried.U.K.
3.14.3-methylbutan-l-ol.U.K.
4.ApparatusU.K.
Mixer (tumbler): approximately 35 to 40 r.p.m.
5.ProcedureU.K.
5.1.Extraction of sampleU.K.
Weigh 2,5 g of the sample to the nearest mg and place in a 250 ml volumetric flask. Add 200 ml of ethanol (3.1) and mix in the tumbler for one hour. Add 5 ml of Carrez solution I (3.2) and stir for approximately 30 seconds. Add 5 ml of Carrez solution II (3.3) and again stir for one minute. Make up to volume with ethanol (3.1), homogenise and filter. Remove 200 ml of the filtrate and evaporate to approximately half volume in order to eliminate most of the ethanol. Transfer the evaporation residue quantitatively to a 200 ml volumetric flask using warm water, cool, bring up to volume with water, homogenise and filter if necessary. This solution will be used to determine the amount of reducing sugars and, after inversion, of total sugars.
5.2.Determination of reducing sugarsU.K.
Using a pipette, remove not more than 25 ml of the solution containing less than 60 mg of reducing sugars expressed as glucose. If necessary, make up to 25 ml with distilled water and determine the content of reducing sugars by the Luff-Schoorl method. The result is expressed as the percentage content of glucose in the sample.
5.3.Determination of total sugars after inversionU.K.
Using a pipette take 50 ml of the solution and transfer to a 100 ml volumetric flask. Add a few drops of methyl orange solution (3.4) then, carefully and stirring continuously, add hydrochloric acid (3.5) until the liquid turns a definite red. Add 15 ml of hydrochloric acid (3.6), immerse the flask in a fast boiling water bath and keep there for 30 minutes. Cool rapidly to approximately 20 oC and add 15 ml of sodium hydroxide solution (3.7). Make up to 100 ml with water and homogenise. Remove not more than 25 ml containing less than 60 mg of reducing sugars expressed as glucose. If necessary, make up to 25 ml with distilled water and determine the content of reducing sugars by the Luff-Schoorl method. The result is expressed as the percentage of glucose or, where appropriate, sucrose, by multiplying by the factor 0,95.
5.4.Titration by the Luff-Schoorl methodU.K.
Using a pipette, take 25 ml of Luff-Schoorl reagent (3.8) and transfer to a 300 ml Erlenmeyer flask; add exactly 25 ml of the clarified sugar solution. Add 2 granules of pumice stone (3.13), heat, stirring by hand, over a free flame of medium height and bring the liquid to the boil in approximately two minutes. Place the Erlenmeyer immediately on an asbestos-coated wire gauze with a hole approximately 6 cm in diameter under which a flame has been lit. The flame shall be regulated in such a way that only the base of the Erlenmeyer is heated. Fit a reflux condenser to the Erlenmeyer flask. Boil for exactly 10 minutes. Cool immediately in cold water and after approximately five minutes titrate as follows:
Add 10 ml of potassium iodide solution (3.12) and immediately afterwards (carefully, because of the risk of abundant foaming), add 25 ml of sulphuric acid (3.11). Titrate with sodium thiosulphate solution (3.9) until a dull yellow colour appears, add the starch indicator (3.10) and complete titration.
Carry out the same titration on an accurately measured mixture of 25 ml of Luff-Schoorl reagent (3.8) and 25 ml of water, after adding 10 ml of potassium iodide solution (3.12) and 25 ml of sulphuric acid (3.11) without boiling.
6.Calculation of resultsU.K.
Using the table establish the amount of glucose in mg which corresponds to the difference between the values of the two titrations, expressed in mg of sodium thiosulphate 0,1 mol/litre. Express the result as a percentage of the sample.
7.Special proceduresU.K.
7.1.In the case of feed which are rich in molasses and other feed which are not particularly homogeneous, weigh out 20 g and place with 500 ml of water in a 1 litre volumetric flask. Mix for one hour in the tumbler. Clarify using Carrez 1 (3.2) and II (3.3) reagents as described under 5.1, this time however using four times the quantities of each reagent. Bring up to volume with 80 % ethanol (v/v).U.K.
Homogenise and filter. Eliminate the ethanol as described under 5.1. If there is no dextrinised starch, bring up to volume with distilled water.
7.2.In the case of molasses and feed materials which are rich in sugar and almost starch-free (carobs, dried beetroot cossettes etc.), weigh out 5 g, place in a 250 ml volumetric flask, add 200 ml of distilled water and mix in the tumbler for one hour, or more if necessary. Clarify using Carrez I (3.2) and II (3.3) reagents as described under 5.1. Bring up to volume with cold water, homogenise and filter. In order to determine the amount of total sugars, continue as described under 5.3.U.K.
8.ObservationsU.K.
8.1.In order to prevent foaming it is advisable to add (irrespective of the volume) approximately 1 ml of 3-methylbutan-l-ol (3.14) before boiling with Luff-Schoorl reagent.U.K.
8.2.The difference between the content of total sugars after inversion, expressed as glucose, and the content of reducing sugars, expressed as glucose, multiplied by 0,95, gives the percentage content of sucrose.U.K.
8.3.In order to determine the content of reducing sugars, excluding lactose, two methods may be adopted:U.K.
8.3.1.For an approximate calculation, multiply by 0,675 the lactose content established by a different method of analysis and subtract the result obtained from the content of reducing sugars.U.K.
8.3.2.For an accurate calculation of reducing sugars, excluding lactose, the same sample must be used for the two final determinations. One of the analyses is carried out on part of the solution obtained under 5.1, the other on part of the solution obtained during the determination of lactose by the method laid down for that purpose (after fermenting the other types of sugar and clarifying).U.K.
In both cases the amount of sugar present is determined by the Luff-Schoorl method and calculated in mg of glucose. One of the values is subtracted from the other and the difference is expressed as a percentage of the sample.
Example:U.K.
The two volumes taken correspond, for each determination, to a sample of 250 mg.
In the first case 17 ml of sodium thiosulphate solution 0,1 mol/litre corresponding to 44,2 mg of glucose is consumed; in the second, 11 ml, corresponding to 27,6 mg of glucose.
The difference is 16,6 mg of glucose.
The content of reducing sugars (excluding lactose), calculated as glucose, is therefore:

Table of values for 25 ml of Luff-Schoorl reagent
ml of Na2 S2 O30,1 mol/litre, two minutes' heating, 10 minutes' boiling
| Na2 S2 O30,1 mol/litre | Glucose, fructose invert sugarsC6 H12 O6 | LactoseC12 H22 O11 | MaltoseC12 H22 O11 | Na2 S2 O30,1 mol/litre | |||
|---|---|---|---|---|---|---|---|
| ml | mg | difference | mg | difference | mg | difference | ml |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 | 2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 | 2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 | 3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 | 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 | 3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 | 3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 | 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K.DETERMINATION OF LACTOSEU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine the level of lactose in feed containing more than 0,5 % of lactose.
2.PrincipleU.K.
The sugars are dissolved in water. The solution is subjected to fermentation by the yeast Saccharomyces cerevisiae which leaves the lactose intact. After clarification and filtration the lactose content of the filtrate is determined by the Luff-Schoorl method.
3.ReagentsU.K.
3.1.Suspension of Saccharomyces cerevisiae: suspend 25 g of fresh yeast in 100 ml of water. The suspension will keep for a maximum period of one week in a refrigerator.U.K.
3.2.Carrez solution I: dissolve in water 21,9 g of zinc acetate, Zn (CH3 COO)2 2H2O and 3 g of glacial acetic acid. Make up to 100 ml with water.U.K.
3.3.Carrez solution II: dissolve in water 10,6 g of potassium ferrocyanide K4Fe (CN)6 3H2O. Make up to 100 ml with water.U.K.
3.4.Luff-Schoorl reagent:U.K.
Stirring carefully, pour the citric acid solution (3.4.2) into the sodium carbonate solution (3.4.3). Add the copper sulphate solution (3.4.1) and make up to 1 litre with water. Leave to settle overnight and filter. Check the concentration of the reagent thus obtained (Cu 0,05 mol/litre; Na2 CO3 1 mol/litre). The solution's pH shall be approximately 9,4.
3.4.1.Copper sulphate solution: dissolve 25 g of copper sulphate Cu SO4 5H2O, free from iron, in 100 ml of water.U.K.
3.4.2.Citric acid solution: dissolve 50 g of citric acid C6H8O7·H2O in 50 ml of water.U.K.
3.4.3.Sodium carbonate solution: dissolve 143,8 g of anhydrous sodium carbonate in approximately 300 ml of warm water. Leave to cool.U.K.
3.5.Granulated pumice stone boiled in hydrochloric acid, washed in water and dried.U.K.
3.6.Potassium iodide, solution 30 % (w/v).U.K.
3.7.Sulphuric acid 3 mol/litre.U.K.
3.8.Solution of sodium thiosulphate 0,1 mol/litre.U.K.
3.9.Starch solution: add a mixture of 5 g of soluble starch in 30 ml of water to 1 litre of boiling water. Boil for three minutes, leave to cool, and if necessary add 10 mg of mercuric iodide as a preservative.U.K.
4.ApparatusU.K.
Water bath with thermostat set at 38-40 oC.
5.ProcedureU.K.
Weigh 1 g of the sample to the nearest mg and place this portion of the sample in a 100 ml volumetric flask. Add 25 to 30 ml of water. Place the flask in a boiling water bath for 30 minutes and then cool to approximately 35 oC. Add 5 ml of yeast suspension (3.1) and homogenise. Leave the flask to stand for two hours in a water bath, at a temperature of 38-40o C. Cool to approximately 20 oC.
Add 2,5 ml of Carrez solution I (3.2) and stir for 30 seconds, then add 2,5 ml of Carrez solution II (3.3) and again stir for 30 seconds. Make up to 100 ml with water, mix and filter. Using a pipette, remove an amount of filtrate which does not exceed 25 ml and which preferably contains from 40 to 80 mg of lactose and transfer it to a 300 ml Erlenmeyer flask. If necessary, make up to 25 ml with water.
Carry out a blank test in the same way with 5 ml of yeast suspension (3.1). Determine the lactose content according to Luff-Schoorl, as follows: add exactly 25 ml of Luff-Schoorl reagent (3.4) and two granules of pumice stone (3.5). Stir by hand-while heating over a free flame of medium height and bring the liquid to the boil in approximately two minutes. Place the Erlenmeyer immediately on an asbestos-coated wire gauze with a hole approximately 6 cm in diameter under which a flame has been lit. The flame shall be regulated in such a way that only the base of the Erlenmeyer is heated. Fit a reflux condenser to the Erlenmeyer flask. Boil for exactly 10 minutes. Cool immediately in cold water and after approximately five minutes titrate as follows:
Add 10 ml of potassium iodide solution (3.6) and immediately afterwards (carefully, because of the risk of abundant foaming) add 25 ml of sulphuric acid (3.7). Titrate with sodium thiosulphate solution (3.8) until a dull yellow colour appears, add the starch indicator (3.9) and complete titration.
Carry out the same titration on an accurately measured mixture of 25 ml of Luff-Schoorl reagent (3.4) and 25 ml of water, after adding 10 ml of potassium iodide solution (3.6) and 25 ml of sulphuric acid (3.7) without boiling.
6.Calculation of resultsU.K.
Using the attached table, establish the amount of lactose in mg which corresponds to the difference between the results of the two titrations, expressed in ml of sodium thiosulphate 0,1 mol/litre.
Express the result of anhydrous lactose as a percentage of the sample.
7.ObservationU.K.
For products containing more than 40 % of fermentable sugar, use more than 5 ml of yeast suspension (3.1).
Table of values for 25 ml of Luff-Schoorl reagent
ml of Na2 S2 O30,1 mol/litre, two minutes' heating, 10 minutes' boiling
| Na2 S2 O30,1 mol/litre | Glucose, fructose invert sugarsC6 H12 O6 | LactoseC12 H22 O11 | MaltoseC12 H22 O11 | Na2 S2 O30,1 mol/litre | |||
|---|---|---|---|---|---|---|---|
| ml | mg | difference | mg | difference | mg | difference | ml |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 | 2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 | 2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 | 3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 | 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 | 3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 | 3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 | 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L.DETERMINATION OF STARCHU.K.
POLARIMETRIC METHODU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine the levels of starch and of high molecular weight starch degradation products in feed for the purpose of checking compliance with the declared energy value (provisions in Annex VII) and Council Directive 96/25/EC(29).
2.PrincipleU.K.
The method comprises two determinations. In the first, the sample is treated with dilute hydrochloric acid. After clarification and filtration the optical rotation of the solution is measured by polarimetry.
In the second, the sample is extracted with 40 % ethanol. After acidifying the filtrate with hydrochloric acid, clarifying and filtering, the optical rotation is measured as in the first determination.
The difference between the two measurements, multiplied by a known factor, gives the starch content of the sample.
3.ReagentsU.K.
3.1.Hydrochloric acid, solution 25 % (w/w) density: 1,126 g/ml.U.K.
3.2.Hydrochloric acid. solution 1,13 % (w/v)U.K.
The concentration must be checked by titration using a sodium hydroxide solution 0,1 mol/litre in the presence of 0,1 % (w/v) methyl red in 94 % (v/v) ethanol. For the neutralisation of 10 ml, 30,94 ml of NaOH 0,1 mol/litre is needed.
3.3.Carrez solution I: dissolve 21,9 g of zinc acetate Zn(CH3COO)2 2H2O and 3 g of glacial acetic acid in water. Make up to 100 ml with water.U.K.
3.4.Carrez solution II: dissolve 10,6 g of potassium ferrocyanide K4 Fe(CN)6 3H2O in water. Make up to 100 ml with water.U.K.
3.5.Ethanol, solution 40 % (v/v), density: 0,948 g/ml at 20 oC.U.K.
4.ApparatusU.K.
4.1.250 ml Erlenmeyer flask with standard ground-glass joint and with reflux condenser.U.K.
4.2.Polarimeter or saccharimeter.U.K.
5.ProcedureU.K.
5.1.Preparation of the sampleU.K.
Crush the sample until it is fine enough for all of it to pass through a 0,5 mm round-meshed sieve.
5.2.Determination of the total optical rotation (P or S) (see observation 7.1)U.K.
Weigh 2,5 g of the crushed sample to the nearest mg and place in a 100 ml graduated flask. Add 25 ml of hydrochloric acid (3.2), shake to obtain even distribution of the test sample and add a further 25 ml of hydrochloric acid (3.2). Immerse the flask in a boiling water bath shaking vigorously and steadily for the first three minutes to prevent the formation of agglomerates. The quantity of water in the water bath must be sufficient for the bath to remain at boiling point when the flask is introduced into it. The flask must not be taken out of the bath whilst being shaken. After exactly 15 minutes, remove from the bath, add 30 ml of cold water and cool immediately to 20 oC.
Add 5 ml of Carrez solution I (3.3) and shake for approximately 30 seconds. Then add 5 ml of Carrez solution II (3.4) and shake again for approximately 30 seconds. Make up to volume with water, mix and filter. If the filtrate is not perfectly clear (which is rare), repeat the determination using a larger quantity of Carrez solutions I and II, for example 10 ml.
Measure the optical rotation of the solution in a 200 mm tube with the polarimeter or saccharimeter.
5.3.Determination of the optical rotation (P' or S') of substances soluble in 40 % ethanolU.K.
Weigh 5 g of the sample to the nearest mg, place in a 100 ml graduated flask and add about 80 ml of ethanol (3.5) (see observation 7.2). Leave the flask to stand for 1 hour at room temperature; during this time, shake vigorously on six occasions so that the test sample is thoroughly mixed with the ethanol. Make up to volume with ethanol (3.5), mix and filter.
Pipette 50 ml of the filtrate (corresponds to 2,5 g of the sample) into a 250 ml Erlenmeyer flask, add 2,1 ml of hydrochloric acid (3.1) and shake vigorously. Fit a reflux condenser to the Erlenmeyer flask and immerse the latter in a boiling water bath. After exactly 15 minutes, remove the Erlenmeyer flask from the bath, transfer the contents to a 100 ml graduated flask, rinsing with a little cold water, and cool to 20 oC.
Clarify using Carrez solutions I (3.3) and II (3.4), make up to volume with water, mix, filter and measure the optical rotation as indicated in the 2nd and 3rd paragraphs of 5.2.
6.Calculation of resultsU.K.
The starch content (%) is calculated as follows:
6.1.Measurement by polarimeterU.K.

P
=
Total optical rotation in angle degrees
P'
=
Optical rotation in angle degrees of the substances soluble in 40 % (V/V) ethanol

=
Specific optical rotation of pure starch. The numerical values conventionally accepted for this factor are the following:
| +185,9o: | rice starch |
| +185,7o: | potato starch |
| +184,6o: | maize starch |
| +182,7o: | wheat starch |
| +181,5o: | barley starch |
| +181,3o: | oat starch |
| +184,0o: | other types of starch and starch mixtures in compound feed |
6.2.Measurement by saccharimeterU.K.

S
=
Total optical rotation in saccharimeter degrees
S'
=
Optical rotation in saccharimeter degrees of the substances soluble in 40 % (v/v) ethanol
N
=
weight (g) of saccharose in 100 ml of water yielding an optical rotation of 100 saccharimeter degrees when measured using a 200 mm tube
16,29 g for the French saccharimeters
26,0 g for the German saccharimeters
20,0 g for mixed saccharimeters.

6.3.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed 0,4 in absolute value for a starch content lower than 40 % and 1 % relative for starch contents equal to or greater than 40 %.
7.ObservationsU.K.
7.1.If the sample contains more than 6 % of carbonates, calculated in terms of calcium carbonate, they must be destroyed by treatment with an exactly appropriate quantity of dilute sulphuric acid before determination of the total optical rotation.U.K.
7.2.In the case of products with a high lactose content, such as powdered milk serum or skimmed milk powder, proceed as follows after adding 80 ml of ethanol (3.5). Fit a reflux condenser to the flask and immerse the latter in a water bath at 50 oC for 30 minutes. Leave to cool and continue the analysis as indicated in 5.3.U.K.
7.3.The following feed materials, where they are present in significant amounts in feed, are known to give rise to interferences when determining the starch content by the polarimetric method and thereby incorrect results could be yielded:U.K.
(sugar) beet products such as (sugar)beet pulp, (sugar) beet molasses, (sugar) beet pulp — molassed, (sugar) beet vinasse, (beet) sugar,
citrus pulp,
linseed; linseed expeller; linseed extracted,
rape seed; rape seed expeller; rape seed extracted; rape seed hulls,
sunflower seed; sunflower seed extracted; sunflower seed, partially decorticated, extracted,
copra expeller; copra extracted,
potato pulp,
dehydrated yeast,
products rich in inulin (e.g. Chips and meal of Jerusalem artichokes),
greaves.
M.DETERMINATION OF CRUDE ASHU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the crude ash content of feed.
2.PrincipleU.K.
The sample is ashed at 550 oC; the residue is weighed.
3.ReagentsU.K.
Ammonium nitrate, solution 20 % (w/v).
4.ApparatusU.K.
4.1.Hot-plate.U.K.
4.2.Electric muffle-furnace with thermostat.U.K.
4.3.Crucibles for ashing made of silica, porcelain or platinum either rectangular (approx. 60 × 40 × 25 mm) or circular (diameter: 60 to 75 mm, height: 20 to 40 mm).U.K.
5.ProcedureU.K.
Weigh out to the nearest mg approximately 5 g of the sample (2,5 in the case of products which have a tendency to swell) and place in a crucible for ashing which has first been heated at 550 oC, cooled down and tared. Place the crucible on the hot-plate and heat gradually until the substance carbonises. Ash according to 5.1 or 5.2.
5.1.Put the crucible into the calibrated muffle furnace set at 550 oC. Keep at this temperature until white, light grey or reddish ash is obtained which appears to be free from carbonaceous particles. Place the crucible in a desiccator, leave to cool and weigh immediately.U.K.
5.2.Put the crucible into the calibrated muffle-furnace set at 550 oC. Ash for 3 hours. Place the crucible in a desiccator, leave to cool and weigh immediately. Ash again for 30 minutes to ensure that the weight of the ash remains constant (loss in weight between two successive weightings must be less than or equal to 1 mg).U.K.
6.Calculation of resultsU.K.
Calculate the weight of the residue by deducting the tare.
Express the result as a percentage of the sample.
7.ObservationsU.K.
7.1.The ash of substances which are difficult to ash must be subjected to an initial ashing of at least three hours, cooled and then a few drops of 20 % solution of ammonium nitrate or water added to it (carefully, to avoid dispersal of the ash or the formation of lumps). Continue calcining after drying in the oven. Repeat the operation as necessary until ashing is complete.U.K.
7.2.In the case of substances resistant to the treatment described under 7.1, proceed as follows: after ashing for three hours, place the ash in warm water and filter through a small, ash-free filter. Ash the filter and its contents in the original crucible. Place the filtrate in the cooled crucible, evaporate until dry, ash and weigh.U.K.
7.3.In the case of oils and fats, weigh accurately a sample of 25 g in a suitably sized crucible. Carbonise by setting light to the substance with a strip of ash-free filter paper. After combustion, moisten with as little water as possible. Dry and ash as described under 5.U.K.
N.DETERMINATION OF ASH WHICH IS INSOLUBLE IN HYDROCHLORIC ACIDU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the level in feed of mineral substances which are insoluble in hydrochloric acid. Two methods can be used, depending on the nature of the sample.
1.1.Method A: applicable to organic feed materials and to most compound feed.U.K.
1.2.Method B: applicable to mineral compounds and mixtures and to compound feed, whose content in substances insoluble in hydrochloric acid, as determined by Method A, is greater than 1 %.U.K.
2.PrincipleU.K.
2.1.Method A: the sample is ashed, the ash boiled in hydrochloric acid and the insoluble residue filtered and weighed.U.K.
2.2.Method B: the sample is treated with hydrochloric acid. The solution is filtered, the residue ashed and the ash thus obtained treated in accordance with Method A.U.K.
3.ReagentsU.K.
3.1.Hydrochloric acid 3 mol/litre.U.K.
3.2.Trichloroacetic acid, solution 20 % solution (w/v).U.K.
3.3.Trichloroacetic acid, solution 1 % (w/v).U.K.
4.ApparatusU.K.
4.1.Hot plate.U.K.
4.2.Electric muffle-furnace with thermostat.U.K.
4.3.Crucibles for ashing made of silica, porcelain or platinum, either rectangular (approx. 60 × 40 × 25 mm) or circular (diameter: 60 to 75 mm, height: 20 to 40 mm).U.K.
5.ProcedureU.K.
5.1.Method AU.K.
Ash the sample using the method described for the determination of crude ash. Ash obtained from that analysis may also be used.
Place the ash in a 250 to 400 ml beaker using 75 ml of hydrochloric acid (3.1). Bring slowly to the boil and boil gently for 15 minutes. Filter the warm solution through an ash-free filter paper and wash the residue with warm water until the acid reaction is no longer visible. Dry the filter containing the residue and ash in a tared crucible at a temperature of not less than 550 oC and not more than 700 oC. Cool in a desiccator and weigh.
5.2.Method BU.K.
Weigh 5 g of the sample to the nearest mg and place in a 250 to 400 ml beaker. Add 25 ml of water and 25 ml of hydrochloric acid (3.1) successively, mix and wait for effervescence to cease. Add a further 50 ml of hydrochloric acid (3.1). Wait for any release of gas to cease then place the beaker in a boiling water bath and keep it there for 30 minutes or longer, if necessary, in order to hydrolyse thoroughly any starch which may be present. Filter while warm through an ash-free filter and wash the filter in 50 ml of warm water (see observation 7). Place the filter containing the residue in a crucible for ashing, dry and ash at a temperature of not less than 550 oC and not more than 700 oC. Place the ash in a 250 to 400 ml beaker using 75 ml of hydrochloric acid (3.1); continue as described in the second subparagraph of 5.1.
6.Calculation of resultsU.K.
Calculate the weight of the residue by deducting the tare. Express the result as a percentage of the sample.
7.ObservationU.K.
If filtration proves difficult recommence the analysis, replacing the 50 ml of hydrochloric acid (3.1) by 50 ml of 20 % trichloroacetic acid (3.2) and washing the filter in a warm solution of 1 % trichloroacetic acid (3.3).
O.DETERMINATION OF CARBONATESU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the amount of carbonates, conventionally expressed as calcium carbonate, in most feed.
However in certain cases (for example, with iron carbonate) a special method must be used.
2.PrincipleU.K.
The carbonates are decomposed in hydrochloric acid; the carbon dioxide released is collected in a graduated tube, and its volume compared with that released under the same conditions by a known quantity of calcium carbonate.
3.ReagentsU.K.
3.1.Hydrochloric acid, density 1,1 g/ml.U.K.
3.2.Calcium carbonate.U.K.
3.3.Sulphuric acid, approximately 0,05 mol/litre, coloured with methyl red.U.K.
4.ApparatusU.K.
Scheibler-Dietrich apparatus (see diagram) or equivalent apparatus.
5.ProcedureU.K.
According to the sample's carbonate content, weigh a portion of the sample as shown below:
0,5 g for products containing from 50 % to 100 % of carbonates, expressed as calcium carbonate,
1 g for products containing from 40 % to 50 % of carbonates, expressed as calcium carbonate,
2 to 3 g for other products.
Place the portion of the sample in the special flask (4) of the apparatus, fitted with a small tube of unbreakable material containing 10 ml of hydrochloric acid (3.1), and connect the flask to the apparatus. Turn the three-way cock (5) so that the tube (1) connects with the outside. Using the mobile tube (2), which is filled with coloured sulphuric acid (3.3) and connected to the graduated tube (1), bring the level of the liquid up to the zero mark. Turn the cock (5) in order to connect up tubes (1) and (3) and check that the level is at zero.
Run the hydrochloric acid (3.1) slowly over the portion of the sample, tilting the flask (4). Make the pressure equal by lowering the tube (2). Shake the flask (4) until the release of carbon dioxide has stopped completely.
Restore pressure by bringing the liquid back to the same level in tubes (1) and (2). After a few minutes, when the volume of gas has become constant, take the reading.
Carry out a control test in the same conditions on 0,5 g of calcium carbonate (3.2).
6.Calculation of resultsU.K.
The content of carbonates, expressed as calcium carbonate, is calculated by using the formula:

where:
X
=
% (w/w) of carbonates in the sample, expressed as calcium carbonate
V
=
ml of CO2 released by the portion of the sample.
V1
=
ml of CO2 released by 0,5 g of CaCO3.
m
=
weight, in grammes, of the portion of the sample.
7.ObservationsU.K.
7.1.When the portion of the sample weighs more than 2 g, first place 15 ml of distilled water in the flask (4) and mix before beginning the test. Use the same volume of water for the control test.U.K.
7.2.If the apparatus used has a different volume from that of the Scheibler-Dietrich apparatus, the portions taken from the sample and from the control substance and the calculation of the results must be adapted accordingly.U.K.
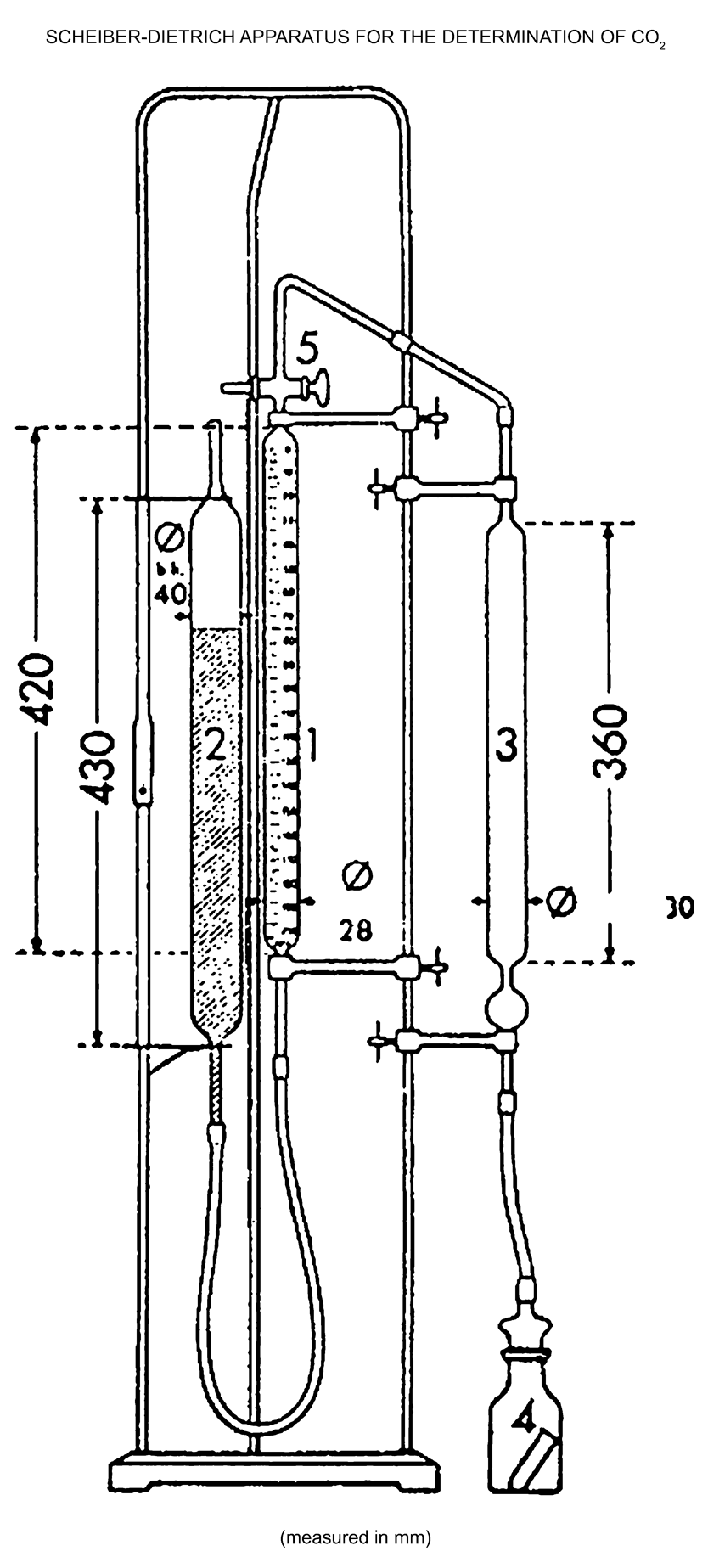
P.DETERMINATION OF TOTAL PHOSPHORUSU.K.
PHOTOMETRIC METHODU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the content of total phosphorus in feed. It is particularly appropriate for the analysis of products low in phosphorus. In certain cases (product rich in phosphorus), a gravimetric method may be used.
2.PrincipleU.K.
The sample is mineralised, either by dry combustion (in the case of organic feed) or by acid digestion (in the case of mineral compounds and liquid feed), and placed in an acid solution. The solution is treated with molybdovanadate reagent. The optical density of the yellow solution thus formed is measured in a spectrophotometer at 430 nm.
3.ReagentsU.K.
3.1.Calcium carbonate.U.K.
3.2.Hydrochloric acid, ρ20 = 1,1 g/ml (approx 6 mol/litre).U.K.
3.3.Nitric acid, ρ20 = 1,045 g/ml.U.K.
3.4.Nitric acid, ρ20 = 1,38 to 1,42 g/ml.U.K.
3.5.Sulphuric acid, ρ20 = 1,84 g/ml.U.K.
3.6.Molybdovanadate reagent: mix 200 ml of ammonium heptamolybdate solution (3.6.1), 200 ml of ammonium monovanadate solution (3.6.2) and 134 ml of nitric acid (3.4) in a 1 litre graduated flask. Make up to volume with water.U.K.
3.6.1.Ammonium heptamolybdate solution: dissolve in hot water 100 g of ammonium heptamolybdate (NH4) 6Mo7O24·4H2O. Add 10 ml of ammonia (density 0,91 g/ml) and make up to 1 litre with water.U.K.
3.6.2.Ammonium monovanadate solution: dissolve 2,35 g of ammonium monovanadate NH4VO3 in 400 ml of hot water. Stirring constantly, slowly add 20 ml of dilute nitric acid (7 ml of HNO3 (3.4) + 13 ml of H2O and make up to 1 litre with water.U.K.
3.7.Standard solution of 1 mg phosphorus per ml: dissolve 4,387 g of potassium dihydrogen phosphate KH2PO4 in water. Make up to 1 litre with water.U.K.
4.ApparatusU.K.
4.1.Silica, porcelain or platinum ashing crucibles.U.K.
4.2.Electric muffle-furnace with thermostat set at 550 oC.U.K.
4.3.250 ml Kjeldahl flask.U.K.
4.4.Graduated flasks and precision pipettes.U.K.
4.5.Spectrophotometer.U.K.
4.6.Test tubes about 16 mm in diameter, with stoppers graded to a diameter of 14,5 mm; capacity: 25 to 30 ml.U.K.
5.ProcedureU.K.
5.1.Preparation of the solutionU.K.
According to the nature of the sample, prepare a solution as indicated in 5.1.1 or 5.1.2.
5.1.1.Usual procedureU.K.
Weigh 1 g or more of the sample to the nearest 1 mg. Place the test sample in a Kjeldahl flask, add 20 ml of sulphuric acid (3.5), shake to impregnate the substance completely with acid and to prevent it from sticking to the sides of the flask, heat and keep at boiling point for 10 minutes. Leave to cool slightly, add 2 ml of nitric acid (3.4), heat gently, leave to cool slightly, add a little more nitric acid (3.4) and bring back to boiling point. Repeat this procedure until a colourless solution is obtained. Cool, add a little water, decant the liquid into a 500 ml graduated flask, rinsing the Kjeldahl flask with hot water. Leave to cool, make up to volume with water, homogenise and filter.
5.1.2.Samples containing organic substances and free from calcium and magnesium dihydrogen phosphatesU.K.
Weigh about 2,5 g of the sample to the nearest 1 mg in an ashing crucible. Mix the test sample until completely merged with 1 g of calcium carbonate (3.1). Ash in the oven at 550 oC until white or grey ash is obtained (a little charcoal does not matter). Transfer the ash into a 250 ml beaker. Add 20 ml of water and hydrochloric acid (3.2) until effervescence ceases. Add a further 10 ml of hydrochloric acid (3.2). Place the beaker on a sand bath and evaporate until dry to make the silica insoluble. Redissolve the residue in 10 ml of nitric acid (3.3) and boil on the sand bath or hot plate for 5 minutes without evaporating until dry. Decant the liquid into a 500 ml graduated flask, rinsing the beaker several times with hot water. Leave to cool, make up to volume with water, homogenise and filter.
5.2.Development of coloration and measurement of optical densityU.K.
Dilute an aliquot part of the filtrate obtained by 5.1.1 or 5.1.2 to obtain a phosphorus concentration of not more than 40 μg/ml. Place 10 ml of this solution in a test tube (4.6) and add 10 ml of molybdovanadate reagent (3.6). Homogenise and leave to stand for at least 10 minutes at 20 oC. Measure the optical density in a spectrophotometer at 430 nm against a solution obtained by adding 10 ml of the molybdovanadate reagent (3.6) to 10 ml of water.
5.3.Calibration curveU.K.
From the standard solution (3.7) prepare solutions containing respectively 5, 10, 20, 30 and 40 μg of phosphorus per ml. Take 10 ml of each of these solutions and add thereto 10 ml of molybdovanadate reagent (3.6). Homogenise and leave to stand for at least 10 minutes at 20 oC. Measure the optical density as indicated in 5.2. Trace the calibration curve by plotting the optical densities against the corresponding quantities of phosphorus. For concentrations between 0 and 40 μg/ml, the curve will be linear.
6.Calculation of resultsU.K.
Determine the amount of phosphorus in the test sample by using the calibration curve.
Express the result as a percentage of the sample.
RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample shall not exceed:
3 %, relative to the higher result, for phosphorus contents of less than 5 %,
0,15 % in absolute value, for phosphorus contents of 5 % or more.
Q.DETERMINATION OF CHLORINE FROM CHLORIDESU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the amount of chlorine in chlorides which are soluble in water, conventionally expressed as sodium chloride. It is applicable to all feed.
2.PrincipleU.K.
The chlorides are dissolved in water. If the product contains organic matter it is clarified. The solution is slightly acidified with nitric acid and the chlorides precipitated in the form of silver chloride by means of a solution of silver nitrate. The excess silver nitrate is titrated with a solution of ammonium thiocyanate, by Volhard's method.
3.ReagentsU.K.
3.1.Solution of ammonium thiocyanate 0,1 mol/litre.U.K.
3.2.Solution of silver nitrate 0,1 mol/litre.U.K.
3.3.Saturated solution of ammonium ferric sulphate (NH4)Fe(SO4)2.U.K.
3.4.Nitric acid, density: 1,38 g/ml.U.K.
3.5.Diethyl ether.U.K.
3.6.Acetone.U.K.
3.7.Carrez I solution: dissolve in water 21,9 g of zinc acetate, Zn (CH3COO)2·2H2O and 3 g of glacial acetic acid. Make up to 100 ml with water.U.K.
3.8.Carrez II solution: dissolve in water 10,6 g of potassium ferrocyanide K4Fe(CN)6·3H2O. Make up to 100 ml with water.U.K.
3.9.Active carbon, free from chlorides and not absorbing them.U.K.
4.ApparatusU.K.
Mixer (tumbler): approximately 35 to 40 r.p.m.
5.ProcedureU.K.
5.1.Preparation of the solutionU.K.
According to the nature of the sample, prepare a solution as shown under 5.1.1, 5.1.2 or 5.1.3.
At the same time carry out a blank test omitting the sample to be analysed.
5.1.1.Samples free from organic matterU.K.
Weigh to the nearest mg a sample of not more than 10 g and containing not more than 3 g of chlorine in the form of chlorides. Place with 400 ml of water in a 500 ml volumetric flask at approximately 20 oC. Mix for 30 minutes in the tumbler, bring up to volume, homogenise and filter.
5.1.2.Samples containing organic matter, excluding the products listed under 5.1.3.U.K.
Weigh approximately 5 g of the sample to the nearest mg and place with 1 g of active carbon in a 500 ml volumetric flask. Add 400 ml of water at approximately 20 oC and 5 ml of Carrez solution I (3.7), stir for 30 seconds then add 5 ml of Carrez solution II (3.8). Mix for 30 minutes in the tumbler, bring up to volume, homogenise and filter.
5.1.3.Cooked feed, flax cakes and flour, products rich in flax flour and other products rich in mucilage or in colloidal substances (for example, dextrinated starch)U.K.
Prepare the solution as described under 5.1.2 but do not filter. Decant (if necessary centrifuge), remove 100 ml of the supernatant liquid and transfer to a 200 ml measuring flask. Mix with acetone (3.6) and bring up to volume with this solvent, homogenise and filter.
5.2.TitrationU.K.
Using a pipette, transfer to an Erlenmeyer flask from 25 ml to 100 ml of the filtrate (according to the assumed chlorine content) obtained as described under 5.1.1, 5.1.2 or 5.1.3. The aliquot portion must not contain more than 150 mg of chlorine (Cl). Dilute if necessary to not less than 50 ml with water, add 5 ml of nitric acid (3.4), 20 ml of saturated solution of ammonium ferric sulphate (3.3) and two drops of ammonium thiocyanate solution (3.1) transferred by means of a burette filled up to the zero mark. Using a burette, transfer the silver nitrate solution (3.2) in such a way that an excess of 5 ml is obtained. Add 5 ml of diethyl ether (3.5) and shake hard to coagulate the precipitate. Titrate the excess silver nitrate with the ammonium thiocyanate solution (3.1) until the reddish-brown tint has lasted for one minute.
6.Calculation of resultsU.K.
The amount of chlorine (X), expressed as % sodium chloride is calculated by using the following formula:

where:
V1
=
ml of silver nitrate solution 0,1 mol/l added
V2
=
ml of ammonium thiocyanate solution 0,1 mol/l used for titration
m
=
weight of sample.
If the blank test indicates that silver nitrate solution 0,1 mol/l has been consumed deduct this value from the volume (V1 – V2).
7.ObservationsU.K.
7.1.Titration may also be carried out by potentiometry.U.K.
7.2.In the case of products which are very rich in oils and fats, first de-fat with diethyl ether or light petroleum.U.K.
7.3.In the case of fish-meal, titration may be carried out by Mohr’s method.U.K.
ANNEX IVU.K.METHODS OF ANALYSIS TO CONTROL THE LEVEL OF AUTHORISED ADDITIVES IN FEED
A.DETERMINATION OF VITAMIN AU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the level of vitamin A (retinol) in feed and premixtures. Vitamin A includes all-trans-retinyl alcohol and its cis-isomers which are determined by this method. The content of vitamin A is expressed in International Units (IU) per kg. One IU corresponds to the activity of 0,3 μg all-trans-vitamin A alcohol or 0,344 μg all-trans-vitamin A acetate or 0,55 μg all-trans-vitamin A palmitate.
The limit of quantification is 2 000 IU vitamin A/kg.
2.PrincipleU.K.
The sample is hydrolysed with ethanolic potassium hydroxide solution and the vitamin A is extracted into light petroleum. The solvent is removed by evaporation and the residue is dissolved in methanol and, if necessary, diluted to the required concentration. The content of vitamin A is determined by reversed phase high performance liquid chromatography (RP-HPLC) using a UV or a fluorescence detector. The chromatographic parameters are chosen so that there is no separation between the all-trans-vitamin A alcohol and its cis isomers.
3.ReagentsU.K.
3.1.Ethanol, σ = 96 %U.K.
3.2.Light petroleum, boiling range 40 oC-60 oCU.K.
3.3.MethanolU.K.
3.4.Potassium hydroxide solution, c = 50 g/100 mlU.K.
3.5.Sodium ascorbate solution, c = 10 g/100 ml (see 7.7 observations)U.K.
3.6.Sodium sulphide, Na2S · x H2O (x = 7-9)U.K.
3.6.1.Sodium sulphide solution, c = 0,5 mol/l in glycerol, ß = 120 g/l (for x = 9) (see 7.8 observations)U.K.
3.7.Phenolphthalein solution, c = 2 g/100 ml in ethanol (3.1)U.K.
3.8.2-PropanolU.K.
3.9.Mobile phase for HPLC: mixture of methanol (3.3) and water, e.g. 980 + 20 (v + v). The exact ratio will be determined by the characteristics of the column employed.U.K.
3.10.Nitrogen, oxygen freeU.K.
3.11.All-trans-vitamin A acetate, extra pure, of certified activity, e.g. 2,8 x 106 IU/gU.K.
3.11.1.Stock solution of all-trans-vitamin A acetate: Weigh to the nearest 0,1 mg, 50 mg of vitamin A acetate (3.11) into a 100 ml graduated flask. Dissolve in 2-propanol (3.8) and make up to the mark with the same solvent. The nominal concentration of this solution is 1 400 IU vitamin A per ml. The exact content has to be determined according to 5.6.3.1.U.K.
3.12.All-trans-vitamin A palmitate, extra pure, of certified activity, e.g. 1,8 x 106 IU/gU.K.
3.12.1.Stock solution of all-trans-vitamin A palmitate: Weigh to the nearest 0,1 mg, 80 mg of vitamin A palmitate (3.12) into a 100 ml graduated flask. Dissolve in 2-propanol (3.8) and make up to the mark with the same solvent. The nominal concentration of this solution is 1 400 IU vitamin A per ml. The exact content has to be determined according to 5.6.3.2.U.K.
3.13.2,6-Di-tert-butyl-4-methylphenol (BHT) (see 7.5 observations)U.K.
4.ApparatusU.K.
4.1.Vacuum rotary evaporatorU.K.
4.2.Amber glasswareU.K.
4.2.1.Flat bottom or conical flasks, 500 ml, with ground-glass socketU.K.
4.2.2.Graduated flasks with ground-glass stoppers, narrow-necked, 10, 25, 100 and 500 mlU.K.
4.2.3.Separating funnels, conical, 1 000 ml, with ground-glass stoppersU.K.
4.2.4.Pear shaped flasks, 250 ml, with ground-glass socketsU.K.
4.3.Allihn condenser, jacket length 300 mm, with ground-glass joint, with adapter for gas feed pipeU.K.
4.4.Pleated filter paper for phase separation, diameter 185 mm (e.g. Schleicher & Schuell 597 HY 1/2)U.K.
4.5.HPLC equipment with injection systemU.K.
4.5.1.Liquid chromatographic column, 250 mm x 4 mm, C18, 5 or 10 μm packing, or equivalent (performance criterion: only a single peak for all retinol isomers under the HPLC-conditions)U.K.
4.5.2.UV or fluorescence detector, with variable wavelength adjustmentU.K.
4.6.Spectrophotometer with 10 mm quartz cellsU.K.
4.7.Water-bath with magnetic stirrerU.K.
4.8.Extraction apparatus (see figure 1) consisting of:U.K.
4.8.1.Glass cylinder of 1 l capacity fitted with a ground glass neck and stopperU.K.
4.8.2.Ground glass insert equipped with a side-arm and an adjustable tube passing through the centre. The adjustable tube shall have a U-shaped lower end and a jet at the opposite end so that the upper liquid layer in the cylinder may be transferred into a separating funnel.U.K.
5.ProcedureU.K.
Note:
Vitamin A is sensitive to (UV-) light and to oxidation. All operations shall be carried out in the absence of light (using amber glassware, or glassware protected with aluminium foil) and oxygen (flush with nitrogen). During extraction air above the liquid shall be replaced by nitrogen (avoid excess pressure by loosening the stopper from time to time).
5.1.Preparation of the sampleU.K.
Grind the sample so that it passes a 1 mm mesh sieve, taking care to avoid generation of heat. Grinding must be carried out immediately before weighing and saponification otherwise there may be losses of vitamin A.
5.2.SaponificationU.K.
Depending on the vitamin A content weigh, to the nearest 1 mg, 2 g to 25 g of the sample into a 500 ml flat bottom or conical flask (4.2.1). Add successively with swirling 130 ml ethanol (3.1), approximately 100 mg BHT (3.13), 2 ml sodium ascorbate solution (3.5) and 2 ml sodium sulphide solution (3.6). Fit a condenser (4.3) to the flask and immerse the flask in a water-bath with magnetic stirrer (4.7). Heat to boiling and allow to reflux for 5 minutes. Then add 25 ml potassium hydroxide solution (3.4) through the condenser (4.3) and allow to reflux for a further 25 min., with stirring under a slow stream of nitrogen. Then rinse the condenser with approximately 20 ml water and cool the content of the flask to room temperature.
5.3.ExtractionU.K.
Transfer by decantation the saponification solution quantitatively by rinsing with a total volume of 250 ml water to a 1 000 ml separating funnel (4.2.3) or to the extraction apparatus (4.8). Rinse the saponification flask successively with 25 ml ethanol (3.1) and 100 ml light petroleum (3.2) and transfer the rinsings to the separating funnel or to the extraction apparatus. The proportion of water and ethanol in the combined solutions must be about 2:1. Shake vigorously for 2 min. and allow to settle for 2 minutes.
5.3.1.Extraction using a separating funnel (4.2.3)U.K.
When the layers have separated (see observation 7.3) transfer the light petroleum layer to another separating funnel (4.2.3). Repeat this extraction twice, with 100 ml light petroleum (3.2) and twice, with 50 ml light petroleum (3.2).
Wash the combined extracts in the separating funnel twice by gently swirling (to avoid formation of emulsions) with 100 ml portions of water and then by repeated shaking with further 100 ml portions of water until the water remains colourless on addition of phenolphthalein solution (3.7) (washing four times is usually sufficient). Filter the washed extract through a dry pleated filter for phase separation (4.4) to remove any suspended water into a 500 ml graduated flask (4.2.2). Rinse the separating funnel and the filter with 50 ml light petroleum (3.2), make up to the mark with light petroleum (3.2) and mix well.
5.3.2.Extraction using an extraction apparatus (4.8)U.K.
When the layers have separated (see observation 7.3) replace the stopper of the glass cylinder (4.8.1) by the ground glass insert (4.8.2) and position the U-shaped lower end of the adjustable tube so that it is just above the level of the interface. By application of pressure from a nitrogen line to the side-arm, transfer the upper light petroleum-layer to a 1 000 ml separating funnel (4.2.3). Add 100 ml light petroleum (3.2) to the glass cylinder, stopper and shake well. Allow the layers to separate and transfer the upper layer to the separating funnel as before. Repeat the extraction procedure with further 100 ml of light petroleum (3.2), then twice with 50 ml portions of light petroleum (3.2) and add the light petroleum layers to the separating funnel.
Wash the combined light petroleum extracts as described in 5.3.1 and proceed as described there.
5.4.Preparation of the sample solution for HPLCU.K.
Pipette an aliquot portion of the light petroleum solution (from 5.3.1 or 5.3.2) into a 250 ml pear shaped flask (4.2.4). Evaporate the solvent nearly to dryness on the rotary evaporator (4.1) with reduced pressure at a bath temperature not exceeding 40 oC. Restore atmospheric pressure by admitting nitrogen (3.10) and remove the flask from the rotary evaporator. Remove the remaining solvent with a stream of nitrogen (3.10) and dissolve the residue immediately in a known volume (10-100 ml) of methanol (3.3) (the concentration of vitamin A must be in the range of 5 IU/ml to 30 IU/ml).
5.5.Determination by HPLCU.K.
Vitamin A is separated on a C18 reversed phase column (4.5.1) and the concentration is measured by means of a UV detector (325 nm) or a fluorescence detector (excitation: 325 nm, emission: 475 nm) (4.5.2).
Inject an aliquot portion (e.g. 20 μl) of the methanolic solution obtained in 5.4 and elute with the mobile phase (3.9). Calculate the mean peak height (area) of several injections of the same sample solution and the mean peak heights (areas) of several injections of the calibration solutions (5.6.2).
HPLC conditionsU.K.
The following conditions are offered for guidance; other conditions may be used provided that they give equivalent results.
| Liquid chromatographic column (4.5.1): | 250 mm × 4 mm, C18, 5 or 10 μm packing, or equivalent |
| Mobile phase (3.9): | Mixture of methanol (3.3) and water e.g. 980 + 20 (v + v). |
| Flow rate: | 1-2 ml/min. |
| Detector (4.5.2): | UV detector (325 nm) or fluorescence detector (excitation: 325 nm/emission: 475 nm) |
5.6.CalibrationU.K.
5.6.1.Preparation of the working standard solutionsU.K.
Pipette 20 ml of the vitamin A acetate stock solution (3.11.1) or 20 ml of the vitamin A palmitate stock solution (3.12.1) into a 500 ml flat bottom or conical flask (4.2.1) and hydrolyse as described under 5.2, but without addition of BHT. Subsequently extract with light petroleum (3.2) according to 5.3 and make up to 500 ml with light petroleum (3.2). Evaporate 100 ml of this extract on the rotary evaporator (see 5.4) nearly to dryness, remove the remaining solvent with a stream of nitrogen (3.10) and redissolve the residue in 10,0 ml of methanol (3.3). The nominal concentration of this solution is 560 IU vitamin A per ml. The exact content has to be determined according to 5.6.3.3. The working standard solution has to be freshly prepared before use.
Pipette 2,0 ml of this working standard solution into a 20 ml graduated flask, make up to the mark with methanol (3.3) and mix. The nominal concentration of this diluted working standard solution is 56 IU vitamin A per ml.
5.6.2.Preparation of the calibration solutions and calibration graphU.K.
Transfer 1,0, 2,0, 5,0 and 10,0 ml of the diluted working standard solution into a series of 20 ml graduated flasks, make up to the mark with methanol (3.3) and mix. The nominal concentrations of these solutions are 2,8, 5,6, 14,0 and 28,0 IU vitamin A per ml.
Inject 20 μl of each calibration solution several times and determine the mean peak heights (areas). Using the mean peak heights (areas) plot a calibration graph considering the results of the UV control (5.6.3.3).
5.6.3.UV standardisation of the standard solutionsU.K.
5.6.3.1.Vitamin A acetate stock solutionU.K.
Pipette 2,0 ml of the vitamin A acetate stock solution (3.11.1) into a 50 ml graduated flask (4.2.2) and make up to the mark with 2-propanol (3.8). The nominal concentration of this solution is 56 IU vitamin A per ml. Pipette 3,0 ml of this diluted vitamin A acetate solution into a 25 ml graduated flask and make up to the mark with 2-propanol (3.8). The nominal concentration of this solution is 6,72 IU vitamin A per ml. Measure the UV spectrum of this solution against 2-propanol (3.8) in the spectrophotometer (4.6) between 300 nm and 400 nm. The extinction maximum must be between 325 nm and 327 nm.
Calculation of the vitamin A content:
IU vitamin A/ml = E326 × 19,0
(
5.6.3.2.Vitamin A palmitate stock solutionU.K.
Pipette 2,0 ml of the vitamin A palmitate stock solution (3.12.1) into a 50 ml graduated flask (4.2.2) and make up to the mark with 2-propanol (3.8). The nominal concentration of this solution is 56 IU vitamin A per ml. Pipette 3,0 ml of this diluted vitamin A palmitate solution into a 25 ml graduated flask and make up to the mark with 2-propanol (3.8). The nominal concentration of this solution is 6,72 IU vitamin A per ml. Measure the UV spectrum of this solution against 2-propanol (3.8) in the spectrophotometer (4.6) between 300 nm and 400 nm. The extinction maximum must be between 325 nm and 327 nm.
Calculation of the vitamin A content:
IU vitamin A/ml = E326 × 19,0
(
5.6.3.3.Vitamin A working standard solutionU.K.
Pipette 3,0 ml of the undiluted vitamin A working standard solution, prepared according to 5.6.1 into a 50 ml graduated flask (4.2.2) and make up to the mark with 2-propanol (3.8). Pipette 5,0 ml of this solution into a 25 ml graduated flask and make up to the mark with 2-propanol (3.8). The nominal concentration of this solution is 6,72 IU vitamin A per ml. Measure the UV spectrum of this solution against 2-propanol (3.8) in the spectrophotometer (4.6) between 300 nm and 400 nm. The extinction maximum must be between 325 nm and 327 nm.
Calculation of the vitamin A content:
IU vitamin A/ml = E325 × 18,3
(
6.Calculation of the resultsU.K.
From the mean height (area) of the vitamin A peaks of the sample solution determine the concentration of the sample solution in IU/ml by reference to the calibration graph (5.6.2).
The vitamin A content w in IU/kg of the sample is given by the following formula:

in which:
c
=
vitamin A concentration of the sample solution (5.4) in IU/ml
V1
=
volume of sample solution (5.4) in ml
V2
=
volume of aliquot taken in 5.4 in ml
m
=
weight of the test portion in g
7.ObservationsU.K.
7.1.For samples with low vitamin A concentration it may be useful to combine the light petroleum-extracts of two saponification-charges (amount weighed: 25 g) to one sample solution for HPLC-determination.U.K.
7.2.The weight of the sample taken for the analysis shall not contain more than 2 g fat.U.K.
7.3.If phase separation does not occur add approximately 10 ml ethanol (3.1) to break the emulsion.U.K.
7.4.With cod-liver oil and other pure fats the saponification time shall be extended to 45-60 minutes.U.K.
7.5.Hydroquinone can be used instead of BHT.U.K.
7.6.Using a normal phase-column the separation of retinol isomers is possible. But in that case, the heights (areas) of all cis and trans isomers peaks have to be summed for calculations.U.K.
7.7.Approximately 150 mg ascorbic acid can be used instead of sodium ascorbate solution.U.K.
7.8.Approximately 50 mg EDTA can be used instead of sodium sulphide solution.U.K.
7.9.In cases of analysis of vitamin A in milk replacers, specific attention has to be paidU.K.
at saponification (5.2): due to the amount of fat present in the sample, increasing of potassium hydroxide solution amount (3.4) may be necessary,
at extraction (5.3): due to the presence of emulsions, adaptation of the water/ethanol 2:1 ratio may be necessary.
To check if the applied method of analysis generates reliable results on this specific matrix (milk replacer), a recovery test shall be applied on an additional test portion. If the recovery rate is lower than 80 %, the analytical result has to be corrected for recovery.
8.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed 15 % relative to the higher result.
9.Results of a collaborative study(30) U.K.
| Premix | Premix feed | Mineral concentrate | Protein feed | Piglet | |
|---|---|---|---|---|---|
| L | 13 | 12 | 13 | 12 | 13 |
| n | 48 | 45 | 47 | 46 | 49 |
| mean [IU/kg] | 17,02 x 106 | 1,21 x 106 | 537 100 | 151 800 | 18 070 |
| Sr [IU/kg] | 0,51 x 106 | 0,039 x 106 | 22 080 | 12 280 | 682 |
| r [IU/kg] | 1,43 x 106 | 0,109 x 106 | 61 824 | 34 384 | 1 910 |
| CVr[%] | 3,0 | 3,5 | 4,1 | 8,1 | 3,8 |
| SR [IU/kg] | 1,36 x 106 | 0,069 x 106 | 46 300 | 23 060 | 3 614 |
| R [IU/kg] | 3,81 x 106 | 0,193 x 106 | 129 640 | 64 568 | 10 119 |
| CVR [%] | 8,0 | 6,2 | 8,6 | 15 | 20 |
L
=
number of laboratories
n
=
number of single values
sr
=
standard deviation of repeatability
SR
=
standard deviation of reproducibility
r
=
repeatability
R
=
reproducibility
CVr
=
coefficient of variation of repeatability
CVR
=
coefficient of variation of reproducibility.

B.DETERMINATION OF VITAMIN EU.K.
1.Purpose and ScopeU.K.
This method makes it possible to determine the level of vitamin E in feed and premixtures. The content of vitamin E is expressed as mg DL-α-tocopherol acetate per kg. 1 mg DL-α-tocopherol acetate corresponds to 0,91 mg DL-α-tocopherol (vitamin E).
The limit of quantification is 2 mg vitamin E/kg. This limit of quantification is only achievable with fluorescence detector. With an UV detector the limit of quantification is 10 mg/kg.
2.PrincipleU.K.
The sample is hydrolysed with ethanolic potassium hydroxide solution and the vitamin E is extracted into light petroleum. The solvent is removed by evaporation and the residue is dissolved in methanol and, if necessary, diluted to the required concentration. The content of vitamin E is determined by reversed phase high performance liquid chromatography (RP-HPLC) using a fluorescence or a UV detector.
3.ReagentsU.K.
3.1.Ethanol, σ = 96 %.U.K.
3.2.Light petroleum, boiling range 40 oC-60 oC.U.K.
3.3.Methanol.U.K.
3.4.Potassium hydroxide solution, c = 50 g/100 ml.U.K.
3.5.Sodium ascorbate solution, c = 10 g/100 ml (see 7.7 observations).U.K.
3.6.Sodium sulphide, Na2S· x H2O (x = 7-9).U.K.
3.6.1.Sodium sulphide solution, c = 0,5 mol/l in glycerol, β = 120 g/l. (for x = 9) (see 7.8 observations)U.K.
3.7.Phenolphthalein solution, c = 2 g/100 ml in ethanol (3.1).U.K.
3.8.Mobile phase for HPLC: mixture of methanol (3.3) and water, e.g. 980 + 20 (v + v). The exact ratio will be determined by the characteristics of the column employed.U.K.
3.9.Nitrogen, oxygen free.U.K.
3.10.DL-α-tocopherol acetate, extra pure, of certified activity.U.K.
3.10.1.Stock solution of DL-α-tocopherol acetate: Weigh to the nearest 0,1 mg, 100 mg of DL-α-tocopherol acetate (3.10) into a 100 ml graduated flask. Dissolve in ethanol (3.1) and make up to the mark with the same solvent. 1 ml of this solution contains 1 mg DL-α-tocopherol acetate. (UV control see 5.6.1.3; stabilisation see 7.4 observations).U.K.
3.11.DL-α-tocopherol, extra pure, of certified activity.U.K.
3.11.1.Stock solution of DL-α-tocopherol: Weigh to the nearest 0,1 mg, 100 mg of DL-α-tocopherol (3.10) into a 100 ml graduated flask. Dissolve in ethanol (3.1) and make up to the mark with the same solvent. 1 ml of this solution contains 1 mg DL-α-tocopherol. (UV control see 5.6.2.3; stabilisation see 7.4 observations).U.K.
3.12.2,6-Di-tert-butyl-4-methylphenol (BHT) (see 7.5 observations).U.K.
4.ApparatusU.K.
4.1.Rotary film evaporator.U.K.
4.2.Amber glassware.U.K.
4.2.1.Flat bottom or conical flasks, 500 ml, with ground-glass socket.U.K.
4.2.2.Graduated flasks with ground-glass stoppers, narrow-necked, 10, 25, 100 and 500 ml.U.K.
4.2.3.Separating funnels, conical, 1 000 ml, with ground-glass stoppers.U.K.
4.2.4.Pear shaped flasks, 250 ml, with ground-glass sockets.U.K.
4.3.Allihn condenser, jacket length 300 mm, with ground-glass joint, with adapter for gas feed pipe.U.K.
4.4.Pleated filter paper for phase separation, diameter 185 mm (e.g. Schleicher & Schuell 597 HY 1/2).U.K.
4.5.HPLC equipment with injection system.U.K.
4.5.1.Liquid chromatographic column, 250 mm × 4 mm, C18, 5 or 10 μm packing, or equivalent.U.K.
4.5.2.Fluorescence or UV detector, with variable wavelength adjustment.U.K.
4.6.Spectrophotometer with 10 mm quartz cells.U.K.
4.7.Water-bath with magnetic stirrer.U.K.
4.8.Extraction apparatus (see figure 1) consisting of:U.K.
4.8.1.Glass cylinder of 1 l capacity fitted with a ground glass neck and stopper.U.K.
4.8.2.Ground glass insert equipped with a side-arm and an adjustable tube passing through the centre. The adjustable tube shall have a U-shaped lower end and a jet at the opposite end so that the upper liquid layer in the cylinder may be transferred into a separating funnel.U.K.
5.ProcedureU.K.
Note:
Vitamin E is sensitive to (UV-) light and to oxidation. All operations shall be carried out in the absence of light (using amber glassware, or glassware protected with aluminium foil) and oxygen (flush with nitrogen). During extraction air above the liquid shall be replaced by nitrogen (avoid excess pressure by loosening the stopper from time to time).
5.1.Preparation of the sampleU.K.
Grind the sample so that it passes a 1 mm mesh sieve, taking care to avoid generation of heat. Grinding must be carried out immediately before weighing and saponification otherwise there may be losses of vitamin E.
5.2.SaponificationU.K.
Depending on the vitamin E content weigh, to the nearest 0,01 g, 2 g to 25 g of the sample into a 500 ml flat bottom or conical flask (4.2.1). Add successively with swirling 130 ml ethanol (3.1), approximately 100 mg BHT (3.12), 2 ml sodium ascorbate solution (3.5) and 2 ml sodium sulphide solution (3.6). Fit the condenser (4.3) to the flask and immerse the flask in a water-bath with magnetic stirrer (4.7). Heat to boiling and allow to reflux for 5 minutes. Then add 25 ml potassium hydroxide solution (3.4) through the condenser (4.3) and allow to reflux for a further 25 min. with stirring under a slow stream of nitrogen. Then rinse the condenser with approximately 20 ml water and cool the content of the flask to room temperature.
5.3.ExtractionU.K.
Transfer by decantation the saponification solution quantitatively by rinsing with a total volume of 250 ml water to a 1 000 ml separating funnel (4.2.3) or to the extraction apparatus (4.8). Rinse the saponification flask successively with 25 ml ethanol (3.1) and 100 ml light petroleum (3.2) and transfer the rinsings to the separating funnel or to the extraction apparatus. The proportion of water and ethanol in the combined solutions must be about 2:1. Shake vigorously for 2 min. and allow to settle for 2 minutes.
5.3.1.Extraction using a separating funnel (4.2.3)U.K.
When the layers have separated (see observation 7.3) transfer the light petroleum layer to another separating funnel (4.2.3). Repeat this extraction twice, with 100 ml light petroleum (3.2) and twice, with 50 ml light petroleum (3.2).
Wash the combined extracts in the separating funnel twice by gently swirling (to avoid formation of emulsions) with 100 ml portions of water and then by repeated shaking with further 100 ml portions of water until the water remains colourless on addition of phenolphthalein solution (3.7) (washing four times is usually sufficient). Filter the washed extract through a dry pleated filter for phase separation (4.4) to remove any suspended water into a 500 ml graduated flask (4.2.2). Rinse the separating funnel and the filter with 50 ml light petroleum (3.2), make up to the mark with light petroleum (3.2) and mix well.
5.3.2.Extraction using an extraction apparatus (4.8)U.K.
When the layers have separated (see observation 7.3) replace the stopper of the glass cylinder (4.8.1) by the ground glass insert (4.8.2) and position the U-shaped lower end of the adjustable tube so that it is just above the level of the interface. By application of pressure from a nitrogen line to the side-arm, transfer the upper light petroleum-layer to a 1 000 ml separating funnel (4.2.3). Add 100 ml light petroleum (3.2) to the glass cylinder, stopper and shake well. Allow the layers to separate and transfer the upper layer to the separating funnel as before. Repeat the extraction procedure with further 100 ml of light petroleum (3.2), then twice with 50 ml portions of light petroleum (3.2) and add the light petroleum layers to the separating funnel.
Wash the combined light petroleum extracts as described in 5.3.1 and proceed as described there.
5.4.Preparation of the sample solution for HPLCU.K.
Pipette an aliquot portion of the light petroleum solution (from 5.3.1 or 5.3.2) into a 250 ml pear shaped flask (4.2.4). Evaporate the solvent nearly to dryness on the rotary evaporator (4.1) with reduced pressure at a bath temperature not exceeding 40 oC. Restore atmospheric pressure by admitting nitrogen (3.9) and remove the flask from the rotary evaporator. Remove the remaining solvent with a stream of nitrogen (3.9) and dissolve the residue immediately in a known volume (10-100 ml) of methanol (3.3) (the concentration of DL-α-tocopherol must be in the range 5 μg/ml to 30 μg/ml).
5.5.Determination by HPLCU.K.
Vitamin E is separated on a C18 reversed phase column (4.5.1) and the concentration is measured using a fluorescence detector (excitation: 295 nm, emission: 330 nm) or a UV detector (292 nm) (4.5.2).
Inject an aliquot portion (e.g. 20 μl) of the methanolic solution obtained in 5.4 and elute with the mobile phase (3.8). Calculate the mean peak heights (areas) of several injections of the same sample solution and the mean peak heights (areas) of several injections of the calibration solutions (5.6.2).
HPLC conditionsU.K.
The following conditions are offered for guidance; other conditions may be used provided that they give equivalent results.
| Liquid chromatographic column (4.5.1): | 250 mm × 4 mm, C18, 5 or 10 μm packing, or equivalent |
| Mobile phase (3.8): | Mixture of methanol (3.3) and water e.g. 980 + 20 (v + v). |
| Flow rate: | 1-2 ml/min. |
| Detector (4.5.2) | Fluorescence detector (excitation: 295 nm/emission: 330 nm) or UV detector (292 nm) |
5.6.Calibration (DL-α-tocopherol acetate or DL-α-tocopherol)U.K.
5.6.1.DL-α-tocopherol acetate standardU.K.
5.6.1.1.Preparation of the working standard solutionU.K.
Transfer by pipette 25 ml of the DL-α-tocopherol acetate stock solution (3.10.1) into a 500 ml flat bottom or conical flask (4.2.1) and hydrolyse as described under 5.2. Subsequently extract with light petroleum (3.2) according to 5.3 and make up to 500 ml with light petroleum. Evaporate 25 ml of this extract on the rotary evaporator (see 5.4) nearly to dryness, remove the remaining solvent with a stream of nitrogen (3.9) and redissolve the residue in 25,0 ml of methanol (3.3). The nominal concentration of this solution is 45,5 μg DL-α-tocopherol per ml, equivalent to 50 μg DL-α-tocopherol acetate per ml. The working standard solution has to be freshly prepared before use.
5.6.1.2.Preparation of the calibration solutions and calibration graphU.K.
Transfer 1,0, 2,0, 4,0 and 10,0 ml of the working standard solution into a series of 20 ml graduated flasks, make up to the mark with methanol (3.3) and mix. The nominal concentrations of these solutions are 2,5, 5,0, 10,0 and 25,0 μg/ml DL-α-tocopherol acetate, i.e. 2,28, 4,55, 9,1 μg/ml and 22,8 μg/ml DL-α-tocopherol.
Inject 20 μl of each calibration solution several times and determine the mean peak heights (areas). Using the mean peak heights (areas) plot a calibration graph.
5.6.1.3.UV standardisation of the DL-α-tocopherol acetate stock solution (3.10.1)U.K.
Dilute 5,0 ml of the DL-α-tocopherol acetate stock solution (3.10.1) to 25,0 ml with ethanol and measure the UV spectrum of this solution against ethanol (3.1) in the spectrophotometer (4.6) between 250 nm and 320 nm.
The absorption maximum shall be at 284 nm:

At this dilution an extinction value of 0,84 to 0,88 must be obtained.
5.6.2.DL-α-tocopherol standardU.K.
5.6.2.1.Preparation of the working standard solutionU.K.
Transfer by pipette 2 ml of the DL-α-tocopherol stock solution (3.11.1) into a 50 ml graduated flask, dissolve in methanol (3.3) and make up to the mark with methanol. The nominal concentration of this solution is 40 μg DL-α-tocopherol per ml, equivalent to 44,0 μg DL-α-tocopherol acetate per ml. The working standard solution has to be freshly prepared before use.
5.6.2.2.Preparation of the calibration solutions and calibration graphU.K.
Transfer 1,0, 2,0, 4,0 and 10,0 ml of the working standard solution into a series of 20 ml graduated flasks, make up to the mark with methanol (3.3) and mix. The nominal concentrations of these solutions are 2,0, 4,0, 8,0 and 20,0 μg/ml DL-α-tocopherol, i.e. 2,2, 4,4, 8,79 μg/ml and 22,0 μg/ml DL-α-tocopherol acetate.
Inject 20 μl of each calibration solution several times and determine the mean peak heights (areas). Using the mean peak heights (areas) plot a calibration graph.
5.6.2.3.UV standardisation of the DL-α-tocopherol stock solution (3.11.1)U.K.
Dilute 2,0 ml of the DL-α-tocopherol stock solution (3.11.1) to 25,0 ml with ethanol and measure the UV spectrum of this solution against ethanol (3.1) in the spectrophotometer (4.6) between 250 nm and 320 nm. The absorption maximum shall be at 292 nm:

At this dilution an extinction value of 0,6 must be obtained.
6.Calculation of the resultsU.K.
From the mean height (area) of the vitamin E peaks of the sample solution determine the concentration of the sample solution in μg/ml (calculated as α-tocopherol acetate) by reference to the calibration graph (5.6.1.2 or 5.6.2.2).
The vitamin E content w in mg/kg of the sample is given by the following formula:

in which:
c
=
vitamin E concentration (as α-tocopherol acetate) of the sample solution (5.4) in μg/ml
V1
=
volume of sample solution (5.4), in ml
V2
=
volume of aliquot taken in (5.4), in ml
m
=
weight of the test portion in g
7.ObservationsU.K.
7.1.For samples with low vitamin E concentration it may be useful to combine the light petroleum-extracts of two saponification-charges (amount weighed: 25 g) to one sample solution for HPLC-determination.U.K.
7.2.The weight of the sample taken for the analysis shall not contain more than 2 g fat.U.K.
7.3.If phase separation does not occur add approximately 10 ml ethanol (3.1) to break the emulsion.U.K.
7.4.After the spectrophotometric measurement of the DL-α-tocopherol acetate or DL-α-tocopherol solution according to 5.6.1.3 or 5.6.2.3 respectively add approximately 10 mg BHT (3.12) to the solution (3.10.1 or 3.10.2) and keep the solution in a refrigerator (storage life max. 4 weeks).U.K.
7.5.Hydroquinone can be used instead of BHT.U.K.
7.6.Using a normal phase-column the separation of α-, β-, γ- and δ-tocopherol is possible.U.K.
7.7.Approximately 150 mg ascorbic acid can be used instead of sodium ascorbate solution.U.K.
7.8.Approximately 50 mg EDTA can be used instead of sodium sulphide solution.U.K.
7.9.Vitamin E acetate hydrolyses very fast under alkaline conditions and is therefore very sensitive to oxidation, especially in the presence of trace elements like iron or copper. In case of the determination of vitamin E in premixtures at levels higher than 5 000 mg/kg, a degradation of vitamin E could be the consequence. Therefore a HPLC method including an enzymatic digestion of the vitamin E formulation without an alkaline saponification step is to be recommended for confirmation.U.K.
8.Repeatability·U.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed 15 % relative to the higher result.
9.Results of a collaborative study(31) U.K.
| Premix | Premix feed | Mineral concentrate | Protein feed | Piglet | |
|---|---|---|---|---|---|
| L | 12 | 12 | 12 | 12 | 12 |
| n | 48 | 48 | 48 | 48 | 48 |
| mean [mg/kg] | 17 380 | 1 187 | 926 | 315 | 61,3 |
| Sr [mg/kg] | 384 | 45,3 | 25,2 | 13,0 | 2,3 |
| r [mg/kg] | 1 075 | 126,8 | 70,6 | 36,4 | 6,4 |
| CVr [%] | 2,2 | 3,8 | 2,7 | 4,1 | 3,8 |
| SR [mg/kg] | 830 | 65,0 | 55,5 | 18,9 | 7,8 |
| R [mg/kg] | 2 324 | 182,0 | 155,4 | 52,9 | 21,8 |
| CVR [%] | 4,8 | 5,5 | 6,0 | 6,0 | 12,7 |
L
=
number of laboratories
n
=
number of single values
sr
=
standard deviation of repeatability
sR
=
standard deviation of reproducibility
r
=
repeatability
R
=
reproducibility
CVr
=
coefficient of variation of repeatability
CVR
=
coefficient of variation of reproducibility

C.DETERMINATION OF THE TRACE ELEMENTS IRON, COPPER, MANGANESE AND ZINCU.K.
1.Purpose and scopeU.K.
The method makes it possible to determine the trace elements iron, copper, manganese and zinc in feed. The limits of quantification are:
iron (Fe): 20 mg/kg
copper (Cu): 10 mg/kg
manganese (Mn): 20 mg/kg
zinc (Zn): 20 mg/kg.
2.PrincipleU.K.
The sample is brought into solution in hydrochloric acid after destruction of organic matter, if any. The elements iron, copper, manganese and zinc are determined, after appropriate dilution, by atomic absorption spectrometry.
3.ReagentsU.K.
Introductory commentsU.K.
For preparation of the reagents and analytical solutions use water free from the cations to be determined, obtained either by double distilling water in a borosilicate glass or quartz still or by double treatment on ion exchange resin.
The reagents must be of at least analytical grade. Freedom from the element to be determined must be checked in a blank experiment. If necessary, the reagents must be further purified.
In place of the standard solutions described below, commercial standard solutions may be used provided that they are guaranteed and have been checked before use.
3.1.Hydrochloric acid (d:1,19 g/ml).U.K.
3.2.Hydrochloric acid (6 mol/litre).U.K.
3.3.Hydrochloric acid (0,5 mol/litre).U.K.
3.4.Hydrofluoric acid 38 % to 40 % (v/v) having an iron (Fe) content of less than 1 mg/litre and a residue after evaporation of less than 10 mg (as sulphate)/litre.U.K.
3.5.Sulphuric acid (d: 1,84 g/ml).U.K.
3.6.Hydrogen peroxide (approximately 100 volumes of oxygen (30 % by weight)).U.K.
3.7.Standard iron solution (1 000 μg Fe/ml) prepared as follows or equivalent commercially available solution: dissolve 1 g of iron wire in 200 ml of 6 mol/litre hydrochloric acid (3.2), add 16 ml of hydrogen peroxide (3.6) and make up to one litre with water.U.K.
3.7.1.Working standard iron solution (100 μg Fe/ml) prepared by diluting one part of the standard solution (3.7) with 9 parts of water.U.K.
3.8.Standard copper solution (1 000 μg Cu/ml) prepared as follows or equivalent commercially available solution:U.K.
dissolve 1 g of copper in powder form in 25 ml of 6 mol/litre hydrochloric acid (3.2), add 5 ml of hydrogen peroxide (3.6) and make up to one litre with water.
3.8.1.Working standard copper solution (10 μg Cu/ml) prepared by diluting 1 part of the standard solution (3.8) with 9 parts of water and then diluting 1 part of the resulting solution with 9 parts of water.U.K.
3.9.Standard manganese solution (1 000 μg Mn/ml) prepared as follows or equivalent commercially available solution:U.K.
dissolve 1 g of manganese in powder form in 25 ml of 6 mol/litre hydrochloric acid (3.2) and make up to one litre with water.
3.9.1.Working standard manganese solution (10 μg Mn/ml) prepared by diluting 1 part of the standard solution (3.9) with 9 parts of water and then diluting 1 part of the resulting solution with 9 parts of water.U.K.
3.10.Standard zinc solution (1 000 μg Zn/ml) prepared as follows or equivalent commercially available solution:U.K.
dissolve 1 g of zinc in strip or leaf form in 25 ml of 6 mol/litre hydrochloric acid (3.2) and make up to one litre with water.
3.10.1.Working standard zinc solution (10 μg Zn/ml) prepared by diluting 1 part of the standard solution (3.10) with 9 parts of water and then diluting 1 part of the resulting solution with 9 parts of water.U.K.
3.11.Lanthanum chloride solution: dissolve 12 g of lanthanum oxide in 150 ml of water, add 100 ml of 6 mol/litre hydrochloric acid (3.2) and make up to one litre with water.U.K.
4.ApparatusU.K.
4.1.Muffle furnace with temperature regulation and preferably recorder.U.K.
4.2.Glassware must be of resistant borosilicate type and it is recommended to use apparatus which is reserved exclusively for trace element determinations.U.K.
4.3.Atomic absorption spectrophotometer meeting the requirements of the method with regard to sensitivity and precision in the required range.U.K.
5.Procedure(32) U.K.
5.1.Samples containing organic matterU.K.
5.1.1.Ashing and preparation of the solution for analysis(33)U.K.
5.1.1.1.Place 5 to 10 g of sample weighed to the nearest 0,2 mg in a quartz or platinum crucible (see Note (b)), dry in an oven at 105 oC and introduce the crucible into the cold muffle furnace (4.1). Close the furnace (see Note (c)) and gradually raise the temperature to 450 to 475 oC over about 90 minutes. Maintain this temperature for 4 to 16 hours (e.g. overnight) to remove carbonaceous material and then open the furnace and allow to cool (see Note (d)).U.K.
Moisten the ashes with water and transfer these in a beaker of 250 ml. Wash the crucible out with a total of about 5 ml of hydrochloric acid (3.1) and add the latter slowly and carefully to the beaker (there may be a vigorous reaction due to CO2 formation). Add hydrochloric acid (3.1) dropwise with agitation until all effervescence has stopped. Evaporate to dryness, occasionally stirring with a glass rod.
Next add 15 ml of 6 mol/litre hydrochloric acid (3.2) to the residue followed by about 120 ml of water. Stir with the glass rod, which shall be left in the beaker, and cover the beaker with a watch-glass. Bring gently to the boil and maintain at boiling point until no more ash can be seen to dissolve. Filter on ash-free filter paper and collect the filtrate in a 250 ml volumetric flask. Wash the beaker and filter with 5 ml of hot 6 mol/litre hydrochloric acid (3.2) and twice with boiling water. Fill the volumetric flask up to the mark with water (HCl concentration about 0,5 mol/litre).
5.1.1.2.If the residue in the filter appears black (carbon), put it back in the furnace and ash again at 450 to 475 oC. This ashing, which only requires a few hours (about three to five hours), is complete when the ash appears white or nearly white. Dissolve the residue with about 2 ml of hydrochloric acid (3.1), evaporate to dryness and add 5 ml of 6 mol/litre hydrochloric acid (3.2). Heat, filter the solution into the volumetric flask and make up to the mark with water (HCl concentration about 0,5 mol/litre.U.K.
Notes:U.K.
(a)
In determining trace elements it is important to be alert to the risks of contamination, particularly by zinc, copper and iron. For this reason, the equipment used in preparing the samples must be free of these metals.
To reduce the general risk of contamination, work in a dust-free atmosphere with scrupulously clean equipment and carefully washed glassware. The determination of zinc is particularly sensitive to many types of contamination, e.g. from glassware, reagents, dust, etc.
(b)
The weight of sample to be ashed is calculated from the approximate trace element content of the feed in relation to the sensitivity of the spectrophotometer used. For certain feed low in trace elements it may be necessary to start with a 10 to 20 g sample and make up the final solution to only 100 ml.
(c)
Ashing must be carried out in a closed furnace without injection of air or oxygen.
(d)
The temperature indicated by the pyrometer must not exceed 475 oC.
5.1.2.Spectrophotometric determinationU.K.
5.1.2.1.Preparation of calibration solutionsU.K.
For each of the elements to be determined, prepare from the working standard solutions given in points 3.7.1, 3.8.1, 3.9.1 and 3.10.1 a range of calibration solutions, each calibration solution having an HCl concentration of about 0,5 mol/litre (and (in the cases of iron, manganese and zinc) a lanthanum chloride concentration equivalent to 0,1 % La (w/v).
The trace element concentrations selected must lie within the range of sensitivity of the spectrophotometer used. The tables below show, by way of example, the compositions of typical ranges of calibration solutions; depending, however, on the type and sensitivity of spectrophotometer used it may be necessary to select other concentrations.
IronU.K.
| μg Fe/ml | 0 | 0,5 | 1 | 2 | 3 | 4 | 5 |
| ml working standard solution (3.7.1) (1 ml = 100 μg Fe) | 0 | 0,5 | 1 | 2 | 3 | 4 | 5 |
| ml HCl (3.2) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml of lanthanum chloride solution (3.11) and make up to 100 ml with water | |||||||
CopperU.K.
| μg Cu/ml | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| ml working standard solution (3.8.1) (1 ml = 10 μg Cu) | 0 | 1 | 2 | 4 | 6 | 8 | 10 |
| ml HCl (3.2) | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
ManganeseU.K.
| μg Mn/ml | 0 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 | 1,0 |
| ml working standard solution (3.9.1) (1 ml = 10 μg Mn) | 0 | 1 | 2 | 4 | 6 | 8 | 10 |
| ml HCl (3.2) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml of lanthanum chloride solution (3.11) and make up to 100 ml with water | |||||||
ZincU.K.
| μg Zn/ml | 0 | 0,05 | 0,1 | 0,2 | 0,4 | 0,6 | 0,8 |
| ml working standard solution (3.10.1) (1 ml = 10 μg Zn) | 0 | 0,5 | 1 | 2 | 4 | 6 | 8 |
| ml HCl (3.2) | 7 | 7 | 7 | 7 | 7 | 7 | 7 |
| + 10 ml of lanthanum chloride solution (3.11) and make up to 100 ml with water | |||||||
5.1.2.2.Preparation of solution for analysisU.K.
For the determination of copper, the solution prepared from point 5.1.1 can normally be used directly. If necessary to bring its concentration within the range of the calibration solutions, an aliquot portion may be pipetted into a 100 ml volumetric flask and made up to the mark with 0,5 mol/litre hydrochloric acid (3.3).
For the determination of iron, manganese and zinc, pipette an aliquot portion of the solution prepared from point 5.1.1 into a 100 ml volumetric flask, add 10 ml of lanthanum chloride solution (3.11) and make up to the mark with 0,5 mol/litre hydrochloric acid (3.3) (see also point 8 ‘Observation’).
5.1.2.3.Blank experimentU.K.
The blank experiment must include all the prescribed steps of the procedure except that the sample material is omitted. The calibration solution ‘0’ must not be used as the blank.
5.1.2.4.Measurement of the atomic absorptionU.K.
Measure the atomic absorption of the calibration solutions and of the solution to be analysed using an oxidising air-acetylene flame at the following wavelengths:
Fe: 248,3 nm
Cu: 324,8 nm
Mn: 279,5 nm
Zn: 213,8 nm
Carry out each measurement four times.
5.2.Mineral feedU.K.
If the sample contains no organic matter, prior ashing is unnecessary. Proceed as described in point 5.1.1.1 starting from the second paragraph. Evaporation with hydrofluoric acid may be omitted.
6.Calculation of resultsU.K.
Using a calibration curve, calculate the trace element concentration in the solution to be analysed and express the result in milligrams of trace element per kilogram of sample (ppm).
7.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample by the same analyst shall not exceed:
5 mg/kg, in absolute value, for contents of the trace element concerned up to 50 mg/kg,
10 % of the higher result for contents of the trace element concerned from 50 and up to 100 mg/kg,
10 mg/kg, in absolute value, for contents of the trace element concerned from 100 and up to 200 mg/kg,
5 % of the higher result for contents of the trace element concerned above 200 mg/kg.
8.ObservationU.K.
The presence of large quantities of phosphates may interfere with the determination of iron, manganese and zinc. Such interference must be corrected by addition of lanthanum chloride solution (3.11). If, however, in the sample the weight ratio Ca + Mg/P is > 2, addition of lanthanum chloride solution (3.11) to the solution for analysis and to the calibration solutions may be omitted.
D.DETERMINATION OF HALOFUGINONEU.K.
DL-trans-7-bromo-6-chloro-3- [3-(3-hydroxy-2-piperidyl)acetonyl]-quinazolin-4-(3H)-one hydrobromide
1.Purpose and scopeU.K.
The method makes it possible to determine the level of halofuginone in feed. The limit of quantification is 1 mg/kg.
2.PrincipleU.K.
After treatment with hot water, halofuginone is extracted as the free base into ethyl acetate and subsequently partitioned as the hydrochloride into an aqueous acid solution. The extract is purified by ion-exchange chromatography. The content of halofuginone is determined by reversed-phase high performance liquid chromatography (HPLC) using an UV detector.
3.ReagentsU.K.
3.1.Acetonitrile, equivalent to HPLC grade.U.K.
3.2.Amberlite XAD-2 resin.U.K.
3.3.Ammonium acetate.U.K.
3.4.Ethyl acetate.U.K.
3.5.Acetic acid, glacial.U.K.
3.6.Halofuginone standard substance (DL-trans-7-brome-6-chloro-3-[3-hydroxy-2-piperidyl)acetonyl] quinazoline-4-(3H)-one hydrobromide, E 764).U.K.
3.6.1.Halofuginone stock standard solution, 100 μg/mlU.K.
Weight to the nearest 0,1 mg, 50 mg of halofuginone (3.6) in a 500 ml graduated flask, dissolve in ammonium acetate buffer solution (3.18), make up to the mark with the buffer solution and mix. This solution is stable for three weeks at 5 oC if stored in the dark.
3.6.2.Calibration solutionsU.K.
Into a series of 100 ml graduated flasks transfer 1,0, 2,0, 3,0, 4,0 and 6,0 ml of the stock standard solution (3.6.1). Make up to the mark with mobile phase (3.21) and mix. These solutions have concentrations of 1,0, 2,0, 3,0, 4,0 and 6,0 μg/ml of halofuginone respectively. These solutions must be freshly prepared before use.
3.7.Hydrochloric acid (ρ20 approximately 1,16 g/ml).U.K.
3.8.Methanol.U.K.
3.9.Silver nitrate.U.K.
3.10.Sodium ascorbate.U.K.
3.11.Sodium carbonate.U.K.
3.12.Sodium chloride.U.K.
3.13.EDTA (ethylenediaminetetraacetic acid, disodium salt).U.K.
3.14.Water, equivalent to HPLC grade.U.K.
3.15.Sodium carbonate solution, c = 10 g/100 ml.U.K.
3.16.Sodium chloride-saturated sodium carbonate solution, c = 5 g/100 ml.U.K.
Dissolve 50 g of sodium carbonate (3.11) in water, dilute to 1 litre and add sodium chloride (3.12) until the solution is saturated.
3.17.Hydrochloric acid, approximately 0,1 mol/l.U.K.
Dilute 10 ml of HCI (3.7) with water to 1 litre.
3.18.Ammonium acetate buffer solution, approximately 0,25 mol/l.U.K.
Dissolve 19,3 g of ammonium acetate (3.3) and 30 ml of acetic acid (3.5) in water (3.14) and dilute to 1 1itre.
3.19.Amberlite XAD-2 resin preparation.U.K.
Wash an appropriate quantity of Amberlite (3.2) with water until all chloride ions have been removed, as indicated by a silver nitrate (3.20) test performed on the discarded aqueous phase. Then wash the resin with 50 ml of methanol (3.8), discard the methanol and store the resin under fresh methanol.
3.20.Silver nitrate solution, approximately 0,1 mol/l.U.K.
Dissolve 0,17 g of silver nitrate (3.9) in 10 ml of water.
3.21.HPLC Mobile phase.U.K.
Mix 500 ml of acetonitrile (3.1) with 300 ml of ammonium acetate buffer solution (3.18) and 1 200 ml of water (3.14). Adjust the pH to 4,3 using acetic acid (3.5). Filter through a 0,22 μm filter (4.8) and degas the solution (e.g. by ultrasonification for 10 minutes). This solution is stable for one month, if stored in the dark in a closed container.
4.ApparatusU.K.
4.1.Ultrasonic bathU.K.
4.2.Rotary film evaporatorU.K.
4.3.CentrifugeU.K.
4.4.HPLC equipment with variable wavelength ultraviolet detector or diode-array detectorU.K.
4.4.1.Liquid chromatographic column, 300 mm x 4 mm, C18, 10 μm packaging, or an equivalent columnU.K.
4.5.Glass column (300 mm x 10 mm) fitted with a sintered-glass filter and a stopcockU.K.
4.6.Glass-fibre filters, diameter 150 mmU.K.
4.7.Membrane filters, 0,45 μmU.K.
4.8.Membrane filters, 0,22 μmU.K.
5.ProcedureU.K.
Note:
Halofuginone as the free base is unstable in alkaline and ethyl acetate solutions. It shall not remain in ethyl acetate for more than 30 minutes.
5.1.GeneralU.K.
5.1.1.A blank feed shall be analysed to check that neither halofuginone nor interfering substances are present.U.K.
5.1.2.A recovery test shall be carried out by analysing the blank feed which has been fortified by addition of a quantity of halofuginone, similar to that present in the sample. To fortify at a level of 3 mg/kg, add 300 μl of the stock standard solution (3.6.1) to 10 g of the blank feed, mix and wait for 10 minutes before proceeding with the extraction step (5.2).U.K.
Note:
for the purpose of this method, the blank feed shall be similar in type to that of the sample and on analysis halofuginone shall not be detected.
5.2.ExtractionU.K.
Weigh to the nearest 0,1 g, 10 g of the prepared sample, into a 200 ml centrifuge tube, add 0,5 g of sodium ascorbate (3.10), 0,5 g of EDTA (3.13) and 20 ml of water and mix. Place the tube for 5 minutes in a water bath (80 oC). After cooling down to room temperature, add 20 ml of sodium carbonate solution (3.15) and mix. Add immediately 100 ml of ethyl acetate (3.4) and shake vigorously by hand for 15 seconds. Then place the tube for three minutes in the ultrasonic bath (4.1) and loosen the stopper. Centrifuge for two minutes and decant the ethyl acetate phase through a glass fibre filter (4.6), into a 500 ml separating funnel. Repeat the extraction of the sample with a second portion of 100 ml of ethyl acetate. Wash the combined extracts for one minute with 50 ml of sodium chloride saturated sodium carbonate solution (3.16) and discard the aqueous layer.
Extract the organic layer for 1 min. with 50 ml of hydrochloric acid (3.17). Run the lower acid layer into a 250 ml separating funnel. Re-extract the organic layer for 1,5 minutes with a further 50 ml of hydrochlorid acid and combine with the first extract. Wash the combined acid extracts by swirling for approximately 10 seconds with 10 ml of ethyl acetate (3.4).
Quantitatively transfer the aqueous layer into a 250 ml round-bottomed flask and discard the organic phase. Evaporate all the remaining ethyl acetate from the acid solution using a rotary film evaporator (4.2). The temperature of the water bath must not exceed 40 oC. Under a vacuum of approximately 25 mbar all of the residual ethyl acetate will be removed within 5 minutes at 38 oC.
5.3.Clean upU.K.
5.3.1.Preparation of the Amberlite columnU.K.
An XAD-2 column is prepared for each sample extract. Transfer 10 g of prepared Amberlite (3.19) into a glass column (4.5) with methanol (3.8). Add a small plug of glass-wool to the top of the resin bed. Drain the methanol from the column and wash the resin with 100 ml of water, stopping the flow as the liquid reaches the top of the resin bed. Allow the column to equilibrate for 10 minutes before use. Never allow the column to run dry.
5.3.2.Sample clean upU.K.
Transfer the extract (5.2) quantitatively to the top of the prepared Amberlite column (5.3.1) and elute, discarding the eluate. The rate of elution must not exceed 20 ml/min. Rinse the round-bottomed flask with 20 ml of hydrochlorid acid (3.17) and use this to wash the resin column. Blow through any remaining acid solution with a stream of air. Discard the washings. Add 100 ml of methanol (3.8) to the column and allow 5 to 10 ml to elute, collecting the eluate in a 250 ml round-bottomed flask. Leave the remaining methanol for 10 minutes to equilibrate with the resin and continue the elution at a rate not exceeding 20 ml/min. collecting the eluate in the same round-bottomed flask. Evaporate the methanol on the rotary film evaporator (4.2), the temperature of the water bath must not exceed 40 oC. Transfer the residue quantitatively into a 10 ml calibrated flask using the mobile phase (3.21). Make up to the mark with mobile phase and mix. An aliquot is filtered through a membrane filter (4.7). Reserve this solution for the HPLC determination (5.4).
5.4.HPLC determinationU.K.
5.4.1.ParametersU.K.
The following conditions are offered for guidance, other conditions may be used provided they yield equivalent results.
Liquid chromatographic column (4.4.1)
HPLC Mobile phase (3.21)
Flow rate: 1,5 to 2 ml/min.
Detection wavelength: 243 nm
Injection volume: 40 to 100 μl.
Check the stability of the chromatographic system, injecting the calibration solution (3.6.2) containing 3,0 μg/ml several times, until constant peak heights (or areas) and retention times are achieved.
5.4.2.Calibration graphU.K.
Inject each calibration solution (3.6.2) several times and measure the peak heights (areas) for each concentration. Plot a calibration graph using the mean peak heights or areas of the calibration solutions as the ordinates and the corresponding concentrations in μg/ml as the abscissae.
5.4.3.Sample solutionU.K.
Inject the sample extract (5.3.2) several times, using the same volume as taken for the calibration solutions and determine the mean peak height (area) of the halofuginone peaks.
6.Calculation of resultsU.K.
Determine the concentration of the sample solution in μg/ml, from the mean height (area) of the halofuginone peaks of the sample solution by reference to the calibration graph (5.4.2).
The content of halofuginone w (mg/kg) of the sample is given by the following formula:

in which:
c
=
halofuginone concentration of the sample solution in μg/ml,
m
=
weight of the test portion in grams.
7.Validation of the resultsU.K.
7.1.IdentityU.K.
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract and the calibration solution (3.6.2) containing 6,0 μg/ml are compared.
7.1.1.Co-chromatographyU.K.
A sample extract is fortified by addition of an appropriate amount of a calibration solution (3.6.2). The amount of added halofuginone must be similar to the estimated amount of halofuginone found in the sample extract.
Only the height of the halofuginone peak shall be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its maximum height, must be within ± 10 % of the original width.
7.1.2.Diode-array detectionU.K.
The results are evaluated according to the following criteria:
(a)
the wavelength of maximum absorption of the sample and of the standard spectra, recorded at the peak apex on the chromatogram, must be the same within a margin determined by the resolving power of the detection system. For diode-array detection, this is typically within ± 2 nm;
(b)
between 225 and 300 nm, the sample and standard spectra recorded at the peak apex on the chromatogram, must not be different for those parts of the spectrum within the range 10 % to 100 % of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15 % of the absorbance of the standard analyte;
(c)
between 225 and 300 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 % to 100 % of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15 % of the absorbance of the spectrum of the apex.
If one of these criteria is not met the presence of the analyte has not been confirmed.
7.2.RepeatabilityU.K.
The difference between results of two parallel determinations carried out on the same sample must not exceed 0,5 mg/kg for halofuginone contents up to 3 mg/kg.
7.3.RecoveryU.K.
For the fortified blank sample the recovery shall be at least 80 %.
8.Results of a collaborative studyU.K.
A collaborative study(34) was arranged in which three samples were analysed by eight laboratories.
Results
| Sample A(blank)On receipt | Sample B (Meal) | Sample C (Pellets) | |||
|---|---|---|---|---|---|
| On receipt | After two months | On receipt | After two months | ||
| Mean [mg/kg] | ND | 2,8 | 2,42 | 2,89 | 2,45 |
| SR [mg/kg] | — | 0,45 | 0,43 | 0,4 | 0,42 |
| CVR [%] | — | 16 | 18 | 14 | 17 |
| Rec. [%] | 86 | 74 | 88 | 75 | |
ND
=
not detected
SR
=
standard deviation of reproducibility
CVR
=
coefficient of variation of reproducibility (%)
Rec.
=
recovery (%)
E.DETERMINATION OF ROBENIDINEU.K.
1,3-bis [(4-chlorobenzylidene)amino]guanidine — hydrochloride
1.Purpose and scopeU.K.
This method makes it possible to determine the levels of robenidine in feed. The limit of quantification is 5 mg/kg.
2.PrincipleU.K.
The sample is extracted with acidified methanol. The extract is dried and an aliquot portion subjected to a clean-up on an aluminium oxide column. Robenidine is eluted from the column with methanol, concentrated, and made up to a suitable volume with mobile phase. The content of robenidine is determined by reversed-phase high-performance liquid chromatography (HPLC) using an UV detector.
3.ReagentsU.K.
3.1.Methanol.U.K.
3.2.Acidified methanol.U.K.
Transfer 4,0 ml hydrochloric acid (ρ20 = 1,18 g/ml) into a 500 ml graduated flask, make up to the mark with methanol (3.1) and mix. This solution shall be freshly prepared before use.
3.3.Acetonitrile, equivalent to HPLC grade.U.K.
3.4.Molecular sieve.U.K.
Type 3A, 8 to 12 mesh beads (1,6-2,5 mm beads, crystalline alumino-silicate, diameter of pores 0,3 mm).
3.5.Aluminium oxide acidic activity grade I for column chromatography.U.K.
Transfer 100 g aluminium oxide into a suitable container and add 2,0 ml of water. Stopper and shake for approximately 20 minutes. Store in a well stoppered container.
3.6.Potassium dihydrogen phosphate solution, c = 0,025 mol/l.U.K.
Dissolve 3,4 g of potassium dihydrogen phosphate in water (HPLC grade) in a 1 000 ml graduated flask, make up to the mark and mix.
3.7.Di-sodium hydrogen phosphate solution, c = 0,025 mol/l.U.K.
Dissolve 3,55 g of anhydrous (or 4,45 g of dihydrate or 8,95 g of dodecahydrate) di-sodium hydrogen phosphate in water (equivalent to HPLC grade) in a 1 litre graduated flask, make up to the mark and mix.
3.8.HPLC mobile phase.U.K.
Mix together the following reagents:
650 ml acetonitrile (3.3),
250 ml water (equivalent to HPLC-grade),
50 ml potassium di-hydrogen phosphate solution (3.6),
50 ml di-sodium hydrogen phosphate solution (3.7).
Filter through a 0,22 μm filter (4.6) and degas the solution, (e.g. by ultrasonification for 10 minutes).
3.9.Standard substance.U.K.
Pure robenidine: 1,3-bis [(4-chlorobenzylidene)amino]guanidine — hydrochloride.
3.9.1.Robenidine stock standard solution: 300 μg/mlU.K.
Weigh to the nearest 0,1 mg, 30 mg of robenidine standard substance (3.9). Dissolve in acidified methanol (3.2) in a 100 ml graduated flask, make up to the mark with the same solvent and mix. Wrap the flask with aluminium foil and store in a dark place.
3.9.2.Robenidine intermediate standard solution: 12 μg/mlU.K.
Transfer 10,0 ml of the stock standard solution (3.9.1) into a 250 ml graduated flask, make up to the mark with the mobile phase (3.8) and mix. Wrap the flask with aluminium foil and store in a dark place.
3.9.3.Calibration solutionsU.K.
Into a series of 50 ml calibrated flasks, transfer 5,0, 10,0, 15,0, 20,0 and 25,0 ml of the intermediate standard solution (3.9.2). Make up to the mark with mobile phase (3.8) and mix. These solutions correspond to 1,2, 2,4, 3,6, 4,8 and 6,0 μg/ml of robenidine respectively. These solutions must be freshly prepared before use.
3.10.Water equivalent to HPLC grade.U.K.
4.ApparatusU.K.
4.1.Glass column.U.K.
Constructed of amber glass fitted with a stopcock and a reservoir of approximately 150 ml capacity, internal diameter 10 to 15 mm, length 250 mm.
4.2.Mechanical shaker or magnetic stirrer.U.K.
4.3.Rotary film evaporator.U.K.
4.4.HPLC equipment with variable wavelength ultraviolet detector or diode array detector operating in the range of 250 to 400 nm.U.K.
4.4.1.Liquid chromatographic column: 300 mm x 4 mm, C18 10 μm packing or equivalent.U.K.
4.5.Glass fibre filter paper (Whatman GF/A or equivalent).U.K.
4.6.Membrane filters, 0,22 μm.U.K.
4.7.Membrane filters, 0,45 μm.U.K.
5.ProcedureU.K.
Note:
Robenidine is light-sensitive. Amber glassware shall be used in all operations.
5.1.GeneralU.K.
5.1.1.A blank feed shall be analysed to check that neither robenidine nor interfering substances are present.U.K.
5.1.2.A recovery test shall be carried out by analysing the blank feed (5.1.1) which has been fortified by addition of a quantity of robenidine, similar to that present in the sample. To fortify at a level of 60 mg/kg, transfer 3,0 ml of the stock standard solution (3.9.1) to a 250 ml conical flask. Evaporate the solution to ca. 0,5 ml in a stream of nitrogen. Add 15 g of the blank feed, mix and wait for 10 minutes before proceeding with the extraction step (5.2).U.K.
Note:
For the purpose of this method, the blank feed shall be similar in type to that of the sample and on analysis robenidine shall not be detected.
5.2.ExtractionU.K.
Weigh to the nearest 0,01 g, approximately 15 g of the prepared sample. Transfer to a 250 ml conical flask and add 100,0 ml of acidified methanol (3.2), stopper and shake for one hour on the shaker (4.2). Filter the solution through a glass fibre filter paper (4.5) and collect the whole filtrate in a 150 ml conical flask. Add 7,5 g molecular sieve (3.4), stopper and shake for five minutes. Filter immediately through a glass-fibre filter paper. Retain this solution for the purification step (5.3).
5.3.PurificationU.K.
5.3.1.Preparation of the aluminium-oxide columnU.K.
Insert a small glass-wool plug into the lower end of a glass column (4.1) and tamp it down using a glass rod. Weigh out 11,0 g of the prepared aluminium oxide (3.5) and transfer to the column. Care shall be taken to minimise the exposure to the atmosphere during this stage. Gently tap the loaded column at its lower end to settle the aluminium oxide.
5.3.2.Sample purificationU.K.
Transfer onto the column by pipette 5,0 ml of the sample extract prepared in (5.2) Rest the pipette tip close to the column wall and allow the solution to be absorbed onto the aluminium oxide. Elute the robenidine from the column using 100 ml methanol (3.1), at a flow rate of 2 to 3 ml/minute and collect the eluate in a 250 ml round bottomed flask. Evaporate the methanol solution to dryness under reduced pressure at 40 oC by means of a rotary film evaporator (4.3). Re-dissolve the residue in 3 to 4 ml of mobile phase (3.8) and transfer quantitatively to a 10 ml graduated flask. Rinse the flask with several 1 to 2 ml portions of mobile phase and transfer these rinsings to the graduated flask. Make up to the mark with the same solvent and mix. An aliquot is filtered through a 0,45 μm membrane filter (4.7). Reserve this solution for HPLC determination (5.4).
5.4.HPLC determinationU.K.
5.4.1.ParametersU.K.
The following conditions are offered for guidance, other conditions may be used provided they yield equivalent results:
Liquid chromatographic column (4.4.1),
HPLC mobile phase (3.8),
Flow rate: 1,5 to 2 ml/minute,
Detector wavelength: 317 nm,
Injection volume: 20 to 50 μl.
Check the stability of the chromatographic system, injecting the calibration solution (3.9.3) containing 3,6 μg/ml several times, until constant peak heights and retention times are achieved.
5.4.2.Calibration graphU.K.
Inject each calibration solution (3.9.3) several times and measure the peak heights (areas) for each concentration. Plot a calibration curve using the mean peak heights or areas of the calibration solutions as the ordinates and corresponding concentrations in μg per ml as abscissae.
5.4.3.Sample solutionU.K.
Inject the sample extract (5.3.2) several times, using the same volume as taken for the calibration solutions and determine the mean peak height (area) of the robenidine peaks.
6.Calculation of resultsU.K.
From the mean height (area) of the robenidine peaks of the sample solution determine the concentration of the sample solution in μg/ml by reference to the calibration graph (5.4.2).
The content of robenidine w (mg/kg) in the sample is given by the following formula:

in which:
c
=
robenidine concentration of the sample solution in μg/ml,
m
=
weight of the test portion in grams.
7.Validation of the resultsU.K.
7.1.IdentityU.K.
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract and the calibration solution (3.9.3) containing 6 μg/ml are compared.
7.1.1.Co-chromatographyU.K.
A sample extract is fortified by addition of an appropriate amount of calibration solution (3.9.3). The amount of added robenidine must be similar to the estimated amount of robenidine found in the sample extract.
Only the height of the robenidine peak shall be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its maximum height, must be within approximately 10 % of the original width.
7.1.2.Diode-array detectionU.K.
The results are evaluated according to the following criteria:
(a)
the wavelength of maximum absorption of the sample and of the standard spectra, recorded at the peak apex on the chromatogram, must be the same within a margin determined by the resolving power of the detection system. For diode-array detection, this is typically within approximately 2 nm;
(b)
between 250 and 400 nm, the sample and standard spectra recorded at the peak apex on the chromatogram, must not be different for those parts of the spectrum within the range 10 % to 100 % of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15 % of the absorbance of the standard analyte;
(c)
between 250 and 400 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 % to 100 % of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15 % of the absorbance of the spectrum of the apex.
If one of these criteria is not met the presence of the analyte has not been confirmed.
7.2.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed 10 % of the higher result for robenidine content higher than 15 mg/kg.
7.3.RecoveryU.K.
For a fortified blank sample the recovery shall be at least 85 %.
8.Results of a collaborative studyU.K.
An EC collaborative study was arranged in which four samples of poultry and rabbit feed, in meal or pelleted form were analysed by 12 laboratories. Duplicate analyses were performed on each sample. The results are given in the table below:
| Poultry | Rabbit | |||
|---|---|---|---|---|
| Meal | Pellet | Meal | Pellet | |
| Mean [mg/kg] | 27,0 | 27,99 | 43,6 | 40,1 |
| sr [mg/kg] | 1,46 | 1,26 | 1,44 | 1,66 |
| CVr [%] | 5,4 | 4,5 | 3,3 | 4,1 |
| SR [mg/kg] | 4,36 | 3,36 | 4,61 | 3,91 |
| CVR [%] | 16,1 | 12,0 | 10,6 | 9,7 |
| Recovery [%] | 90,0 | 93,3 | 87,2 | 80,2 |
sr
=
standard deviation of repeatability,
CVr
=
coefficient of variation of repeatability, %
SR
=
standard deviation of reproducibility,
CVR
=
coefficient of variation of reproducibility. %
F.DETERMINATION OF DICLAZURILU.K.
(+)-4-chlorphenyl [2,6-dichloro-4-(2,3,4,5-tetrahydro-3,5-dioxo-1,2,4-triazin-2-yl)phenyl] acetonitrile
1.Purpose and scopeU.K.
The method makes it possible to determine the level of diclazuril in feed and premixtures. The limit of detection is 0,1 mg/kg, the limit of quantification is 0,5 mg/kg.
2.PrincipleU.K.
After addition of an internal standard, the sample is extracted with acidified methanol. For feed, an aliquot of the extract is purified on a C18 solid phase extraction cartridge. Diclazuril is eluted from the cartridge with a mixture of acidified methanol and water. After evaporation, the residue is dissolved in DMF/water. For premixtures, the extract is evaporated and the residue is dissolved in DMF/water. The content of diclazuril is determined by ternary gradient reversed-phase high-performance liquid chromatography (HPLC) using a UV detector.
3.ReagentsU.K.
3.1.Water, equivalent to HPLC-gradeU.K.
3.2.Ammonium acetateU.K.
3.3.Tetrabutylammonium hydrogen sulphate (TBHS)U.K.
3.4.Acetonitrile, equivalent to HPLC gradeU.K.
3.5.Methanol, equivalent to HPLC gradeU.K.
3.6.N, N-dimethylformamide (DMF)U.K.
3.7.Hydrochloric acid, ρ20 = 1,19 g/mlU.K.
3.8.Standard substance: diclazuril II-24: (+)-4-chlorphenyl [2,6-dichloro-4-(2,3,4,5-tetrahydro-3,5-dioxo-1,2,4-triazin-2-yl) phenyl] acetonitrile with guaranteed purity, E771U.K.
3.8.1.Diclazuril stock standard solution, 500 μg/mlU.K.
Weigh to the nearest 0,1 mg, 25 mg of diclazuril standard substance (3.8) in a 50 ml graduated flask. Dissolve in DMF (3.6), make up to the mark with DMF (3.6) and mix. Wrap the flask with aluminium foil or use amber flask and store in the refrigerator. At a temperature of ≤ 4 oC the solution is stable for 1 month.
3.8.2.Diclazuril standard solution, 50 μg/mlU.K.
Transfer 5,0 ml of the stock standard solution (3.8.1) into a 50 ml graduated flask, make up to the mark with DMF (3.6) and mix. Wrap the flask with aluminium foil or use amber flask and store in the refrigerator. At a temperature of ≤ 4 oC the solution is stable for 1 month.
3.9.Internal standard substance: 2,6 dichloro-α-(4-chlorophenyl)-4-(4,5 dihydro-3,5-dioxo-1,2,4-triazine-2 (3H) — yl) α-methylbenzene-acetonitrileU.K.
3.9.1.Internal stock standard solution, 500 μg/mlU.K.
Weigh to the nearest 0,1 mg 25 mg of internal standard substance (3.9) in a 50 ml graduated flask. Dissolve in DMF (3.6), make up to the mark with DMF (3.6) and mix. Wrap the flask with aluminium foil or use amber flask and store in the refrigerator. At a temperature of ≤ 4 oC the solution is stable for 1 month.
3.9.2.Internal standard solution, 50 μg/mlU.K.
Transfer 5,0 ml of the internal stock standard solution (3.9.1) into a 50 ml graduated flask, make up to the mark with DMF (3.6) and mix. Wrap the flask with aluminium foil or use amber flask and store in the refrigerator. At a temperature of ≤ 4 oC the solution is stable for 1 month.
3.9.3.Internal standard solution for premixtures, p/1 000 mg/mlU.K.
(p = nominal content of diclazuril in the premixture in mg/kg)
Weigh to the nearest 0,1 mg p/10 mg of the internal standard substance in a 100 ml graduated flask, dissolve in DMF (3.6) in a ultrasonic bath (4.6), make up to the mark with DMF and mix. Wrap the flask with aluminium foil or use amber flask and store in a refrigerator. At a temperature of ≤ 4 oC the solution is stable for 1 month.
3.10.Calibration solution, 2 μg/ml.U.K.
Pipet 2,0 ml diclazuril standard solution (3.8.2) and 2,0 ml internal standard solution (3.9.2) into a 50 ml graduated flask. Add 16 ml DMF (3.6), make up to the mark with water and mix. This solution must be prepared freshly before use.
3.11.C18 solid phase extraction cartridge, e.g. Bond Elut, size: 1 cc, sorbent weight: 100 mg.U.K.
3.12.Extraction solvent: acidified methanol.U.K.
Pipet 5,0 ml hydrochloric acid (3.7) into 1 000 ml of methanol (3.5), and mix.
3.13.Mobile phase for HPLCU.K.
3.13.1.Eluent A: ammonium acetate — tetrabutylammonium hydrogen sulphate solution.U.K.
Dissolve 5 g ammonium acetate (3.2) and 3,4 g TBHS (3.3) in 1 000 ml water (3.1) and mix.
3.13.2.Eluent B: acetonitrile (3.4).U.K.
3.13.3.Eluent C: methanol (3.5).U.K.
4.ApparatusU.K.
4.1.Mechanical shakerU.K.
4.2.Equipment for ternary gradient HPLCU.K.
4.2.1.Liquid chromatographic column, Hypersil ODS, 3 μm packing, 100 mm x 4,6 mm, or equivalentU.K.
4.2.2.UV detector with variable wavelength adjustment or diode array detectorU.K.
4.3.Rotary film evaporatorU.K.
4.4.Membrane filter, 0,45 μmU.K.
4.5.Vacuum manifoldU.K.
4.6.Ultrasonic bathU.K.
5.ProcedureU.K.
5.1.GeneralU.K.
5.1.1.Blank feedU.K.
A blank feed shall be analysed to check that neither diclazuril nor interfering substances are present. The blank feed shall be similar in type to that of the sample and on analysis diclazuril or interfering substances shall not be detected.
5.1.2.Recovery testU.K.
A recovery test shall be carried out by analysing the blank feed which has been fortified by addition of a quantity of diclazuril similar to that present in the sample. To fortify at a level of 1 mg/kg add 0,1 ml of the stock standard solution (3.8.1) to 50 g of a blank feed, mix thoroughly and leave for 10 min. mixing again several times before proceeding (5.2).
Alternatively, if a blank feed similar in type to that of the sample is not available (see 5.1.1), a recovery test can be performed by means of the standard addition method. In this case, the sample to be analysed is fortified with a quantity of diclazuril, similar to that already present in the sample. This sample is analysed, together with the unfortified sample and the recovery can be calculated by subtraction.
5.2.ExtractionU.K.
5.2.1.FeedU.K.
Weigh to the nearest 0,01 g approximately 50 g of the sample. Transfer to a 500 ml conical flask, add 1,0 ml internal standard solution (3.9.2), 200 ml extraction solvent (3.12) and stopper the flask. Shake the mixture on the shaker (4.1) overnight. Allow to settle for 10 minutes. Transfer a 20 ml aliquot of the supernatant to a suitable glass container and dilute with 20 ml water. Transfer this solution on an extraction cartridge (3.11), and pass through by applying vacuum (4.5). Wash the cartridge with 25 ml of a mixture of extraction solvent (3.12) and water, 65 + 35 (V + V). Discard the collected fractions and elute the compounds with 25 ml of a mixture of extraction solvent (3.12) and water, 80 + 20 (V + V). Evaporate this fraction until it had just reached dryness by means of the rotary evaporator (4.3) at 60 oC. Dissolve the residue in 1,0 ml DMF (3.6), add 1,5 ml of water (3.1) and mix. Filter through a membrane filter (4.4). Proceed to the HPLC determination (5.3).
5.2.2.PremixturesU.K.
Weigh to the nearest 0,001 g approximately 1 g of the sample. Transfer to a 500 ml conical flask, add 1,0 ml internal standard solution (3.9.3), 200 ml extraction solvent (3.12) and stopper the flask. Shake the mixture overnight on the shaker (4.1). Allow to settle for 10 minutes. Transfer an aliquot of 10 000/p ml (p = nominal content of diclazuril in the premix in mg/kg) of the supernatant to a round bottomed flask of suitable size. Evaporate until it had just reached dryness, under reduced pressure at 60 oC by means of the rotary evaporator (4.3). Redissolve the residue in 10,0ml DMF (3.6), add 15,0 ml water (3.1) and mix. Proceed to the HPLC determination (5.3).
5.3.HPLC determinationU.K.
5.3.1.ParametersU.K.
The following conditions are offered for guidance, other conditions may be used provided that they give equivalent results.
| Liquid chromatographic column (4.2.1) | 100 mm × 4,6 mm, Hypersil ODS, 3 μm packing, or equivalent | |
| Mobile phase: | Eluent A (3.13.1): | Aqueous solution of ammonium acetate and tetrabutyl-ammonium hydrogen sulphate |
| Eluent B (3.13.2): | acetonitrile | |
| Eluent C (3.13.3): | methanol | |
| Elution mode: |
Flush with B during 10 min. | |
| Flow rate: | 1,5-2 ml/min. | |
| Injection volume: | 20 μl | |
| Detector wavelength: | 280 nm. | |
Check the stability of the chromatographic system, injecting several times the calibration solution (3.10), containing 2 μg/ml, until constant peak heights and retention times are achieved.
5.3.2.Calibration solutionU.K.
Inject 20 μl of the calibration solution (3.10) several times and determine the mean peak height (area) of the diclazuril and internal standard peaks.
5.3.3.Sample solutionU.K.
Inject 20 μl of the sample solution (5.2.1 or 5.2.2) several times and determine the mean peak height (area) of the diclazuril and internal standard peaks.
6.Calculation of the resultsU.K.
6.1.FeedsU.K.
The diclazuril content w (mg/kg) in the sample is given by the following formula:

where:
hd,s
=
peak height (area) of diclazuril in the sample solution (5.2.1)
hi,s
=
peak height (area) of the internal standard in the sample solution (5.2.1)
hd,c
=
peak height (area) of diclazuril in the calibration solution (3.10)
hi,c
=
peak height (area) of the internal standard in the calibration solution (3.10)
cd,c
=
diclazuril concentration in the calibration solution in μg/ml (3.10)
m
=
weight of the test portion in g
V
=
volume of the sample extract according to 5.2.1 (i.e. 2,5 ml)
6.2.PremixturesU.K.
The diclazuril content w (mg/kg) in the sample is given by the following formula:

where:
hd,c
=
peak height (area) of diclazuril in the calibration solution (3.10)
hi,c
=
peak height (area) of the internal standard in the calibration solution (3.10)
hd,s
=
peak height (area) of diclazuril in the sample solution (5.2.2)
hi,s
=
peak height (area) of the internal standard in the sample solution (5.2.2)
cd,c
=
diclazuril concentration in the calibration solution in μg/ml (3.10)
m
=
weight of the test portion in g
V
=
volume of the sample extract according to 5.2.2 (i.e. 25 ml)
p
=
nominal content of diclazuril in mg/kg in the premixture
7.Validation of the resultsU.K.
7.1.IdentityU.K.
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract (5.2.1 or 5.2.2) and the calibration solution (3.10) are compared.
7.1.1.Co-chromatographyU.K.
A sample extract (5.2.1 or 5.2.2) is fortified by addition of an appropriate amount of calibration solution (3.10). The amount of added diclazuril must be similar to the amount of diclazuril found in the sample extract.
Only the height of the diclazuril peak and the internal standard peak shall be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its height, must be within ± 10 % of the original width of the diclazuril peak or the internal standard peak of the unfortified sample extract.
7.1.2.Diode-array detectionU.K.
The results are evaluated according to the following criteria:
(a)
The wavelength of maximum absorption of the sample and of the standard spectra, recorded at the peak apex on the chromatogram, must be the same within a margin determined by the resolving power of the detection system. For diode-array detection this is typically within ± 2 nm.
(b)
Between 230 and 320 nm, the sample and standard spectra recorded at the peak apex of the chromatogram, must not be different for those parts of the spectrum within the range 10 % 100 % of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15 % of the absorbance of the standard analyte.
(c)
Between 230 and 320 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 % 100 % of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15 % of the absorbance of the spectrum of the peak apex.
If one of these criteria is not met the presence of the analyte has not been confirmed.
7.2.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed:
30 % relative, to the higher value for diclazuril contents from 0,5 mg/kg to 2,5 mg/kg,
0,75 mg/kg for diclazuril contents between 2,5 mg/kg and 5 mg/kg,
15 % relative to the higher value for diclazuril contents of more than 5 mg/kg.
7.3.RecoveryU.K.
For a fortified (blank) sample the recovery shall be at least 80 %.
8.Results of a collaborative studyU.K.
A collaborative study was arranged in which 5 samples were analysed by 11 laboratories. These samples consisted of two premixtures; one was mixed with an organic matrix (O 100) and the other with an inorganic matrix (A 100). The theoretical content is 100 mg diclazuril per kg. The three mixed feeds for poultry were made by 3 different producers (NL) (L1/Z1/K1). The theoretical content is 1 mg diclazuril per kg. The laboratories were instructed to analyse each of the samples once or in duplicate. (More detailed information on this collaborative study can be found in the Journal of AOAC International, Volume 77, No 6, 1994, p. 1359-1361). The results are given in the following table.
| Sample 1A 100 | Sample 2O 100 | Sample 3L1 | Sample 4Z1 | Sample 5K1 | |
|---|---|---|---|---|---|
| L | 11 | 11 | 11 | 11 | 6 |
| n | 19 | 18 | 19 | 19 | 12 |
| Mean | 100,8 | 103,5 | 0,89 | 1,15 | 0,89 |
| Sr (mg/kg) | 5,88 | 7,64 | 0,15 | 0,02 | 0,03 |
| CVr (%) | 5,83 | 7,38 | 17,32 | 1,92 | 3,34 |
| SR (mg/kg) | 7,59 | 7,64 | 0,17 | 0,11 | 0,12 |
| CVR (%) | 7,53 | 7,38 | 18,61 | 9,67 | 13,65 |
| Nominal content (mg/kg) | 100 | 100 | 1 | 1 | 1 |
L
=
number of laboratories
n
=
number of single values
Sr
=
standard deviation of repeatability
CVr
=
coefficient of variation of repeatability
SR
=
standard deviation of reproducibility
CVR
=
coefficient of variation of reproducibility
9.ObservationsU.K.
The diclazuril response must have been previously demonstrated to be linear over the range of concentrations being measured.
G.DETERMINATION OF LASALOCID SODIUMU.K.
Sodium salt of a polyether monocarboxylic acid produced by Streptomyces lasaliensis
1.Purpose and scopeU.K.
The method makes it possible to determine the level of lasalocid sodium in feed and premixtures. The limit of detection is 5 mg/kg, the limit of quantification is 10 mg/kg.
2.PrincipleU.K.
Lasalocid sodium is extracted from the sample into acidified methanol and determined by reversed-phase high performance liquid chromatography (HPLC) using a spectrofluorometric detector.
3.ReagentsU.K.
3.1.Potassium dihydrogen phosphate (KH2PO4).U.K.
3.2.Orthophosphoric acid, w (w/w) = 85 %.U.K.
3.3.Orthophosphoric acid solution, c = 20 %.U.K.
Dilute 23,5 ml of orthophosphoric acid (3.2) to 100 ml with water.
3.4.6-Methyl-2-heptylamine (1,5-dimethylhexylamine), w (w/w) = 99 %.U.K.
3.5.Methanol, equivalent to HPLC grade.U.K.
3.6.Hydrochloric acid, density = 1,19 g/ml.U.K.
3.7.Phosphate buffer solution, c = 0,01 mol/l.U.K.
Dissolve 1,36 g of KH2PO4 (3.1) in 500 ml of water (3.11), add 3,5 ml of orthophosphoric acid (3.2) and 10,0 ml of 6-methyl-2-heptylamine (3.4). Adjust the pH to 4,0 with orthophosphoric acid solution (3.3) and dilute to 1 000 ml with water (3.11).
3.8.Acidified methanol.U.K.
Transfer 5,0 ml of hydrochloric acid (3.6) into a 1 000 ml graduated flask, make up to the mark with methanol (3.5) and mix. This solution must be prepared freshly before use.
3.9.HPLC mobile phase, phosphate buffer-methanol solution 5 + 95 (V + V).U.K.
Mix 5 ml of phosphate buffer solution (3.7) with 95 ml of methanol (3.5).
3.10.Lasalocid sodium standard substance with guaranteed purity, C34H53O8Na (sodium salt of a polyether monocarboxylic acid produced by Streptomyces lasaliensis), E763.U.K.
3.10.1.Lasalocid sodium stock standard solution, 500 μg/mlU.K.
Weigh to the nearest 0,1 mg, 50 mg of lasalocid sodium (3.10) into a 100 ml graduated flask, dissolve in acidified methanol (3.8), make up to the mark with the same solvent and mix. This solution must be freshly prepared before use.
3.10.2.Lasalocid sodium intermediate standard solution, 50 μg/mlU.K.
Pipette 10,0 ml of stock standard solution (3.10.1) into a 100 ml graduated flask, make up to the mark with acidified methanol (3.8) and mix. This solution must be prepared freshly before use.
3.10.3.Calibration solutionsU.K.
Into a series of 50 ml graduated flasks transfer 1,0, 2,0, 4,0, 5,0 and 10,0 ml of the intermediate standard solution (3.10.2). Make up to the mark with acidified methanol (3.8) and mix. These solutions correspond to 1,0, 2,0, 4,0, 5,0 and 10,0 μg of lasalocid sodium per ml respectively. These solutions must be prepared freshly before use.
3.11.Water, equivalent to HPLC grade.U.K.
4.ApparatusU.K.
4.1.Ultrasonic bath (or shaking water-bath) with temperature control.U.K.
4.2.Membrane filters, 0,45 μm.U.K.
4.3.HPLC equipment with injection system, suitable for injecting volumes of 20 μl.U.K.
4.3.1.Liquid chromatographic column 125 mm x 4 mm, reversed-phase C18, 5 μm packing or equivalent.U.K.
4.3.2.Spectrofluorometer with variable wavelength adjustment of excitation and emission wavelengths.U.K.
5.ProcedureU.K.
5.1.GeneralU.K.
5.1.1.Blank feedU.K.
For the performance of the recovery test (5.1.2) a blank feed shall be analysed to check that neither lasalocid sodium nor interfering substances are present. The blank feed shall be similar in type to that of the sample and lasalocid sodium or interfering substances shall not be detected.
5.1.2.Recovery testU.K.
A recovery test shall be carried out by analysing the blank feed which has been fortified by addition of a quantity of lasalocid sodium, similar to that present in the sample. To fortify at a level of 100 mg/kg, transfer 10,0 ml of the stock standard (3.10.1) to a 250 ml conical flask and evaporate the solution to approximately 0,5 ml. Add 50 g of the blank feed, mix thoroughly and leave for 10 minutes mixing again several times before proceeding with the extraction step (5.2).
Alternatively, if a blank feed similar in type to that of the sample is not available (see 5.1.1), a recovery test can be performed by means of the standard addition method. In this case the sample to be analysed is fortified with a quantity of lasalocid sodium similar to that already present in the sample. This sample is analysed together with the unfortified sample and the recovery calculated by subtraction.
5.2.ExtractionU.K.
5.2.1.FeedU.K.
Weigh to the nearest 0,01 g, from 5 g to 10 g of the sample into a 250 ml conical flask with stopper. Add 100,0 ml of acidified methanol (3.8) by pipette. Stopper loosely and swirl to disperse. Place the flask in an ultrasonic bath (4.1) at approximately 40 oC for 20 minutes, then remove and cool to room temperature. Allow to stand for about 1 hour until the suspended matter has settled, then filter an aliquot portion through a 0,45 μm membrane filter (4.2) into a suitable vessel. Proceed to the HPLC determination (5.3).
5.2.2.PremixturesU.K.
Weigh to the nearest 0,001 g about 2 g of the unground premix into a 250 ml graduated flask. Add 100,0 ml of acidified methanol (3.8) and swirl to disperse. Place the flask and contents in an ultrasonic bath (4.1) at approximately 40 oC for 20 minutes, then remove and cool to room temperature. Dilute to the mark with acidified methanol (3.8) and mix thoroughly. Allow to stand for 1 hour until the suspended matter has settled, then filter an aliquot portion through a 0,45 μm membrane filter (4.2). Dilute an appropriate volume of the clear filtrate with acidified methanol (3.8) to produce a final test solution containing about 4 μg/ml of lasalocid sodium. Proceed to the HPLC determination (5.3).
5.3.HPLC determinationU.K.
5.3.1.ParametersU.K.
The following conditions are offered for guidance; other conditions may be used, provided they yield equivalent results:
| Liquid chromatographic column (4.3.1): | 125 mm × 4 mm, reversed-phase C18, 5 μm packing or equivalent |
| Mobile phase (3.9): | Mixture of phosphate buffer solution (3.7) and methanol (3.5), 5+95 (V+V) |
| Flow rate: | 1,2 ml/min. |
| Detection wavelengths: | |
| Excitation: | 310 nm |
| Emission: | 419 nm |
| Injection volume: | 20 μl |
Check the stability of the chromatographic system, injecting the calibration solution (3.10.3) containing 4,0 μg/ml several times, until constant peak heights (or areas) and retention times are achieved.
5.3.2.Calibration graphU.K.
Inject each calibration solution (3.10.3) several times and determine the mean peak heights (areas) for each concentration. Plot a calibration graph using the mean peak heights (areas) as the ordinates and the corresponding concentrations in μg/ml as the abscissae.
5.3.3.Sample solutionU.K.
Inject the sample extracts obtained in 5.2.1 or 5.2.2 several times, using the same volume as taken for the calibration solution and determine the mean peak heights (areas) of the lasalocid sodium peaks.
6.Calculation of resultsU.K.
From the mean peak height (area) produced by injection of the sample solution (5.3.3) determine the concentration of lasalocid sodium (μg/ml) by reference to the calibration graph.
6.1.FeedU.K.
The lasalocid sodium content, w (mg/kg) in the sample is given by the following formula:

where:
c
=
lasalocid sodium concentration of the sample solution (5.2.1) in μg/ml
V1
=
volume of the sample extract according to 5.2.1 in ml (i.e. 100)
m
=
weight of the test portion in g
6.2.PremixturesU.K.
The lasalocid sodium content, w (mg/kg) in the sample is given by the following formula:

where:
c
=
lasalocid sodium concentration of the sample solution (5.2.2) in μg/ml
V2
=
volume of the sample extract according to 5.2.2 in ml (i.e. 250)
f
=
dilution factor according to 5.2.2
m
=
weight of the test portion in g
7.Validation of the resultsU.K.
7.1.IdentityU.K.
Methods based on spectrofluorometry are less subject to interference than those in which UV detection is used. The identity of the analyte can be confirmed by co-chromatography.
7.1.1.Co-chromatographyU.K.
A sample extract (5.2.1 or 5.2.2) is fortified by the addition of an appropriate amount of a calibration solution (3.10.3). The amount of added lasalocid sodium must be similar to the amount of lasalocid sodium found in the sample extract. Only the height of the lasalocid sodium peak shall be enhanced after taking into account the amount of lasalocid sodium added and the dilution of the extract. The peak width, at half height, must be within ± 10 % of the original peak width produced by the unfortified sample extract.
7.2.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed:
15 % relative to the higher value for lasalocid sodium contents from 30 mg/kg to 100 mg/kg,
15 mg/kg for lasalocid sodium contents from 100 mg/kg to 200 mg/kg,
7,5 % relative to the higher value for lasalocid sodium contents of more than 200 mg/kg.
7.3.RecoveryU.K.
For the fortified (blank) feed sample, the recovery shall be at least 80 %. For the fortified premixture samples, the recovery shall be at least 90 %.
8.Results of a collaborative studyU.K.
A collaborative study(35) was arranged in which 2 premixtures (samples 1 and 2) and 5 feeds (samples 3-7) were analysed by 12 laboratories. Duplicate analyses were performed on each sample. The results are given in the following table:
a Content declared by manufacturer. | |||||||
b Feed prepared in the laboratory. | |||||||
| Sample 1Chicken premix | Sample 2Turkey premix | Sample 3Turkey pellets | Sample 4Chicken crumbs | Sample 5Turkey Feed | Sample 6Poultry Feed A | Sample 7Poultry Feed B | |
|---|---|---|---|---|---|---|---|
| L | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| n | 23 | 23 | 23 | 23 | 23 | 23 | 23 |
| Mean [mg/kg] | 5 050 | 16 200 | 76,5 | 78,4 | 92,9 | 48,3 | 32,6 |
| sr [mg/kg] | 107 | 408 | 1,71 | 2,23 | 2,27 | 1,93 | 1,75 |
| CVr [%] | 2,12 | 2,52 | 2,24 | 2,84 | 2,44 | 4,0 | 5,37 |
| sR [mg/kg] | 286 | 883 | 3,85 | 7,32 | 5,29 | 3,47 | 3,49 |
| CVR [%] | 5,66 | 5,45 | 5,03 | 9,34 | 5,69 | 7,18 | 10,7 |
| Nominal content [mg/kg] | 5 000a | 16 000a | 80a | 105a | 120a | 50b | 35b |
L
=
number of laboratories
n
=
number of single results
sr
=
standard deviation of repeatability
sR
=
standard deviation of reproducibility
CVr
=
coefficient of variation of repeatability, %
CVR
=
coefficient of variation of reproducibility, %.
ANNEX VU.K.METHODS OF ANALYSIS TO CONTROL UNDESIRABLE SUBSTANCES IN FEED
A.DETERMINATION OF FREE AND TOTAL GOSSYPOLU.K.
1.Purpose and scopeU.K.
This method makes it possible to determine the levels of free gossypol, total gossypol and chemically related substances in cottonseed, cottonseed meal and cottonseed cake and in compound feed containing these feed materials where more than 20 mg/kg of free gossypol, total gossypol and chemically related substances are present.
2.PrincipleU.K.
The gossypol is extracted in the presence of 3-aminopropan-1-ol, either with a mixture of propan-2-ol and hexane, for the determination of free gossypol, or with dimethylformamide, for the determination of total gossypol. The gossypol is converted by aniline into gossypol-dianiline, the optical density of which is measured at 440 nm.
3.ReagentsU.K.
3.1.Propan-2-ol-hexane mixture: mix 60 parts by volume of propan-2-ol with 40 parts by volume of n-hexane.U.K.
3.2.Solvent A: Place in a 1 litre graduated flask approximately 500 ml of propan-2-ol-hexane mixture (3.1), 2 ml of 3-aminopropan-1-ol, 8 ml of glacial acetic acid and 50 ml of water. Make up to volume with the propan-2-ol-hexane mixture (3.1). This reagent is stable for one week.U.K.
3.3.Solvent B: Pipette 2 ml of 3-aminopropan-1-ol and 10 ml of glacial acetic acid into a 100 ml graduated flask. Cool to room temperature and make up to volume with N, N-dimethylformamide. This reagent is stable for one week.U.K.
3.4.Aniline: If the optical density in the blank test exceeds 0,022, distil the aniline over zinc dust, discarding the first and last 10 % fractions of the distillate. Refrigerated and stored in a brown, stoppered glass flask, this reagent will keep for several months.U.K.
3.5.Standard gossypol solution A: Place 27,9 mg of gossypol acetate in a 250 ml graduated flask. Dissolve and make up to volume with solvent A (3.2). Pipette 50 ml of this solution into a 250 ml graduated flask and make up to volume with solvent A. The gossypol concentration of this solution is 0,02 mg/ml. Leave to stand for one hour at room temperature before use.U.K.
3.6.Standard gossypol solution B: Place 27,9 mg of gossypol acetate in a 50 ml graduated flask, Dissolve and make up to volume with solvent B (3.3). The gossypol concentration of this solution is 0,5 mg/ml.U.K.
Standard gossypol solutions A and B will remain stable for 24 hours if protected from the light.
4.ApparatusU.K.
4.1.Mixer (tumbler): approximately 35 r.p.m.U.K.
4.2.Spectrophotometer.U.K.
5.ProcedureU.K.
5.1.Test sampleU.K.
The amount of test sample used depends on the presumed gossypol content of the sample. It is preferable to work with a small test sample and a relatively large aliquot part of the filtrate, so as to obtain sufficient gossypol for precise photometric measurement to be possible. For the determination of free gossypol in cottonseed, cottonseed meal and cottonseed cake, the test sample shall not exceed 1 g; for compound feed, it may be as much as 5 g. A 10 ml aliquot part of filtrate is suitable in most cases; it shall contain 50 to 100 μg of gossypol. For the determination of total gossypol, the test sample shall be between 0,5 and 5 g, that a 2 ml aliquot part of filtrate will contain 40 to 200 μg of gossypol.
The analysis shall be carried out at a room temperature of about 20 oC.
5.2.Determination of free gossypolU.K.
Place the test sample in a ground-necked 250 ml flask, the bottom of the flask having been covered with crushed glass. Using a pipette, add 50 ml of solvent A (3.2), stopper the flask and mix for one hour in the mixer. Filter through a dry filter and collect the filtrate in a small ground-necked flask. During filtration, cover the funnel with a watch glass.
Pipette identical aliquot parts of filtrate containing 50 to 100 μg of gossypol into each of two 25 ml graduated flasks (A and B). If necessary, make up the volume to 10 ml with solvent A (3.2). Then make the contents of flask (A) up to volume with the propan-2-ol-hexane mixture (3.1). This solution will be used as a reference solution against which to measure the sample solution.
Pipette 10 ml of solvent A (3.2) into each of two other 25 ml graduated flasks (C and D). Make the contents of flask (C) up to volume with the propan-2-ol-hexane mixture (3.1). This solution will be used as a reference solution against which to measure the blank test solution.
Add 2 ml of aniline (3.4) to each of flasks (D) and (B). Heat for 30 minutes over a boiling water bath to develop the colour. Cool to room temperature, make up to volume with the propan-2-ol-hexane mixture (3.1), homogenise and leave to stand for one hour.
Determine the optical density of the blank test solution (D) by comparison with the reference solution (C), and the optical density of the sample solution (B) by comparison with the reference solution (A), in the spectrophotometer at 440 nm using 1 cm glass cells.
Subtract the optical density of the blank test solution from that of the sample solution (= corrected optical density). From this value calculate the free gossypol content as indicated in 6.
5.3.Determination of total gossypolU.K.
Place a test sample containing 1 to 5 mg of gossypol in a 50 ml graduated flask and add 10 ml of solvent B (3.3). At the same time, prepare a blank test, placing 10 ml of solvent B (3.3) in another 50 ml graduated flask. Heat the two flasks for 30 minutes over a boiling water bath. Cool to room temperature and make the contents of each flask up to volume with the propan-2-ol-hexane mixture (3.1). Homogenise and leave to settle for 10 to 15 minutes, then filter and collect the filtrates in ground-necked flasks.
Pipette 2 ml of the sample filtrate into each of two 25 ml graduated flasks, and 2 ml of the blank test filtrate into each of two other 25 ml flasks. Make the contents of one flask from each series up to 25 ml with the propan-2-ol-hexane mixture (3.1). These solutions will be used as reference solutions.
Add 2 ml of aniline (3.4) to each of the other two flasks. Heat for 30 minutes over a boiling water bath to develop the colour. Cool to room temperature, make up to 25 ml with the propan-2-ol-hexane mixture (3.1), homogenise and leave to stand for one hour.
Determine the optical density as indicated in 5.2 for free gossypol. From this value calculate the total gossypol content as indicated in 6.
6.Calculation of resultsU.K.
Results may be calculated either from the specific optical density (6.1), or by reference to a calibration curve (6.2).
6.1.From the specific optical densityU.K.
The specific optical densities, under the conditions described, will be the following:

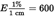
The free or total gossypol content of the sample is calculated by using the following formula:

where:
E
=
corrected optical density, determined as indicated in 5.2,
p
=
test sample in g,
a
=
aliquot part of the filtrate in ml.
6.2.From a calibration curveU.K.
6.2.1.Free gossypolU.K.
Prepare 2 series of five 25 ml graduated flasks. Pipette aliquots of 2,0, 4,0, 6,0, 8,0 and 10,0 ml of standard gossypol solution A (3.5) into each series of flasks. Make up the volumes to 10 ml with solvent A (3.2). Complete each series with a 25 ml graduated flask containing only 10 ml of solvent A (3.2) (blank test).
Make the volume of the flasks in the first series (including the flask for the blank test) up to 25 ml with the propan-2-ol-hexane mixture (3.1) (reference series).
Add 2 ml of aniline (3.4) to each flask in the second series (including the flask for the blank test). Heat for 30 minutes over a boiling water bath to develop the colour. Cool to room temperature, make up to volume with the propan-2-ol-hexane mixture (3.1), homogenise and leave to stand for one hour (standard series).
Determine as indicated in 5.2 the optical density of the solutions in the standard series by comparison with the corresponding solutions in the reference series. Trace the calibration curve by plotting the optical densities against the quantities of gossypol (in μg).
6.2.2.Total gossypolU.K.
Prepare six 50 ml graduated flasks. In the first flask place 10 ml of solvent B (3.3), and in the others 2,0, 4,0, 6,0, 8,0 and 10,0 ml of standard gossypol solution B (3.6) respectively. Make the contents of each flask up to 10 ml with solvent B (3.3). Heat for 30 minutes over a boiling water bath. Cool to room temperature, make up to volume with the propan-2-ol-hexane mixture (3.1) and homogenise.
Place 2,0 ml of these solutions in each of two series of six 25 ml graduated flasks. Make the contents of the flasks in the first series up to 25 ml with the propan-2-ol-hexane mixture (3.1) (reference series).
Add 2 ml of aniline (3.4) to each flask in the second series. Heat for 30 minutes over a boiling water bath. Cool to room temperature, make up to volume with the propan-2-ol-hexane mixture (3.1), homogenise and leave to stand for one hour (standard series).
Determine as indicated in 5.2 the optical density of the solutions in the standard series by comparison with the corresponding solutions in the reference series. Trace the calibration curve by plotting the optical densities against the quantities of gossypol (in μg).
6.3.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed:
15 %, in relative value to the higher level, for gossypol contents of less than 500 ppm,
75 ppm, in absolute value, for contents of not less than 500 ppm and not more than 750 ppm,
10 %, in relative value to the higher value, for contents of more than 750 ppm.
[F17B. DETERMINATION OF THE LEVELS OF DIOXINS (PCDD/PCDF) AND PCBs U.K.
CHAPTER I U.K. Methods of sampling and interpretation of analytical results
1. Scope and definitions U.K.
The samples intended for the official control of the levels of polychlorinated dibenzo-p-dioxins (PCDDs), polychlorinated dibenzofurans (PCDFs), dioxin-like polychlorinated biphenyls (PCBs)(36) and non dioxin-like PCBs in feed shall be taken in accordance with the provisions of Annex I. The quantitative requirements in relation to the control of substances or products uniformly distributed throughout the feed as provided for in point 5.1. of Annex I shall be applied. Aggregate samples thus obtained shall be considered representative for the lots or sublots from which they are taken. Compliance with maximum levels laid down by Directive 2002/32/EC shall be established on the basis of the levels determined in the laboratory samples.
For the purposes of this Part B, the definitions laid down in Annex I to Commission Decision 2002/657/EC(37) shall apply.
In addition to those definitions, the following definitions shall apply for the purpose of this Part B:
‘Screening methods’ means methods used for selection of those samples with levels of PCDD/Fs and dioxin-like PCBs that exceed the maximum levels or the action thresholds. They shall allow a cost-effective high sample-throughput, thus increasing the chance to discover new incidents with high exposure and health risks to consumers. Screening methods shall be based on bioanalytical or GC-MS methods. Results from samples exceeding the cut-off value used to check compliance with the maximum level shall be verified by a full re-analysis from the original sample using a confirmatory method.
‘Confirmatory methods’ means methods that provide full or complementary information enabling the PCDD/Fs and dioxin-like PCBs to be identified and quantified unequivocally at the maximum or in case of need at the action threshold. Such methods utilize gas chromatography/high resolution mass spectrometry (GC-HRMS) or gas chromatography/tandem mass spectrometry (GC-MS/MS).
2. Compliance of the lot or sublot with the maximum level U.K.
2.1. As regards non-dioxin-like PCBs U.K.
The lot or sublot complies with the maximum level if the analytical result for the sum of PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 and PCB 180 (hereafter referred to as non-dioxin-like PCBs) does not exceed the maximum level laid down by Directive 2002/32/EC, taking into account the expanded measurement uncertainty(38). The lot or sublot does not comply with the maximum level as laid down by Directive 2002/32/EC, if the mean of two upper-bound(39) analytical results obtained from duplicate analysis(40), taking into account the expanded measurement uncertainty, exceeds the maximum level beyond reasonable doubt, i.e. the analysed concentration after deduction of the expanded measurement uncertainty is used to assess compliance.
The expanded measurement uncertainty is calculated using a coverage factor of 2 which gives a level of confidence of approximately 95 %. A lot or sublot is non-compliant if the mean of the measured values minus the expanded uncertainty of the mean is above the maximum level.
The rules, mentioned in the paragraphs above under this point, shall apply for the analytical result obtained on the sample for official control. In case of analysis for defence or reference purposes, [F18enactments other than EU-derived domestic legislation apply].
Textual Amendments
F18Words in Annex 5 Pt. B Ch. 1 point 2.1 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 89(a)(i); 2020 c. 1, Sch. 5 para. 1(1)
2.2. As regards PCDD/Fs and dioxin-like PCBs U.K.
The lot or sublot complies with the maximum level if the result of a single analysis
performed by a screening method with a false-compliant rate below 5 %, indicates that the level does not exceed the respective maximum level of PCDD/Fs and the sum of PCDD/Fs and dioxin-like PCBs laid down by Directive 2002/32/EC,
performed by a confirmatory method, does not exceed the respective maximum level of PCDD/Fs and the sum of PCDD/Fs and dioxin-like PCBs laid down by Directive 2002/32/EC, taking into account the expanded measurement uncertainty.
For screening assays a cut-off value shall be established for decisions on sample compliance with the respective maximum levels set for either PCDD/Fs, or for the sum of PCDD/Fs and dioxin-like PCBs.
The lot or sublot does not comply with the maximum level as laid down by Directive 2002/32/EC if the mean of two upper-bound(41) analytical results obtained from duplicate analysis(42) using a confirmatory method, taking into account the expanded measurement uncertainty, exceeds the maximum level beyond reasonable doubt, i.e. the analysed concentration after deduction of the expanded measurement uncertainty is used to assess compliance.
The expanded measurement uncertainty is calculated using a coverage factor of 2 which gives a level of confidence of approximately 95 %. A lot or sublot is non-compliant if the mean of the measured values minus the expanded uncertainty of the mean is above the maximum level.
The sum of the estimated expanded uncertainties of the separate analytical results of PCDD/Fs and dioxin-like PCBs shall be used for the sum of PCDD/Fs and dioxin-like PCBs.
The rules, mentioned in the paragraphs above under this point, shall apply for the analytical result obtained on the sample for official control. In case of analysis for defence or reference purposes, [F19enactments other than EU-derived domestic legislation apply].
Textual Amendments
F19Words in Annex 5 Pt. B Ch. 1 point 2.2 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 89(a)(ii); 2020 c. 1, Sch. 5 para. 1(1)
3. Results exceeding action thresholds as laid down in Annex II to Directive 2002/32/EC U.K.
Action thresholds serve as a tool for the selection of samples in those cases where it is necessary to identify a source of contamination and to take measures for its reduction or elimination. Screening methods shall establish the appropriate cut-off values for selection of those samples. Where significant efforts are necessary to identify a source and to reduce or eliminate the contamination, it is appropriate to confirm exceedance of the action thresholds by duplicate analysis using a confirmatory method and taking into account the expanded measurement uncertainty(43).
CHAPTER II U.K. Sample preparation and requirements for methods of analysis used in offical control of the levels of dioxins (PCDD/Fs) and dioxin-like PCBs in feed
1. Field of application U.K.
The requirements set out in this Chapter shall be applied where feed is analysed for the official control of the levels of 2,3,7,8-substituted PCDD/Fs and dioxin-like PCBs and as regards sample preparation and analytical requirements for other regulatory purposes, which includes the controls performed by the feed business operator to ensure compliance with the provisions of Regulation (EC) No 183/2005 of the European Parliament and of the Council(44).
Monitoring for the presence of PCDD/Fs and dioxin-like PCBs in feed may be performed with two different types of analytical methods:
(a)
Screening methods
The goal of screening methods is to select those samples with levels of PCDD/Fs and dioxin-like PCBs that exceed the maximum levels or the action thresholds. Screening methods shall ensure cost-effective high sample-throughput, thus increasing the chance to discover new incidents with high exposure and health risks of consumers. Their application shall aim to avoid false-compliant results. They may comprise bioanalytical and GC-MS methods.
Screening methods compare the analytical result with a cut-off value, providing a yes/no-decision over the possible exceedance of the maximum level or action threshold. The concentration of PCDD/Fs and the sum of PCDD/Fs and dioxin-like PCBs in samples suspected to be non-compliant with the maximum level shall be determined or confirmed by a confirmatory method.
In addition, screening methods may give an indication of the levels of PCDD/Fs and dioxin-like PCBs present in the sample. In case of application of bioanalytical screening methods the result is expressed as Bioanalytical Equivalents (BEQ), whereas in case of application of physico-chemical GC-MS methods it is expressed as Toxic Equivalents (TEQ). The numerically indicated results of screening methods are suitable for demonstrating compliance or suspected noncompliance or exceedance of action thresholds and give an indication of the range of levels in case of follow-up by confirmatory methods. They are not suitable for purposes such as evaluation of background levels, estimation of intake, following of time trends in levels or re-evaluation of action thresholds and maximum levels.
(b)
Confirmatory methods
Confirmatory methods allow the unequivocal identification and quantification of PCDD/Fs and dioxin-like PCBs present in a sample and provide full information on congener level. Therefore, those methods allow the control of maximum levels and action thresholds, including the confirmation of results obtained by screening methods. Furthermore, results may be used for other purposes such as determination of low background levels in feed monitoring, following of time trends, exposure assessment and building of a database for possible re-evaluation of action thresholds and maximum levels. They are also important for establishing congener patterns in order to identify the source of a possible contamination. Such methods utilise GC-HRMS. For confirming compliance or non-compliance with the maximum level, also GC-MS/MS can be used.
2. Background U.K.
For calculation of TEQ concentrations, the concentrations of the individual substances in a given sample shall be multiplied by their respective Toxic Equivalency Factor (TEF) (see footnote 1 of Chapter I) and subsequently summed to give the total concentration of dioxin-like compounds expressed as TEQs.
For the purposes of this Part B, the accepted specific limit of quantification of an individual congener means the lowest content of the analyte that can be measured with reasonable statistical certainty, fulfilling the identification criteria as described in internationally recognised standards, for example, in standard EN 16215:2012 (Animal feed — Determination of dioxins and dioxin-like PCBs by GC-HRMS and of indicator PCBs by GC-HRMS) and/or in EPA methods 1613 and 1668 as revised.
The limit of quantification of an individual congener may be identified as
(a)
the concentration of an analyte in the extract of a sample which produces an instrumental response at two different ions to be monitored with a S/N (signal/noise) ratio of 3:1 for the less intensive raw data signal; or
(b)
if for technical reasons the signal-to-noise calculation does not provide reliable results, the lowest concentration point on a calibration curve that gives an acceptable (≤ 30 %) and consistent (measured at least at the start and at the end of an analytical series of samples) deviation to the average relative response factor calculated for all points on the calibration curve in each series of samples. The limit of quantification (LOQ) is calculated from the lowest concentration point taking into account the recovery of internal standards and sample intake.
Bioanalytical screening methods will not give results at the congener level but merely an indication(45) of the TEQ level, expressed in BEQ to acknowledge the fact that not all compounds present in a sample extract that produce a response in the test may fulfill or meet all requirements of the TEQ-principle.
Screening and confirmatory methods may only be applied for control of a certain matrix if the methods are sensitive enough to detect levels reliably at the action threshold or maximum level.
3. Quality assurance requirements U.K.
3.1.Measures shall be taken to avoid cross-contamination at each stage of the sampling and analysis procedure.U.K.
3.2.The samples shall be stored and transported in glass, aluminum, polypropylene or polyethylene containers suitable for storage without any influence on the levels of PCDD/Fs and dioxin-like PCBs in the samples. Traces of paper dust shall be removed from the sample container.U.K.
3.3.The sample storage and transportation shall be performed in a way that maintains the integrity of the feed sample.U.K.
3.4.Insofar as relevant, each laboratory sample shall be finely grinded and mixed thoroughly using a process that has been demonstrated to achieve complete homogenisation (for example, ground to pass a 1 mm sieve). Samples shall be dried before grinding if the moisture content is too high.U.K.
3.5.Control of reagents, glassware and equipment for possible influence of TEQ- or BEQ-based results shall be carried out.U.K.
3.6.A blank analysis shall be performed by carrying out the entire analytical procedure omitting only the sample.U.K.
3.7.For bioanalytical methods, all glassware and solvents used in analysis shall be tested to be free of compounds that interfere with the detection of target compounds in the working range. Glassware shall be rinsed with solvents or heated at temperatures suitable to remove traces of PCDD/Fs, dioxin-like compounds and interfering compounds from its surface.U.K.
3.8.Sample quantity used for the extraction shall be sufficient to fulfill the requirements with respect to a sufficiently low working range including the concentrations of maximum levels or action threshold.U.K.
3.9.The specific sample preparation procedures used for the products under consideration shall follow internationally accepted guidelines.U.K.
4. Requirements for laboratories U.K.
4.1.In accordance with the provisions of Regulation (EC) No 882/2004, laboratories shall be accredited by a recognised body operating in accordance with ISO Guide 58 to ensure that they are applying analytical quality assurance. Laboratories shall be accredited following the EN ISO/IEC 17025 standard. The principles as described in the Technical Guidelines for the estimation of measurement uncertainty and limits of quantification for PCDD/F and PCB analysis shall be followed when applicable(46)U.K.
4.2.Laboratory proficiency shall be proven by the continuous successful participation in inter-laboratory studies for the determination of PCDD/Fs and dioxin-like PCBs in relevant feed matrices and concentration ranges.U.K.
4.3.Laboratories applying screening methods for the routine control of samples shall establish a close cooperation with laboratories applying the confirmatory method, both for quality control and confirmation of the analytical result of suspected samples.U.K.
5. Basic requirements to be met by analytical procedure for dioxins (PCDD/Fs) and dioxin-like PCBs U.K.
5.1. Low working range and limits of quantification U.K.
For PCDD/Fs, detectable quantities shall be in the upper femtogram (10 – 15 g) range because of extreme toxicity of some of these compounds. For most PCB congeners a limit of quantification in the nanogram (10 – 9 g) range is already sufficient. For the measurement of the more toxic dioxin-like PCB congeners (in particular non-ortho-substituted congeners), the lower end of the working range shall reach the low picogram (10 – 12 g) levels. For all other PCB congeners a limit of quantification in the nanogram (10 – 9 g) range is sufficient.
5.2. High selectivity (specificity) U.K.
5.2.1.A distinction is required between PCDD/Fs and dioxin-like PCBs and a multitude of other, coextracted and possibly interfering compounds present at concentrations up to several orders of magnitude higher than those of the analytes of interest. For GC-MS methods, a differentiation among various congeners is required, such as between toxic (for example, the seventeen 2,3,7,8-substituted PCDD/Fs, and twelve dioxin-like PCBs) and other congeners.U.K.
5.2.2.Bioanalytical methods shall be able to detect the target compounds as the sum of PCDD/Fs, and/or dioxin-like PCBs. Sample clean-up shall aim at removing compounds causing false non-compliant results or compounds that may decrease the response, causing false compliant results.U.K.
5.3. High accuracy (trueness and precision, bioassay apparent recovery) U.K.
5.3.1.For GC-MS methods, the determination shall provide a valid estimate of the true concentration in a sample. High accuracy is required to avoid the rejection of a sample analysis result on the basis of poor reliability of the determined TEQ level. Accuracy is expressed as trueness (difference between the mean value measured for an analyte in a certified material and its certified value, expressed as a percentage of this value) and precision (RSD R relative standard deviation calculated from results generated under reproducibility conditions).U.K.
5.3.2.For bioanalytical methods, the bioassay apparent recovery shall be determined. Bioassay apparent recovery means the BEQ level calculated from the TCDD or PCB 126 calibration curve corrected for the blank and then divided by the TEQ level determined by the confirmatory method. It aims at correcting factors like the loss of PCDD/Fs and dioxin-like compounds during the extraction and clean-up steps, co-extracted compounds increasing or decreasing the response (agonistic and antagonistic effects), the quality of the curve fit, or differences between the TEF values and the Relative Potency (REP) values. The bioassay apparent recovery is calculated from suitable reference samples with representative congener patterns around the level of interest.U.K.
5.4. Validation in the range of maximum level and general quality control measures U.K.
5.4.1.Laboratories shall demonstrate the performance of a method in the range of the maximum level, for example, 0,5x, 1x and 2x the maximum level with an acceptable coefficient of variation for repeated analysis, during the validation procedure and during routine analysis.U.K.
5.4.2.Regular blank controls and spiking experiments or analysis of control samples (preferably, if available, certified reference material) shall be performed as internal quality control measures. Quality control charts for blank controls, spiking experiments or analysis of control samples shall be recorded and checked to make sure the analytical performance is in accordance with the requirements.U.K.
5.5. Limit of quantification U.K.
5.5.1.For a bioanalytical screening method, the establishment of the limit of quantification (LOQ) is not an indispensable requirement but the method shall prove that it can differentiate between the blank and the cut-off value. When providing a BEQ level, a reporting level shall be established to deal with samples showing a response below this level. The reporting level shall be demonstrated to be different from procedure blank samples at least by a factor of three, with a response below the working range. It shall therefore be calculated from samples containing the target compounds around the required minimum level, and not from an S/N ratio or an assay blank.U.K.
5.5.2.The LOQ for a confirmatory method shall be about one fifth of the maximum level.U.K.
5.6. Analytical criteria U.K.
For reliable results from confirmatory or screening methods, the following criteria shall be met in the range of the maximum level for the TEQ or BEQ value, respectively, whether determined as total TEQ or total BEQ (as the sum of PCDD/Fs and dioxin-like PCBs) or separately for PCDD/Fs and dioxin-like PCBs:
a With respect to the maximum levels. | ||
| Screening with bioanalytical or physico-chemical methods | Confirmatory methods | |
|---|---|---|
| False-compliant rate a | < 5 % | |
| Trueness | – 20 % to + 20 % | |
| Repeatability (RSD r ) | < 20 % | |
| Intermediate precision (RSD R ) | < 25 % | < 15 % |
5.7. Specific requirements for screening methods U.K.
5.7.1.Both GC-MS and bioanalytical methods may be used for screening. For GC-MS methods the requirements laid down in point 6 shall be met. For cell based bioanalytical methods specific requirements are laid down in point 7.U.K.
5.7.2.Laboratories applying screening methods for the routine control of samples shall establish a close cooperation with laboratories applying the confirmatory method.U.K.
5.7.3.Performance verification of the screening method is required during routine analysis, by analytical quality control and on-going method validation. There shall be a continuous programme for the control of compliant results.U.K.
5.7.4.Check on possible suppression of the cell response and cytotoxicity:U.K.
20 % of the sample extracts shall be measured in routine screening without and with 2,3,7,8-TCDD added corresponding to the maximum level or action threshold, to check if the response is possibly suppressed by interfering substances present in the sample extract. The measured concentration of the spiked sample shall be compared to the sum of the concentration of the unspiked extract plus the spiking concentration. If this measured concentration is more than 25 % lower than the calculated (sum) concentration, this is an indication of potential signal suppression and the respective sample shall be submitted to GC-HRMS confirmatory analysis. Results shall be monitored in quality control charts.
5.7.5.Quality control on compliant samples:U.K.
Approximately 2 to 10 % of the compliant samples, depending on sample matrix and laboratory experience, shall be confirmed by GC/HRMS.
5.7.6.Determination of false-compliant rates from quality control data:U.K.
The rate of false-compliant results from screening of samples below and above the maximum level or the action threshold shall be determined. Actual false-compliant rates shall be below 5 %. When a minimum of 20 confirmed results per matrix/matrix group is available from the quality control of compliant samples, conclusions on the false compliant rate shall be drawn from this database. The results from samples analysed in ring trials or during contamination incidents, covering a concentration range up to for example 2x the maximum level (ML), may also be included in the minimum of 20 results for evaluation of the false-compliant rate. The samples shall cover most frequent congener patterns, representing various sources.
Although screening assays shall preferentially aim to detect samples exceeding the action threshold, the criterion for determining false-compliant rates is the maximum level, taking into account the expanded measurement uncertainty of the confirmatory method.
5.7.7.Potential non-compliant samples from screening shall always be verified by a full re-analysis of the original sample by a confirmatory method of analysis. These samples may also be used to evaluate the rate of false non-compliant results. For screening methods, the rate of false non-compliant results shall be the fraction of results confirmed to be compliant from confirmatory analysis, while in previous screening the sample has been declared to be potentially non-compliant. Evaluation of the advantages of the screening method shall be based on comparison of false-non-compliant samples with the total number of samples checked. This rate shall be low enough to make the use of a screening tool advantageous.U.K.
5.7.8.Under validation conditions, bioanalytical methods shall provide a valid indication of the TEQ level, calculated and expressed as BEQ.U.K.
Also for bioanalytical methods carried out under repeated conditions, the intra-laboratory RSD r would typically be smaller than under reproducibility conditions(RSD R )
6. SPECIFIC requirements for GC-MS methods to be complied with for screening or confirmatory purposes U.K.
6.1. Acceptable differences between upper-bound and lower-bound WHO-TEQ results U.K.
The difference between upper-bound level and lower-bound level shall not exceed 20 % for confirmation of exceedance of maximum level or in case of need of action thresholds.
6.2. Control of recoveries U.K.
6.2.1.Addition of 13 C-labelled 2,3,7,8-chlorine-substituted internal PCDD/F standards and of 13 C-labelled internal dioxin-like PCB standards shall be carried out at the very beginning of the analytical method e.g. prior to extraction in order to validate the analytical procedure. At least one congener for each of the tetra- to octa-chlorinated homologous groups for PCDD/Fs and at least one congener for each of the homologous groups for dioxin-like PCBs shall be added (alternatively, at least one congener for each mass spectrometric selected ion recording function used for monitoring PCDD/Fs and dioxin-like PCBs). In the case of confirmatory methods, all 17 13 C-labelled 2,3,7,8-substituted internal PCDD/F standards and all 12 13 C-labelled internal dioxin-like PCB standards shall be used.U.K.
6.2.2.Relative response factors shall also be determined for those congeners for which no 13 C-labelled analogue is added by using appropriate calibration solutions.U.K.
6.2.3.For feed of plant origin and feed of animal origin containing less than 10 % fat, the addition of the internal standards shall be mandatory prior to extraction. For feed of animal origin containing more than 10 % fat, the internal standards shall be added either before or after fat extraction. An appropriate validation of the extraction efficiency shall be carried out, depending on the stage at which internal standards are introduced.U.K.
6.2.4.Prior to GC-MS analysis, 1 or 2 recovery (surrogate) standard(s) shall be added.U.K.
6.2.5.Control of recovery is required. For confirmatory methods, the recoveries of the individual internal standards shall be in the range of 60 to 120 %. Lower or higher recoveries for individual congeners, in particular for some hepta- and octa- chlorinated dibenzo-p-dioxins and dibenzofurans, shall be acceptable on the condition that their contribution to the TEQ value does not exceed 10 % of the total TEQ value (based on sum of PCDD/F and dioxin-like PCBs). For GC-MS screening methods, the recoveries shall be in the range of 30 to 140 %.U.K.
6.3. Removal of interfering substances U.K.
Separation of PCDD/Fs from interfering chlorinated compounds such as non-dioxin-like PCBs and chlorinated diphenyl ethers shall be carried out by suitable chromatographic techniques (preferably with a florisil, alumina and/or carbon column).
Gas-chromatographic separation of isomers shall be < 25 % peak to peak between 1,2,3,4,7,8-HxCDF and 1,2,3,6,7,8-HxCDF.
6.4. Calibration with standard curve U.K.
The range of the calibration curve shall cover the relevant range of maximum level or action thresholds.
6.5. Specific criteria for confirmatory methods U.K.
For GC-HRMS:
In HRMS, the resolution shall typically be greater than or equal to 10 000 for the entire mass range at 10 % valley.
Fulfilment of further identification and confirmation criteria as described in internationally recognised standards, for example, in standard EN 16215:2012 (Animal feed — Determination of dioxins and dioxin-like PCBs by GC-HRMS and of indicator PCBs by GC-HRMS) and/or in EPA methods 1613 and 1668 as revised.
For GC-MS/MS:
Monitoring of at least 2 specific precursor ions, each with one specific corresponding transition product ion for all labelled and unlabelled analytes in the scope of analysis.
Maximum permitted tolerance of relative ion intensities of ± 15 % for selected transition product ions in comparison to calculated or measured values (average from calibration standards), applying identical MS/MS conditions, in particular collision energy and collision gas pressure, for each transition of an analyte.
Resolution for each quadrupole to be set equal to or better than unit mass resolution (unit mass resolution: sufficient resolution to separate two peaks one mass unit apart) in order to minimise possible interferences on the analytes of interest.
Fulfilment of the further criteria as described in internationally recognised standards, for example, in standard EN 16215:2012 (Animal feed — Determination of dioxins and dioxin-like PCBs by GC-HRMS and of indicator PCBs by GC-HRMS) and/or in EPA methods 1613 and 1668 as revised, except the obligation to use GC-HRMS.
7. Specific requirements for bioanalytical methods U.K.
Bioanalytical methods are methods based on the use of biological principles like cell-based assays, receptor-assays or immunoassays. This point 7 establishes requirements for bioanalytical methods in general.
A screening method in principle classifies a sample as compliant or suspected to be non-compliant. For this, the calculated BEQ level is compared to the cut-off value (see point 7.3). Samples below the cut-off value are declared compliant, samples equal or above the cut-off value are suspected to be non-compliant, requiring analysis by a confirmatory method. In practice, a BEQ level corresponding to two-thirds of the maximum level may serve as cut-off value provided that a false-compliant rate below 5 % and an acceptable rate for false non-compliant results are ensured. With separate maximum levels for PCDD/Fs and for the sum of PCDD/Fs and dioxin-like PCBs, checking compliance of samples without fractionation requires appropriate bioassay cut-off values for PCDD/Fs. For checking of samples exceeding the action thresholds, an appropriate percentage of the respective action threshold shall suit as cut-off value.
If an indicative level is expressed in BEQs, sample results shall be in the working range and shall exceed the reporting limit (see points 7.1.1 and 7.1.6).
7.1. Evaluation of the test response U.K.
7.1.1. General requirements U.K.
When calculating the concentrations from a TCDD calibration curve, values at the higher end of the curve will show a high variation (high coefficient of variation (CV)). The working range is the area where this CV is smaller than 15 %. The lower end of the working range (reporting limit) shall be set at least by a factor of three above the procedure blanks. The upper end of the working range is usually represented by the EC 70 value (70 % of maximal effective concentration), but lower if the CV is higher than 15 % in this range. The working range shall be established during validation. Cut-off values (see point 7.3) shall be well within the working range.
Standard solutions and sample extracts shall be tested in triplicate or at least in duplicate. When using duplicates, a standard solution or a control extract tested in four to six wells divided over the plate shall produce a response or concentration (only possible in the working range) based on a CV < 15 %.
7.1.2. Calibration U.K.
7.1.2.1. Calibration with standard curve U.K.
Levels in samples shall be estimated by comparison of the test response with a calibration curve of TCDD (or PCB 126 or a PCDD/PCDF/dioxin-like PCB standard mixture) to calculate the BEQ level in the extract and subsequently in the sample.
Calibration curves shall contain 8 to 12 concentrations (at least in duplicates), with enough concentrations in the lower part of the curve (working range). Special attention shall be paid to the quality of the curve-fit in the working range. As such, the R 2 value is of little or no value in estimating the goodness of fit in non-linear regression. A better fit shall be achieved by minimising the difference between calculated and observed levels in the working range of the curve, for example by minimising the sum of squared residuals.
The estimated level in the sample extract shall be subsequently corrected for the BEQ level calculated for a matrix or solvent blank sample (to account for impurities from solvents and chemicals used), and the apparent recovery (calculated from the BEQ level of suitable reference samples with representative congener patterns around the maximum level or action threshold). To perform a recovery correction, the apparent recovery shall be within the required range (see point 7.1.4). Reference samples used for recovery correction shall comply with the requirements laid down in point 7.2.
7.1.2.2. Calibration with reference samples U.K.
Alternatively, a calibration curve prepared from at least four reference samples (see point 7.2.4): one matrix blank, plus three reference samples at 0,5x, 1x and 2x the maximum level or action threshold may be used, eliminating the need to correct for blank and recovery if matrix properties of the reference samples match those of the unknown samples. In this case, the test response corresponding to two-thirds of the maximum level (see point 7.3) may be calculated directly from these samples and used as cut-off value. For checking of samples exceeding the action thresholds, an appropriate percentage of these action thresholds shall suit as cut-off value.
7.1.3. Separate determination of PCDD/Fs and dioxin-like PCBs U.K.
Extracts may be split into fractions containing PCDD/Fs and dioxin-like PCBs, allowing a separate indication of PCDD/Fs and dioxin-like PCB TEQ levels (in BEQ). A PCB 126 standard calibration curve shall preferentially be used to evaluate results for the fraction containing dioxin-like PCBs.
7.1.4. Bioassay apparent recoveries U.K.
The ‘bioassay apparent recovery’ shall be calculated from suitable reference samples with representative congener patterns around the maximum level or action threshold and expressed as percentage of the BEQ level in comparison to the TEQ level. Depending on the type of assay and TEFs(47) used, the differences between TEF and REP factors for dioxin-like PCBs can cause low apparent recoveries for dioxin-like PCBs in comparison to PCDD/Fs. Therefore, if a separate determination of PCDD/Fs and dioxin-like PCBs is performed, bioassay apparent recoveries shall be: for dioxin-like PCBs 20 % to 60 %, for PCDD/Fs 50 % to 130 % (ranges apply for the TCDD calibration curve). As the contribution of dioxin-like PCBs to the sum of PCDD/Fs and dioxin-like PCBs can vary between different matrices and samples, bioassay apparent recoveries for the sum of PCDD/Fs and dioxin-like PCBs reflect these ranges and shall be between 30 % and 130 %. Any implication of substantially revised TEF values for [F20retained EU law] for PCDD/Fs and dioxin-like PCBs requires the revision of these ranges.
Textual Amendments
F20Words in Annex 5 Pt. B Ch. 2 point 7.1.4 substituted (31.12.2020) by The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 89(b); 2020 c. 1, Sch. 5 para. 1(1)
7.1.5. Control of recoveries for clean-up U.K.
The loss of compounds during the clean-up shall be checked during validation. A blank sample spiked with a mixture of the different congeners shall be submitted to clean-up (at least n = 3) and the recovery and variability checked by a confirmatory method. The recovery shall be within 60 % to 120 % especially for congeners contributing more than 10 % to the TEQ-level in various mixtures.
7.1.6. Reporting limit U.K.
When reporting BEQ levels, a reporting limit shall be determined from relevant matrix samples involving typical congener patterns, but not from the calibration curve of the standards due to low precision in the lower range of the curve. Effects from extraction and clean-up shall be taken into account. The reporting limit shall be set at least by a factor of three above the procedure blanks.
7.2. Use of reference samples U.K.
7.2.1.Reference samples shall represent sample matrix, congener patterns and concentration ranges for PCDD/Fs and dioxin-like PCBs around the maximum level or action threshold.U.K.
7.2.2.A matrix blank, and where it is not possible, a procedure blank, and a reference sample at the maximum level or action threshold shall be included in each test series. These samples shall be extracted and tested at the same time under identical conditions. The reference sample shall show a clearly elevated response in comparison to the blank sample, thus ensuring the suitability of the test. Those samples may be used for blank and recovery corrections.U.K.
7.2.3.Reference samples chosen to perform a recovery correction shall be representative for the test samples, meaning that congener patterns may not lead to an underestimation of levels.U.K.
7.2.4.Extra reference samples at e.g. 0,5x and 2x the maximum level or action threshold may be included to demonstrate the proper performance of the test in the range of interest for the control of the maximum level or action threshold. Combined, these samples may be used for calculating the BEQ levels in test samples (see point 7.1.2.2).U.K.
7.3. Determination of cut-off values U.K.
The relationship between bioanalytical results in BEQ and results from the confirmatory method in TEQ shall be established, for example by matrix-matched calibration experiments, involving reference samples spiked at 0, 0,5x, 1x and 2x the ML, with 6 repetitions on each level (n = 24). Correction factors (blank and recovery) may be estimated from this relationship but shall be checked in accordance with point 7.2.2.
Cut-off values shall be established for decisions over sample compliance with maximum levels or for the control of action thresholds, if relevant, with the respective maximum levels or action threshold set for either PCDD/Fs and dioxin-like PCBs alone, or for the sum of PCDD/Fs and dioxin-like PCBs. They are represented by the lower end-point of the distribution of bioanalytical results (corrected for blank and recovery) corresponding to the decision limit of the confirmatory method based on a 95 % level of confidence, implying a false-compliant rate < 5 %, and on a RSD R < 25 %. The decision limit of the confirmatory method is the maximum level, taking into account the expanded measurement uncertainty.
The cut-off value (in BEQ) may be calculated in accordance with one of the approaches set out in points 7.3.1, 7.3.2 and 7.3.3. (see Figure 1).
7.3.1.Use of the lower band of the 95 % prediction interval at the decision limit of the confirmatory method:U.K.
with:
BEQ DL
BEQ corresponding to the decision limit of the confirmatory method, being the maximum level taking into account the expanded measurement uncertainty
s y,x
residual standard deviation
t α,f = m – 2
student factor (α = 5 %, f = degrees of freedom, single-sided)
m
total number of calibration points (index j)
n
number of repetitions on each level
x i
sample concentration (in TEQ) of calibration point i determined by a confirmatory method


7.3.2.Calculation from bioanalytical results (corrected for blank and recovery) of multiple analyses of samples (n ≥ 6) contaminated at the decision limit of the confirmatory method, as the lower endpoint of the data distribution at the corresponding mean BEQ value:U.K.
Cut-off value = BEQ DL – 1,64 × SD R
with:
SD R
standard deviation of bioassay results at BEQ DL , measured under within-laboratory reproducibility conditions
7.3.3.Calculation as mean value of bioanalytical results (in BEQ, corrected for blank and recovery) from multiple analysis of samples (n ≥ 6) contaminated at two-thirds of the maximum level or action threshold, based on the observation that this level will be around the cut-off value determined under point 7.3.1 or point 7.3.2:U.K.
Calculation of cut-off values based on a 95 % level of confidence implying a false-compliant rate < 5 %, and a RSD R < 25 %:
(1)
from the lower band of the 95 % prediction interval at the decision limit of the confirmatory method.
(2)
from multiple analysis of samples (n ≥ 6) contaminated at the decision limit of the confirmatory method as the lower end-point of the data distribution (represented in the figure by a bell-shaped curve) at the corresponding mean BEQ value.
Figure 1 U.K.
7.3.4. Restrictions to cut-off values U.K.
BEQ-based cut-off values calculated from the RSD R achieved during validation using a limited number of samples with different matrix/congener patterns may be higher than the TEQ-based maximum levels or action thresholds due to a better precision than attainable in routine when an unknown spectrum of possible congener patterns has to be controlled. In such cases, cut-off values shall be calculated from an RSD R = 25 %, or two-thirds of the maximum level or action threshold shall be preferred.
7.4. Performance characteristics U.K.
7.4.1.Since no internal standards can be used in bioanalytical methods, tests on the repeatability of bioanalytical methods shall be carried out to obtain information on the standard deviation within and between test series. Repeatability shall be below 20 % and intra-laboratory reproducibility shall be below 25 %. This shall be based on the calculated levels in BEQ after blank and recovery correction.U.K.
7.4.2.As part of the validation process, the test shall be shown to discriminate between a blank sample and a level at the cut-off value, allowing the identification of samples above the corresponding cut-off value (see point 7.1.2).U.K.
7.4.3.Target compounds, possible interferences and maximum tolerable blank levels shall be defined.U.K.
7.4.4.The percent standard deviation in the response or concentration calculated from the response (only possible in working range) of a triplicate determination of a sample extract may not be above 15 %.U.K.
7.4.5.The uncorrected results of the reference sample(s) expressed in BEQ (blank and at the maximum level or action threshold) shall be used for evaluation of the performance of the bioanalytical method over a constant time period.U.K.
7.4.6.Quality control charts for procedure blanks and each type of reference sample shall be recorded and checked to make sure the analytical performance is in accordance with the requirements, in particular for the procedure blanks with regard to the requested minimum difference to the lower end of the working range and for the reference samples with regard to within-laboratory reproducibility. Procedure blanks shall be controlled in a manner to avoid false-compliant results when subtracted.U.K.
7.4.7.The results from the confirmatory methods of suspected samples and 2 to 10 % of the compliant samples (minimum of 20 samples per matrix) shall be collected and used to evaluate the performance of the screening method and the relationship between BEQ and TEQ. This database may be used for the re-evaluation of cut-off values applicable to routine samples for the validated matrices.U.K.
7.4.8.Successful method performance may also be demonstrated by participation in ring trials. The results from samples analysed in ring trials, covering a concentration range up to e.g. 2 × maximum level, may be included in the evaluation of the false-compliant rate, if a laboratory is able to demonstrate its successful performance. The samples shall cover most frequent congener patterns, representing various sources.U.K.
7.4.9.During incidents, the cut-off values may be re-evaluated, reflecting the specific matrix and congener patterns of this single incident.U.K.
8. Reporting of the results U.K.
8.1. Confirmatory methods U.K.
8.1.1.The analytical results shall contain the levels of the individual PCDD/F and dioxin-like PCB congeners and TEQ-values shall be reported as lower-bound, upper-bound and medium-bound in order to include a maximum of information in the reporting of the results and thereby enabling the interpretation of the results according to specific requirements.U.K.
8.1.2.The report shall include the method used for extraction of PCDD/Fs and dioxin-like PCBs.U.K.
8.1.3.The recoveries of the individual internal standards shall be made available in case the recoveries are outside the range referred to in point 6.2.5, in case the maximum level is exceeded (in this case, the recoveries for one of the two duplicate analysis) and in other cases upon request.U.K.
8.1.4.As the expanded measurement uncertainty is to be taken into account when deciding about the compliance of a sample, this parameter shall be made available. Thus, analytical results shall be reported as x +/– U whereby x is the analytical result and U is the expanded measurement uncertainty using a coverage factor of 2 which gives a level of confidence of approximately 95 %. In the case of a separate determination of PCDD/Fs and dioxin-like-PCBs, the sum of the estimated expanded uncertainty of the separate analytical results of PCDD/Fs and dioxin-like PCBs shall be used for the sum of PCDD/Fs and dioxin-like PCBs.U.K.
8.1.5.The results shall be expressed in the same units and with at least the same number of significant figures as the maximum levels laid down by Directive 2002/32/ECU.K.
8.2. Bioanalytical screening methods U.K.
8.2.1.The result of the screening shall be expressed as ‘compliant’ or ‘suspected to be non-compliant’ (‘suspected’).U.K.
8.2.2.In addition, an indicative result for PCDD/Fs and/or dioxin-like PCBs expressed in BEQ, and not TEQ, may be given.U.K.
8.2.3.Samples with a response below the reporting limit shall be expressed as ‘lower than the reporting limit’. Samples with a response above the working range shall be reported as ‘exceeding the working range’ and the level corresponding to the upper end of the working range shall be given in BEQ.U.K.
8.2.4.For each type of sample matrix, the report shall mention the maximum level or action threshold on which the evaluation is based.U.K.
8.2.5.The report shall mention the type of the test applied, the basic test principle and the kind of calibration.U.K.
8.2.6.The report shall include the method used for extraction of PCDD/Fs and dioxin-like PCBs.U.K.
8.2.7.In case of samples suspected to be non-compliant, the report needs to include a note on the action to be taken. The concentration of PCDD/Fs and the sum of PCDD/Fs and dioxin-like PCBs in those samples with elevated levels has to be determined/confirmed by a confirmatory method.U.K.
8.2.8.Non-compliant results shall only be reported from confirmatory analysis.U.K.
8.3. Physico-chemical screening methods U.K.
8.3.1.The result of the screening shall be expressed as ‘compliant’ or ‘suspected to be non-compliant’ (‘suspected’).U.K.
8.3.2.For each type of sample matrix, the report shall mention the maximum level or action threshold on which the evaluation is based.U.K.
8.3.3.In addition, levels for individual PCDD/F and/or dioxin-like PCB congeners and TEQ-values reported as lower-bound, upper-bound and medium-bound may be given. The results shall be expressed in the same units and with at least the same number of significant figures as the maximum levels laid down by Directive 2002/32/EC.U.K.
8.3.4.The recoveries of the individual internal standards shall be made available in case the recoveries are outside the range referred to in point 6.2.5, in case the maximum level is exceeded (in this case, the recoveries for one of the two duplicate analysis) and in other cases upon request.U.K.
8.3.5.The report shall mention the GC-MS method applied.U.K.
8.3.6.The report shall include the method used for extraction of PCDD/Fs and dioxin-like PCBs.U.K.
8.3.7.In case of samples suspected to be non-compliant, the report needs to include a note on the action to be taken. The concentration of PCDD/Fs and the sum of PCDD/Fs and dioxin-like PCBs in those samples with elevated levels has to be determined/confirmed by a confirmatory method.U.K.
8.3.8.Non-compliance can only be decided after confirmatory analysis.U.K.
CHAPTER III U.K. Sample preparation and requirements for methods of analysis used in offical control of the levels of non dioxin-like PCBs in feed
1. Field of application U.K.
The requirements set out in this Chapter shall be applied where feed is analysed for the official control of the levels of non-dioxin-like PCBs and as regards sample preparation and analytical requirements for other regulatory purposes, which includes the controls performed by the feed business operator to ensure compliance with the provisions of Regulation (EC) No 183/2005.
2. Applicable detection methods U.K.
Gas chromatography/Electron Capture Detection (GC-ECD), GC-LRMS, GC-MS/MS, GC-HRMS or equivalent methods.
3. Identification and confirmation of analytes of interest U.K.
3.1.Relative retention time in relation to internal standards or reference standards (acceptable deviation of +/– 0,25 %).U.K.
3.2.Gas chromatographic separation of the non-dioxin-like PCBs from interfering substances, especially co-eluting PCBs, in particular if levels of samples are in the range of legal limits and non-compliance is to be confirmed(48).U.K.
3.3.Requirements for GC-MS techniquesU.K.
Monitoring of at least the following number of molecular ions or characteristic ions from the molecular cluster:
(a)
two specific ions for HRMS;
(b)
three specific ions for LRMS;
(c)
two specific precursor ions, each with one specific corresponding transition product ion for for MS-MS.
Maximum permitted tolerances for abundance ratios for selected mass fragments:
Relative deviation of abundance ratio of selected mass fragments from theoretical abundance or calibration standard for target ion (most abundant ion monitored) and qualifier ion(s): ± 15 %
3.4.Requirements for GC-ECD techniquesU.K.
Results exceeding the maximum level shall be confirmed with two GC columns with stationary phases of different polarity.
4. Demonstration of performance of method U.K.
The performance of the method shall be validated in the range of the maximum level (0,5 to 2 times the maximum level) with an acceptable coefficient of variation for repeated analysis (see requirements for intermediate precision in point (9).
5. Limit of quantification U.K.
The sum of the LOQs(49) of non-dioxin-like PCBs shall not be higher than one-third of the maximum level(50).
6. Quality control U.K.
Regular blank controls, analysis of spiked samples, quality control samples, participation in inter-laboratory studies on relevant matrices.
7. Control of recoveries U.K.
7.1.Suitable internal standards with physico-chemical properties comparable to analytes of interest shall be used.U.K.
7.2.Addition of internal standards:U.K.
Addition to products (before extraction and clean-up process).
7.3.Requirements for methods using all six isotope-labelled non-dioxin-like PCB congenersU.K.
(a)
results shall be corrected for recoveries of internal standards;
(b)
recoveries of isotope-labelled internal standards shall be between 60 and 120 %;
(c)
lower or higher recoveries for individual congeners with a contribution to the sum of non-dioxin-like PCBs below 10 % are acceptable.
7.4.Requirements for methods using not all six isotope-labelled internal standards or other internal standards:U.K.
(a)
recovery of internal standard(s) shall be controlled for every sample;
(b)
recoveries of internal standard(s) shall be between 60 and 120 %;
(c)
results shall be corrected for recoveries of internal standards.
7.5.The recoveries of unlabelled congeners shall be checked by spiked samples or quality control samples with concentrations in the range of the maximum level. Recoveries for these congeners shall be considered acceptable, if they are between 60 and 120 %.U.K.
8. Requirements for laboratories U.K.
In accordance with the provisions of Regulation (EC) No 882/2004, laboratories shall be accredited by a recognised body operating in accordance with ISO Guide 58 to ensure that they are applying analytical quality assurance. Laboratories shall be accredited following the EN ISO/IEC 17025 standard. In addition, the principles as described in Technical Guidelines for the estimation of measurement uncertainty and limits of quantification for PCB analysis shall be followed when applicable(51).
9. Performance characteristics: criteria for the sum of non-dioxin-like PCBs at the maximum level U.K.
a Use of all six 13 C-labelled analogues as internal standards required. | ||
| Isotope dilution mass spectrometry a | Other techniques | |
|---|---|---|
| Trueness | – 20 to + 20 % | – 30 to + 30 % |
| Intermediate precision (RSD %) | ≤ 15 % | ≤ 20 % |
| Difference between upper and lower-bound calculation | ≤ 20 % | ≤ 20 % |
10. Reporting of the results U.K.
10.1.The analytical results shall contain the levels of the individual non-dioxin-like PCBs and the sum of those PCB congenersreported as lower-bound, upper-bound and medium-bound in order to include a maximum of information in the reporting of the results and thereby enabling the interpretation of the results according to specific requirements.U.K.
10.2.The report shall include the method used for the extraction of PCBs.U.K.
10.3.The recoveries of the individual internal standards shall be made available in case the recoveries are outside the range referred to in point 7, in case the maximum level is exceeded and in other cases upon request.U.K.
10.4.As the expanded measurement uncertainty is to be taken into account when deciding about the compliance of a sample, that parameter shall also be made available. Thus, analytical results shall be reported as x +/– U whereby x is the analytical result and U is the expanded measurement uncertainty using a coverage factor of 2 which gives a level of confidence of approximately 95 %.U.K.
10.5.The results shall be expressed in the same units and with at least the same number of significant figures as the maximum levels laid down by Directive 2002/32/EC.]U.K.
Textual Amendments
[F21ANNEX VIU.K. METHODS OF ANALYSIS FOR THE DETERMINATION OF CONSTITUENTS OF ANIMAL ORIGIN FOR THE OFFICIAL CONTROL OF FEED
Textual Amendments
1. PURPOSE AND SCOPE U.K.
The determination of constituents of animal origin in feed shall be performed by light microscopy or polymerase chain reaction (PCR) in accordance with the provisions laid down in this Annex.
These two methods make it possible to detect the presence of constituents of animal origin in feed materials and compound feed. However, they do not make it possible to calculate the amount of such constituents in feed materials and compound feed. Both methods have a limit of detection below 0,1 % (w/w).
The PCR method makes it possible to identify the taxonomic group of constituents of animal origin present in feed materials and compound feed.
These methods shall apply for the control of the application of the prohibitions laid down in Article 7(1) and Annex IV to Regulation (EC) No 999/2001 and in Article 11(1) of Regulation (EC) No 1069/2009.
Depending on the type of feed being tested, these methods may be used, within one single operational protocol, either on their own or combined together in accordance with the standard operating procedures (SOP) established by the EU reference laboratory for animal proteins in feedingstuffs (EURL-AP) and published on its website(52).
2. METHODS U.K.
2.1. Light microscopy U.K.
2.1.1. [F22Principle U.K.
The constituents of animal origin which may be present in feed materials and compound feed sent for analysis are identified on the basis of typical and microscopically identifiable characteristics like muscle fibres and other meat particles, cartilage, bones, horn, hair, bristles, blood, milk globules, lactose crystals, feathers, egg shells, fish bones and scales.]
Textual Amendments
2.1.2. Reagents and equipment U.K.
2.1.2.1. Reagents U.K.
2.1.2.1.1. Concentrating agent U.K.
2.1.2.1.1.1. Tetrachloroethylene (specific gravity 1,62) U.K.
2.1.2.1.2. Staining reagent U.K.
2.1.2.1.2.1.Alizarin Red solution (dilute 2,5 ml 1M hydrochloric acid in 100 ml water and add 200 mg Alizarin Red to this solution)U.K.
2.1.2.1.3. Mounting media U.K.
2.1.2.1.3.1. Lye (NaOH 2,5 % w/v or KOH 2,5 % w/v) U.K.
2.1.2.1.3.2. [F22Glycerol (undiluted, viscosity: 1 490 cP) or a mounting medium with equivalent properties for non-permanent slide preparation] U.K.
2.1.2.1.3.3. Norland ® Optical Adhesive 65 (viscosity: 1 200 cP) or a resin with equivalent properties for permanent slide preparation U.K.
2.1.2.1.4. Mounting media with staining properties U.K.
2.1.2.1.4.1.Lugol solution (dissolve 2 g potassium iodide in 100 ml water and add 1 g iodine while frequently shaking)U.K.
2.1.2.1.4.2. Cystine reagent (2 g lead acetate, 10 g NaOH/100 ml water) U.K.
2.1.2.1.4.3.Fehling’s reagent (prepared before use from equals parts (1/1) of two stock solutions A and B. Solution A: dissolve 6,9 g copper (II) sulphate pentahydrate in 100 ml water. Solution B: dissolve 34,6 g potassium sodium tartrate tetrahydrate and 12 g NaOH in 100 ml water)U.K.
2.1.2.1.4.4.Tetramethylbenzidine/Hydrogen peroxide. (dissolve 1 g 3,3’,5,5’ tetramethylbenzidine (TMB) in 100 ml glacial acetic acid and 150 ml water. Before use, mix 4 parts of this TMB solution with 1 part 3 % hydrogen peroxide)U.K.
2.1.2.1.5. Rinsing agents U.K.
2.1.2.1.5.1. Ethanol ≥ 96 % (technical grade) U.K.
2.1.2.1.5.2. Acetone (technical grade) U.K.
2.1.2.1.6. Bleaching reagent U.K.
2.1.2.1.6.1. Commercial sodium hypochlorite solution (9 - 14 % active chlorine) U.K.
2.1.2.2. Equipment U.K.
2.1.2.2.1. Analytical balance with an accuracy of 0,001 g U.K.
2.1.2.2.2. [F22Grinding equipment: knife or rotor mill. If a rotor mill is used, mill sieves ≤ 0,5 mm shall be prohibited] U.K.
2.1.2.2.3. [F22Sieves with square meshes of 0,25 mm and 1 mm width. With the exception of sample pre-sieving, the diameter of the sieves should not exceed 10 cm to avoid loss of materials. Calibration of sieves is not required] U.K.
2.1.2.2.4.Conical glass separation funnel with a content of 250 ml with Teflon or ground glass stopcock at the base of the cone. Stopcock opening diameter shall be ≥ 4mm. Alternatively, a conical bottomed settling beaker may be used provided the laboratory has demonstrated that detection levels are equivalent to that obtained using the conical glass separation funnel.U.K.
Separation funnel U.K.
2.1.2.2.5.Stereomicroscope covering at least a 6,5× to 40× final magnification rangeU.K.
2.1.2.2.6.Compound microscope covering at least a 100× to 400× final magnification range with transmitted light bright field. Polarised light and differential interferential contrast can additionally be usedU.K.
2.1.2.2.7. Standard laboratory glassware U.K.
2.1.2.2.8. Equipment for slide preparation: classical microscope slides, hollow slides, coverslips (20 × 20 mm), tweezers, fine spatula U.K.
[F232.1.2.2.9. Laboratory oven U.K.
Textual Amendments
2.1.2.2.10. Centrifuge U.K.
2.1.2.2.11. Filter paper: qualitative cellulose filter (pore size 4-11 μm)] U.K.
2.1.3. Sampling and sample preparation U.K.
2.1.3.1. [F22Sampling U.K.
A representative sample, taken in accordance with the provisions laid down in Annex I to this Regulation shall be used.]
2.1.3.2. Precautions to be taken U.K.
In order to avoid laboratory cross-contamination, all reusable equipment shall be carefully cleaned before use. Separation funnel pieces shall be disassembled before cleaning. Separation funnel pieces and glassware shall be pre-washed manually and then washed in a washing machine. Sieves shall be cleaned by using a brush with stiff synthetic hairs. A final cleaning of sieves with acetone and compressed air is recommended after sieving of fatty material like fishmeal.
2.1.3.3. Preparation of samples other than fat or oil U.K.
2.1.3.3.1. [F22Sample drying : samples with a moisture content > 14 % shall be dried prior to handling according to Annex III to this Regulation.]U.K.
2.1.3.3.2. [F22Sample pre-sieving : in order to collect information on possible environmental contamination of the feed, it is recommended to pre-sieve at 1 mm pelleted feeds and kernels and to subsequently prepare, analyse, and report separately on the two resulting fractions, which must be considered as distinct samples.]U.K.
2.1.3.3.3. Sub-sampling and grinding : at least 50 g of the sample shall be sub-sampled for analysis and subsequently ground.U.K.
2.1.3.3.4. Extraction and preparation of the sediment : a portion of 10 g (accurate to 0,01 g) of the ground sub-sample shall be transferred into the separation funnel or conical bottomed settling beaker and 50 ml of tetrachloroethylene shall be added. The portion transferred into the funnel shall be limited to 3 g in case of fishmeal or other pure animal products, mineral ingredients or premixes which generate more than 10 % of sediment. The mixture shall be vigorously shaken for at least 30 s and at least 50 ml more of tetrachloroethylene shall be added cautiously while washing down the inside surface of the funnel to remove any adhering particles. The resulting mixture shall be left to stand for at least 5 minutes before the sediment is separated off by opening the stopcock.U.K.
If a conical bottomed settling beaker is used then the mixture shall be vigorously stirred for at least 15 s and any particles adhering to the side of the beaker shall be carefully washed down the inside surface with at least 10 ml of clean tetrachloroethylene. The mixture shall be left to stand for 3 minutes and then stirred again for 15 seconds and any particles adhering to the side of the beaker shall be carefully washed down the inside surface with at least 10 ml of clean tetrachloroethylene. The resulting mixture shall be left to stand for at least 5 minutes and then the liquid fraction is removed and discarded by careful decanting, taking care not to lose any of the sediment.
[F22The sediment shall be collected on a filter paper placed into a funnel to allow the separation of the remaining TCE while avoiding fat deposition into the sediment. The sediment shall be dried. It is recommended to subsequently weigh the sediment (accurate to 0,001 g) to control the sedimentation step. Lastly, the sediment shall be sieved at 0,25 mm and the two resulting fractions shall be examined, unless sieving is not deemed necessary.]
2.1.3.3.5. Extraction and preparation of the flotate : after recovery of the sediment with the method described above, two phases should remain in the separation funnel: a liquid one consisting of tetrachloroethylene and a solid one made of floating material. This solid phase is the flotate and shall be recovered by pouring off completely tetrachloroethylene from the funnel by opening the stopcock. By inverting the separation funnel, the flotate shall be transferred into a large Petri dish and air dried in a fumehood. If more than 5 % of the flotate consists of particles > 0,50 mm, it shall be sieved at 0,25 mm and the two resulting fractions shall be examined.U.K.
2.1.3.3.6. Preparation of raw material : a portion of at least 5 g of the ground sub-sample shall be prepared. If more than 5 % of the material consists of particles > 0,50 mm, it shall be sieved at 0,25 mm and the two resulting fractions shall be examined.U.K.
2.1.3.4. Preparation of samples consisting of fat or oil U.K.
The following protocol shall be followed for the preparation of samples consisting of fat or oil:
if the fat is solid, it shall be warmed in a oven until it is liquid.
by using a pipette, 40 ml of fat or oil shall be transferred from the bottom of the sample to a centrifugation tube.
centrifuge during 10 minutes at 4 000 r.p.m.
if the fat is solid after centrifugation, it shall be warmed in an oven until it is liquid.
repeat the centrifugation during 5 minutes at 4 000 r.p.m.
by using a small spoon or a spatula, one half of the decanted impurities shall be transferred to microscopic slides for examination, Glycerol is recommended as mounting medium.
the remaining impurities shall be used for preparing the sediment as described in point 2.1.3.3.
2.1.3.5. Use of staining reagents U.K.
In order to facilitate the correct identification of the constituents of animal origin, the operator may use staining reagents during the sample preparation in accordance with guidelines issued by the EURL-AP and published on its website.
In case Alizarin Red solution is used to colour the sediment, the following protocol shall apply:
the dried sediment shall be transferred into a glass test tube and rinsed twice with approximately 5 ml of ethanol (each time a vortex of 30 s shall be used, the solvent shall be let settle about 1 min 30 s and poured off).
the sediment shall be bleached by adding at least 1 ml sodium hypochlorite solution. The reaction shall be allowed to continue for 10 min. The tube shall be filled with water, the sediment shall be let settle 2-3 min, and the water and the suspended particles shall be poured off gently.
the sediment shall be rinsed twice more with about 10 ml of water (a vortex shall be used for 30 s, let settle, and pour off the water each time).
2 to 10 drops of the Alizarin Red solution shall be added and the mixture shall be vortexed. The reaction shall be let occur for 30 s and the coloured sediment shall be rinsed twice with approximately 5 ml ethanol followed by one rinse with acetone (each time a vortex of 30 s shall be used, the solvent shall be let settle about 1 min and poured off).
the coloured sediment shall be dried.
2.1.4. Microscopic examination U.K.
2.1.4.1. Slide preparation U.K.
[F22Microscopic slides shall be prepared from the sediment and, depending on the operator’s choice, from either the flotate or the raw material.]
A sufficient number of slides shall be prepared in order to ensure that a complete examination protocol as laid down in point 2.1.4.2 can be carried-out.
Microscopic slides shall be mounted with the adequate mounting medium in accordance with the SOP established by the EURL-AP and published on its website. The slides shall be covered with coverslips.
[F222.1.4.2. Observation flowchart for the detection of animal particles in compound feed and feed material U.K.
The prepared microscopic slides shall be observed in accordance with the observation flowcharts laid down in diagrams 1 and 2.
The microscopic observations shall be conducted using the compound microscope on the sediment and, depending on the operator’s choice, either on the flotate or on the raw material. The stereomicroscope may be used in addition to the compound microscope for the coarse fractions. Each slide shall be screened entirely at various magnifications. Precise explanations on how to use the observation flowcharts are detailed by a SOP established by the EURL-AP and published on its website.
The minimum numbers of slides to be observed at each step of the observation flowcharts shall be strictly respected, unless the entire fraction material does not permit to reach the stipulated slide number, for instance when no sediment is obtained. No more than 6 slides per determination shall be used for recording of the number of particles.
When additional slides are prepared on the flotate or the raw material using a more specific mounting medium with staining properties, as laid down in point 2.1.2.1.4, to further characterise structures (e.g. feathers, hairs, muscle or blood particles) which have been detected on slides prepared by other mounting media, as laid down in point 2.1.2.1.3, the number of particles shall be counted based on a number of slides per determination not exceeding 6, including the additional slides with a more specific mounting medium.
In order to facilitate the identification of the particles’ nature and origin, the operator may use support tools like decision support systems, image libraries and reference samples.]


2.1.4.3. [F22Number of determinations U.K.
Determinations shall be performed on different sub-samples of 50 g each.
If following the first determination carried out in accordance with the observation flowchart laid down in diagram 1, no animal particles are detected, no additional determination is necessary and the result of the analysis shall be reported using the terminology laid down in point 2.1.5.1.
If, following the first determination carried out in accordance with the observation flowchart laid down in diagram 1, one or more animal particles of a given nature (i.e. terrestrial vertebrate or fish) are detected, and the nature of the particles found confirms the declared content of the sample, no second determination is necessary. If the number of the animal particles of a given nature detected during this first determination is higher than 5, the result of the analysis shall be reported per animal nature using the terminology laid down in point 2.1.5.3. Otherwise, the result of the analysis shall be reported per animal nature using the terminology laid down in point 2.1.5.2.
In other cases, including when no declaration of content has been provided to the laboratory a second determination shall be carried out from a new sub-sample.
If, following the second determination carried out in accordance with the observation flowchart laid down in diagram 2, the sum of the animal particles of a given nature detected over the two determinations is higher than 10, the result of the analysis shall be reported per animal nature using the terminology laid down in point 2.1.5.3. Otherwise, the result of the analysis shall be reported per animal nature using the terminology laid down in point 2.1.5.2.]
2.1.5. [F22Expression of the results U.K.
When reporting the results, the laboratory shall indicate on which type of material the analysis has been carried-out (sediment, flotate or raw material). The reporting shall clearly indicate how many determinations have been carried-out and if sieving of the fractions prior to slide preparation, in accordance with the last paragraph of point 2.1.3.3.4., was not performed.
The laboratory report shall at least contain information on the presence of constituents derived from terrestrial vertebrates and from fish.
The different situations shall be reported in the following ways.
2.1.5.1.
No animal particle of a given nature detected:
‘As far as was discernible using a light microscope, no particle derived from terrestrial vertebrates was detected in the submitted sample.’
‘As far as was discernible using a light microscope, no particle derived from fish was detected in the submitted sample.’
2.1.5.2.
Between 1 and 5 animal particles of a given nature detected when only one determination has been performed, or between 1 and 10 particles of a given nature detected in case of two determinations (the number of detected particles is below the decision limit established in the standard operating procedures (SOP) of the EU reference laboratory for animal proteins in feedingstuffs (EURL-AP) and published on its website(53)):
When only one determination has been performed:
‘As far as was discernible using a light microscope, no more than 5 particles derived from terrestrial vertebrates were detected in the submitted sample. The particles were identified as … [bone, cartilage, muscle, hair, horn…]. This low level presence is below the decision limit established for this microscopic method.’
‘As far as was discernible using a light microscope, no more than 5 particles derived from fish were detected in the submitted sample. The particles were identified as … [fishbone, fish scale, cartilage, muscle, otolith, gill…]. This low level presence, is below the decision limit established for this microscopic method.’
When two determinations have been performed:
‘As far as was discernible using a light microscope, no more than 10 particles derived from terrestrial vertebrates were detected over the two determinations in the submitted sample. The particles were identified as … [bone, cartilage, muscle, hair, horn…]. This low level presence is below the decision limit established for this microscopic method.’
‘As far as was discernible using a light microscope, no more than 10 particles derived from fish were detected over the two determinations in the submitted sample. The particles were identified as … [fishbone, fish scale, cartilage, muscle, otolith, gill…]. This low level presence is below the decision limit established for this microscopic method.’
Additionally:
In case of sample pre-sieving, the laboratory report shall mention in which fraction (sieved fraction, pelleted fraction or kernels) the animal particles have been detected insofar as the detection of animal particles only in the sieved fraction may be the sign of an environmental contamination.
When only animal particles which cannot be categorised as either terrestrial vertebrates or fish are detected (e.g. muscle fibres), the report shall mention that only such animal particles were detected and that it cannot be excluded that they originate from terrestrial vertebrates
2.1.5.3.
More than 5 animal particles of a given nature detected when only one determination has been performed, or more than 10 particles of a given nature detected in case of two determinations:
When only one determination has been performed:
‘As far as was discernible using a light microscope, more than 5 particles derived from terrestrial vertebrates were detected in the submitted sample. The particles were identified as … [bone, cartilage, muscle, hair, horn…].’
‘As far as was discernible using a light microscope, more than 5 particles derived from fish were detected in the submitted sample. The particles were identified as … [fishbone, fish scale, cartilage, muscle, otolith, gill…].’
When two determinations have been performed:
‘As far as was discernible using a light microscope, more than 10 particles derived from terrestrial vertebrates were detected over the two determinations in the submitted sample. The particles were identified as … [bone, cartilage, muscle, hair, horn…].’
‘As far as was discernible using a light microscope, more than 10 particles derived from fish were detected over the two determinations in the submitted sample. The particles were identified as … [fishbone, fish scale, cartilage, muscle, otolith, gill…].’
Additionally:
In case of sample pre-sieving, the laboratory report shall mention in which fraction (sieved fraction, pelleted fraction or kernels) the animal particles have been detected insofar as the detection of animal particles only in the sieved fraction may be the sign of an environmental contamination.
When only animal particles which cannot be categorised as either terrestrial vertebrates or fish are detected (e.g. muscle fibres), the report shall mention that only such animal particles were detected and that it cannot be excluded that they originate from terrestrial vertebrates.]
2.2. PCR U.K.
2.2.1. Principle U.K.
Deoxyribonucleic acid (DNA) fragments of animal origin which may be present in feed materials and compound feed are detected by a genetic amplification technique through PCR, targeting species-specific DNA sequences.
The PCR method first requires a DNA extraction step. The amplification step shall be applied afterwards to the so-obtained DNA extract, in order to detect the animal species targeted by the assay.
2.2.2. Reagents and equipment U.K.
2.2.2.1. Reagents U.K.
2.2.2.1.1. Reagents for DNA extraction step U.K.
Only reagents approved by the EURL-AP and published on its website shall be used.
2.2.2.1.2. Reagents for genetic amplification step U.K.
2.2.2.1.2.1. Primers and probes U.K.
Only primers and probes with sequences of oligonucleotides validated by the EURL-AP shall be used(54).
2.2.2.1.2.2. Master Mix U.K.
Only Master Mix solutions which do not contain reagents susceptible to lead to false results due to presence of animal DNA shall be used(55).
2.2.2.1.2.3. Decontamination reagents U.K.
2.2.2.1.2.3.1. Hydrochloric acid solution (0,1 N) U.K.
2.2.2.1.2.3.2. Bleach (solution of sodium hypochlorite at 0,15 % of active chlorine) U.K.
2.2.2.1.2.3.3. Non-corrosive reagents for decontaminating costly devices like analytical balances (e.g. DNA Erase TM of MP Biomedicals) U.K.
2.2.2.2. Equipment U.K.
2.2.2.2.1. Analytical balance with an accuracy of 0,001 g U.K.
2.2.2.2.2. Grinding equipment U.K.
2.2.2.2.3. Thermocycler enabling real-time PCR U.K.
2.2.2.2.4. Microcentrifuge for microfuge tubes U.K.
2.2.2.2.5. Set of micropipettes allowing to pipet from 1 μl up to 1 000 μl U.K.
2.2.2.2.6.Standard molecular biology plastic-ware: microfuge tubes, filtered plastic tips for micropipettes, plates suitable for the thermocycler.U.K.
2.2.2.2.7. Freezers to store samples and reagents U.K.
2.2.3. Sampling and sample preparation U.K.
2.2.3.1. Sampling U.K.
A representative sample, taken in accordance with the provisions laid down in Annex I, shall be used.
2.2.3.2. Sample preparation U.K.
The preparation of laboratory samples up to DNA extraction shall comply with the requirements set out in Annex II. At least 50 g of the sample shall be sub-sampled for analysis and subsequently ground.
The sample preparation shall be performed in a room different from the ones dedicated to DNA extraction and to genetic amplification reactions as described by ISO 24276.
Two test portions of at least 100 mg each shall be prepared.
2.2.4. DNA extraction U.K.
The DNA extraction shall be performed on each test portion prepared using the SOP established by the EURL-AP and published on its website.
Two extraction controls shall be prepared for each extraction series as described by ISO 24276.
an extraction blank control,
a positive DNA extraction control.
2.2.5. Genetic amplification U.K.
The genetic amplification shall be performed using the methods validated for each species requiring identification. These methods are laid down in the SOP established by the EURL-AP and published on its website. Each DNA extract shall be analysed at least at two different dilutions in order to evaluate inhibition.
Two amplification controls shall be prepared per species target as described by ISO 24276.
a positive DNA target control shall be used for each plate or series of PCR assays,
an amplification reagent control (also called no template control) shall be used for each plate or series of PCR assays.
2.2.6. Interpretation and expression of results U.K.
When reporting the results, the laboratory shall indicate at least the weight of the test portions used, the extraction technique used, the number of determinations carried-out and the limit of detection of the method.
Results shall not be interpreted and reported if the positive DNA extraction control and the positive DNA target controls do not provide positive results for the target under assay while the amplification reagent control is negative.
In case results from the two test portions are not consistent, at least the genetic amplification step shall be repeated. If the laboratory suspects that the DNA extracts can be the cause of the inconsistency, a new DNA extraction and a subsequent genetic amplification shall be performed before interpreting the results.
The final expression of the results shall be based on the integration and the interpretation of the results of the two test portions in accordance with the SOP established by the EURL-AP and published on its website.
2.2.6.1. Negative result U.K.
A negative result shall be reported as follows:
No DNA from X was detected in the submitted sample (with X being the animal species or group of animal species that is targeted by the assay).
2.2.6.2. Positive result U.K.
A positive result shall be reported as follows:
DNA from X was detected in the submitted sample (with X being the animal species or group of animal species that is targeted by the assay).]
ANNEX VIIU.K.METHOD OF CALCULATING THE ENERGY VALUE OF POULTRYFEED
1.Method of calculation and expression of energy valueU.K.
The energy value of compound poultry feed must be calculated in accordance with the formula set out below on the basis of the percentages of certain analytical components of the feed. This value is to be expressed in megajoules (MJ) of metabolisable energy (ME), corrected for nitrogen, per kilogram of compound feed:
MJ/kg of ME = 0,1551 × % crude protein + 0,3431 × % crude fat + 0,1669 × % starch + 0,1301 × % total sugar (expressed as sucrose).
2.Tolerances applicable to declared valuesU.K.
If the official inspection reveals a discrepancy (increased or reduced energy value of the feed) between the result of the inspection and the declared energy value, a minimum tolerance of 0,4 MJ/kg of ME shall be permitted.
3.Expression of resultU.K.
After application of the above formula, the result obtained must be given to one decimal place.
4.Sampling and analysis methodsU.K.
Sampling of the compound feed and determination of the content of analytical components indicated in the method of calculation must be performed in accordance with the F24... sampling methods and analysis methods for the official control of feed respectively.
Textual Amendments
F24Word in Annex 7 point 4 omitted (31.12.2020) by virtue of The Animal Feed (Amendment) (EU Exit) Regulations 2019 (S.I. 2019/654), regs. 1, 91; 2020 c. 1, Sch. 5 para. 1(1)
The following are to be applied:
for determining the crude fat content: procedure B of the method for the determination of crude oils and fats, laid down in Part H of Annex III.
for determining the starch content: the polarimetric method, laid down in Part L of Annex III.
ANNEX VIIIU.K.METHODS OF ANALYSIS TO CONTROL ILLEGAL PRESENCE OF NO LONGER AUTHORISED ADDITIVES IN FEED
Important notes:U.K.
More sensitive methods of analysis than the methods of analysis mentioned in this Annex can be used to detect the illegal presence of no longer authorised additives in feed.
The methods of analysis mentioned in this Annex shall be used for confirmatory purposes.
A.DETERMINATION OF METHYL BENZOQUATEU.K.
7-benzyloxy-6-butyl-3-methoxycarbonyl-4-quinolone
1.Purpose and scopeU.K.
This method makes it possible to determine the level of methyl benzoquate in feed. The limit of quantification is 1 mg/kg.
2.PrincipleU.K.
Methyl benzoquate is extracted from the sample with methanolic methanesulfonic acid solution. The extract is purified with dichloromethane, by ion-exchange chromatography and then again with dichloromethane. The methyl benzoquate content is determined by reversed-phase high-performance liquid chromatography (HPLC) with an UV detector.
3.ReagentsU.K.
3.1.DichloromethaneU.K.
3.2.Methanol, equivalent to HPLC gradeU.K.
3.3.HPLC mobile phaseU.K.
Mixture of methanol (3.2) and water (equivalent to HPLC grade) 75 + 25 (v + v).
Filter through a 0,22 μm filter (4.5) and degas the solution (e.g. by ultrasonification for 10 minutes).
3.4.Methanesulfonic acid solution, c = 2 %U.K.
Dilute 20,0 ml methanesulfonic acid to 1 000 ml with methanol (3.2).
3.5.Hydrochloric acid solution, c = 10 %U.K.
Dilute 100 ml hydrochloric acid (ρ201,18 g/ml) to 1 000 ml with water.
3.6.Cation-exchange resin Amberlite CG-120 (Na), 100 to 200 meshU.K.
The resin is pretreated before use. Slurry 100 g resin with 500 ml hydrochloric acid solution (3.5) and heat on a hot plate to boiling, stirring continuously. Allow to cool and decant off the acid. Filter through a filter paper under vacuum. Wash the resin twice with 500 ml portions of water and then with 250 ml of methanol (3.2). Rinse the resin with a further 250 ml portion of methanol and dry by passing air through the filter cake. Store the dried resin in a stoppered bottle.
3.7.Standard substance: pure methyl benzoquate (7-benzyloxy-6-butyl-3-methoxycarbonyl-4-quinolone)U.K.
3.7.1.Methyl benzoquate stock standard solution, 500 μg/mlU.K.
Weigh to the nearest 0,1 mg, 50 mg of standard substance (3.7), dissolve in methanesulfonic acid solution (3.4) in a 100 ml graduated flask, make up to the mark and mix.
3.7.2.Methyl benzoquate intermediate standard solution, 50 μg/mlU.K.
Transfer 5,0 ml of methyl benzoquate stock standard solution (3.7.1) into a 50 ml graduated flask, make up to the mark with methanol (3.2) and mix.
3.7.3.Calibration solutionsU.K.
Transfer 1,0, 2,0, 3,0, 4,0 and 5,0 ml of methyl benzoquate intermediate standard solution (3.7.2) into a series of 25 ml graduated flasks. Make up to the mark with the mobile phase (3.3) and mix. These solutions have concentrations of 2,0, 4,0, 6,0, 8,0 and 10,0 μg/ml methyl benzoquate respectively. These solutions must be freshly prepared before use.
4.ApparatusU.K.
4.1.Laboratory shakerU.K.
4.2.Rotary film evaporatorU.K.
4.3.Glass column (250 mm × 15 mm) fitted with a stopcock and reservoir of approximately 200 ml capacityU.K.
4.4.HPLC equipment with variable wavelength ultraviolet detector or diode-array detectorU.K.
4.4.1.Liquid chromatographic column: 300 mm × 4 mm, C18, 10 μm packing or equivalentU.K.
4.5.Membrane filters, 0,22 μmU.K.
4.6.Membrane filters, 0,45 μmU.K.
5.ProcedureU.K.
5.1.GeneralU.K.
5.1.1.A blank feed shall be analysed to check that neither methyl benzoquate nor interfering substances are present.U.K.
5.1.2.A recovery test shall be carried out by analysing the blank feed which has been fortified by addition of a quantity of methyl benzoquate, similar to that present in the sample. To fortify at a level of 15 mg/kg, add 600 μl of the stock standard solution (3.7.1) to 20 g of the blank feed, mix and wait for 10 minutes before proceeding with the extraction step (5.2).U.K.
Note for the purpose of this method, the blank feed shall be similar in type to that of the sample and on analysis methyl benzoquate must not be detected.
5.2.ExtractionU.K.
Weigh to the nearest 0,01 g, approximately 20 g of the prepared sample and transfer to a 250 ml conical flask. Add 100,0 ml of methanesulfonic acid solution (3.4) and shake mechanically (4.1) for 30 minutes. Filter the solution through a filter paper and retain the filtrate for the liquid-liquid partition step (5.3).
5.3.Liquid-liquid partitionU.K.
Transfer into a 500 ml separating funnel containing 100 ml of hydrochloric acid solution (3.5), 25,0 ml of the filtrate obtained in (5.2). Add 100 ml dichloromethane (3.1) to the funnel and shake for one minute. Allow the layers to separate and run off the lower (dichloromethane) layer into a 500 ml round-bottomed flask. Repeat the extraction of the aqueous phase with two further 40-ml portions of dichloromethane and combine these with the first extract in the round-bottomed flask. Evaporate the dichloromethane extract down to dryness on the rotary evaporator (4.2) operating under reduced pressure at 40 oC. Dissolve the residue in 20 to 25 ml methanol (3.2), stopper the flask and retain the whole of the extract for ion-exchange chromatography (5.4).
5.4.Ion-exchange chromatographyU.K.
5.4.1.Preparation of the cation-exchange columnU.K.
Insert a plug of glass wool into the lower end of a glass column (4.3). Prepare a slurry of 5,0 g of the treated cation-exchange resin (3.6) with 50 ml of hydrochloric acid (3.5), pour into the glass column and allow to settle. Run out the excess acid to just above the resin surface and wash the column with water until the effluent is neutral to litmus. Transfer 50 ml methanol (3.2) onto the column and allow to drain down to the resin surface.
5.4.2.Column chromatographyU.K.
By means of a pipette, carefully transfer the extract obtained in (5.3) onto the column. Rinse the round-bottomed flask with two portions of 5 to 10 ml methanol (3.2) and transfer these washings to the column. Run the extract down to the resin surface and wash the column with 50 ml methanol, ensuring that the flow rate does not exceed 5 ml per minute. Discard the effluent. Elute the methyl benzoquate from the column using 150 ml of methanesulfonic acid solution (3.4) and collect the column eluate in a 250 ml conical flask.
5.5.Liquid-liquid partitionU.K.
Transfer the eluate obtained in (5.4.2) into a 1 litre separating funnel. Rinse the conical flask with 5 to 10 ml methanol (3.2) and combine the washings with the contents of the separating funnel. Add 300 ml of hydrochloric acid solution (3.5) and 130 ml of dichloromethane (3.1). Shake for 1 minute and allow the phases to separate. Run off the lower (dichloromethane) layer into a 500 ml round bottomed flask. Repeat the extraction of the aqueous phase with two further 70 ml portions of dichloromethane and combine these extracts with the first in the round-bottomed flask.
Evaporate the dichloromethane extract down to dryness on the rotary evaporator (4.2) operating under reduced pressure at 40 oC. Dissolve the residue in the flask with approximately 5 ml of methanol (3.2) and transfer this solution quantitatively to a 10 ml graduated flask. Rinse the round-bottomed flask with a further two portions of 1 to 2 ml of methanol and transfer these to the graduated flask. Make up to the mark with methanol and mix. An aliquot portion is filtered through a membrane filter (4.6). Reserve this solution for HPLC-determination (5.6).
5.6.HPLC determinationU.K.
5.6.1.ParametersU.K.
The following conditions are offered for guidance, other conditions may be used provided that they give equivalent results:
liquid chromatographic column (4.4.1),
HPLC mobile phase: methanol-water mixture (3.3),
flow rate: 1 to 1,5 ml/minute,
detection wavelength: 265 nm,
Injection volume: 20 to 50 μl.
Check the stability of the chromatographic system, injecting the calibration solution (3.7.3) containing 4 μg/ml several times, until constant peak heights or areas and retention times are achieved.
5.6.2.Calibration graphU.K.
Inject each calibration solution (3.7.3) several times and measure the peak heights (areas) for each concentration. Plot a calibration graph using the mean peak heights or areas of the calibration solutions as the ordinates and the corresponding concentrations in μg/ml as the abscissae.
5.6.3.Sample solutionU.K.
Inject the sample extract (5.5) several times, using the same volume as taken for the calibration solutions and determine the mean peak height (area) of the methyl benzoquate peaks.
6.Calculation of resultsU.K.
Determine the concentration of the sample solution in μg/ml from the mean height (area) of the methyl benzoquate peaks of the sample solution by reference to the calibration graph (5.6.2).
The content of methyl benzoquate w (mg/kg) of the sample is given by the following formula:

in which:
c
=
methyl benzoquate concentration of the sample solution in μg/ml,
m
=
weight of the test portion in grams.
7.Validation of the resultsU.K.
7.1.IdentityU.K.
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract and the calibration solution (3.7.3) containing 10 μg/ml are compared.
7.1.1.Co-chromatographyU.K.
A sample extract is fortified by addition of an appropriate amount of the intermediate standard solution (3.7.2). The amount of added methyl benzoquate must be similar to the estimated amount of methyl benzoquate in the sample extract.
Only the height of the methyl-benzoquate peak shall be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its maximum height, must be within approximately 10 % of the original width.
7.1.2.Diode-array detectionU.K.
The results are evaluated according to the following criteria:
(a)
the wavelength of maximum absorption of the sample and of the standard spectra recorded at the peak apex on the chromatogram must be the same within a margin determined by the resolving power of the detection system. For diode-array detection, this is typically within approximately 2 nm;
(b)
between 220 and 350 nm, the sample and standard spectra recorded at the peak apex on the chromatogram must not be different for those parts of the spectrum within the range 10 % to 100 % of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15 % of the absorbance of the standard analyte;
(c)
between 220 and 350 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 % to 100 % of relative absorbance. This criterion is met when the same maxima are present and when at no observed points the deviation between the spectra does not exceed 15 % of the absorbance of the spectrum of the apex.
If one of these criteria is not met the presence of the analyte has not been confirmed.
7.2.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed: 10 % relative to the higher result for methyl benzoquate contents between 4 and 20 mg/kg.
7.3.RecoveryU.K.
For a fortified blank sample the recovery shall be at least 90 %.
8.Results of a collaborative studyU.K.
Five samples were analysed by 10 laboratories. Duplicate analyses were performed on each sample.
| Blank | Meal 1 | Pellet 1 | Meal 2 | Pellet2 | |
|---|---|---|---|---|---|
| Mean [mg/kg] | ND | 4,5 | 4,5 | 8,9 | 8,7 |
| sr [mg/kg] | — | 0,3 | 0,2 | 0,6 | 0,5 |
| CVr [%] | — | 6,7 | 4,4 | 6,7 | 5,7 |
| sR [mg/kg] | — | 0,4 | 0,5 | 0,9 | 1,0 |
| CVR [%] | — | 8,9 | 11,1 | 10,1 | 11,5 |
| Recovery [%] | — | 92,0 | 93,0 | 92,0 | 89,0 |
ND
=
Not detected
sr
=
standard deviation of repeatability
CVr
=
coefficient of variation of repeatability, %
sR
=
standard deviation of reproducibility
CVR
=
coefficient of variation of reproducibility, %.
B.DETERMINATION OF OLAQUINDOXU.K.
2-[N-2'-(hydroxyethyl)carbamoyl]-3-methylquinoxaline-N1,N4-dioxide
1.Purpose and scopeU.K.
This method makes it possible to determine the level olaquindox in feed. The limit of quantification is 5 mg/kg.
2.PrincipleU.K.
The sample is extracted by a water-methanol mixture. The content of olaquindox is determined by reversed-phase high-performance liquid chromatography (HPLC) using an UV detector.
3.ReagentsU.K.
3.1.Methanol.U.K.
3.2.Methanol, equivalent to HPLC grade.U.K.
3.3.Water, equivalent to HPLC grade.U.K.
3.4.Mobile phase for HPLC.U.K.
Water (3.3)-methanol (3.2) mixture, 900 +100 (V + V).
3.5.Standard substance: pure olaquindox 2-[N-2'-(hydroxyethyl)carbamoyl]-3-methylquinoxaline-N1,N4-dioxide, E 851.U.K.
3.5.1.Olaquindox stock standard solution, 250 μg/mlU.K.
Weigh to the nearest 0,1 mg 50 mg of olaquindox (3.5) in a 200 ml graduated flask and add ca. 190 ml water. Then place the flask for 20 min. into an ultrasonic bath (4.1). After ultrasonic treatment bring the solution to room temperature, make up to the mark with water and mix. Wrap the flask with aluminium foil and store in a refrigerator. This solution must be prepared fresh each month.
3.5.2.Olaquindox intermediate standard solution, 25 μg/mlU.K.
Transfer 10,0 ml of the stock standard solution (3.5.1) into a 100 ml graduated flask, make up to the mark with the mobile phase (3.4) and mix. Wrap the flask with aluminium foil and store in a refrigerator. This solution must be prepared fresh each day.
3.5.3.Calibration solutionsU.K.
Into a series of 50 ml graduated flasks transfer 1,0, 2,0, 5,0, 10,0, 15,0 and 20,0 ml of the intermediate standard solution (3.5.2). Make up to the mark with the mobile phase (3.4) and mix. Wrap the flasks with aluminium foil. These solutions correspond to 0,5, 1,0, 2,5, 5,0, 7,5 and 10,0 μg of olaquindox per ml respectively.
These solutions must be prepared fresh each day.
4.ApparatusU.K.
4.1.Ultrasonic bathU.K.
4.2.Mechanical shakerU.K.
4.3.HPLC equipment with variable wavelength ultraviolet detector or diode array detectorU.K.
4.3.1.Liquid chromatographic column, 250 mm × 4 mm, C18, 10 μm packing, or equivalentU.K.
4.4.Membrane filters, 0,45 μmU.K.
5.ProcedureU.K.
Note:
Olaquindox is light sensitive. Carry out all procedures under subdued light or use amber glassware.
5.1.GeneralU.K.
5.1.1.A blank feed shall be analysed to check that neither olaquindox nor interfering substances are present.U.K.
5.1.2.A recovery test shall be carried out by analysing the blank feed which has been fortified by addition of a quantity of olaquindox, similar to that present in the sample. To fortify at a level of 50 mg/kg, transfer 10,0 ml of the stock standard solution (3.5.1) to a 250 ml conical flask and evaporate the solution to ca. 0,5 ml. Add 50 g of the blank feed, mix thoroughly and leave for 10 min. mixing again several times before proceeding with the extraction step (5.2).U.K.
Note:
For the purpose of this method the blank feed shall be similar in type to that of the sample and olaquindox must not be detected.
5.2.ExtractionU.K.
Weigh to the nearest 0,01 g, approximately 50 g of the sample. Transfer to a 1 000 ml conical flask, add 100 ml of methanol (3.1) and place the flask for 5 min. in the ultrasonic bath (4.1). Add 410 ml water and leave in the ultrasonic bath for further 15 min. Remove the flask from the ultrasonic bath, shake it for 30 min. on the shaker (4.2) and filter through a folded filter. Transfer 10,0 ml of the filtrate into a 20 ml graduated flask, make up to the mark with water and mix. An aliquot is filtered through a membrane filter (4.4). (see 9. Observation) Proceed to the HPLC determination (5.3).
5.3.HPLC determinationU.K.
5.3.1.Parameters:U.K.
The following conditions are offered for guidance, other conditions may be used provided that they give equivalent results.
| Analytical column (4.3.1) | |
| Mobile Phase (3.4): | water (3.3)-methanol (3.2) mixture, 900 + 100 (V + V) |
| Flow rate: | 1,5-2 ml/min. |
| Detection wavelength: | 380 nm |
| Injection volume: | 20 μl –100 μl |
Check the stability of the chromatographic system, injecting several times the calibration solution (3.5.3) containing 2,5 μg/ml, until constant peak heights and retention times are achieved.
5.3.2.Calibration graphU.K.
Inject each calibration solution (3.5.3) several times and determine the mean peak heights (areas) for each concentration. Plot a calibration graph using the mean peak heights (areas) of the calibration solutions as the ordinates and the corresponding concentrations in μg/ml as the abscissae.
5.3.3.Sample solutionU.K.
Inject the sample extract (5.2) several times using the same volume as taken for the calibration solutions and determine the mean peak height (area) of the olaquindox peaks.
6.Calculation of the resultsU.K.
From the mean height (area) of the olaquindox peaks of the sample solution determine the concentration of the sample solution in μg/ml by reference to the calibration graph (5.3.2).
The olaquindox content w in mg/kg of the sample is given by the following formula:

in which:
c
=
olaquindox concentration of the sample extract (5.2) in μg/ml
m
=
weight of the test portion in g (5.2).
7.Validation of the resultsU.K.
7.1.IdentityU.K.
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract (5.2) and the calibration solution (3.5.3) containing 5,0 μg/ml are compared.
7.1.1.Co-chromatographyU.K.
A sample extract (5.2) is fortified by addition of an appropriate amount of calibration solution (3.5.3). The amount of added olaquindox must be similar to the amount of olaquindox found in the sample extract.
Only the height of the olaquindox peak shall be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its height, must be within ± 10 % of the original width of the olaquindox peak of the unfortified sample extract.
7.1.2.Diode array detectionU.K.
The results are evaluated according to the following criteria:
(a)
The wavelength of maximum absorption of the sample and of the standard spectra, recorded at the peak apex on the chromatogram, must be the same within a margin determined by the resolving power of the detection system. For diode-array detection this is typically within ± 2 nm.
(b)
Between 220 and 400 nm, the sample and standard spectra recorded at the peak apex of the chromatogram, must not be different for those parts of the spectrum within the range 10 %-100 % of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15 % of the absorbance of the standard analyte.
(c)
Between 220 and 400 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 %-100 % of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15 % of the absorbance of the spectrum of the peak apex.
If one of these criteria is not met the presence of the analyte has not been confirmed.
7.2.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed 15 % relative to the higher result for olaquindox contents between 10 and 200 mg/kg.
7.3.RecoveryU.K.
For a fortified blank sample the recovery shall be at least 90 %.
8.Results of a collaborative studyU.K.
An EC collaborative study was arranged in which four piglet feed samples including one blank feed were analysed by up to 13 laboratories. The results are given below:
| Sample 1 | Sample 2 | Sample 3 | Sample 4 | |
|---|---|---|---|---|
| L | 13 | 10 | 11 | 11 |
| n | 40 | 40 | 44 | 44 |
| mean [mg/kg] | — | 14,6 | 48,0 | 95,4 |
| Sr [mg/kg] | — | 0,82 | 2,05 | 6,36 |
| SR [mg/kg] | — | 1,62 | 4,28 | 8,42 |
| CVr [%] | — | 5,6 | 4,3 | 6,7 |
| CVR [%] | — | 11,1 | 8,9 | 8,8 |
| Nominal content | ||||
| [mg/kg] | — | 15 | 50 | 100 |
| recovery % | — | 97,3 | 96,0 | 95,4 |
L
=
number of laboratories
n
=
number of single values
Sr
=
standard deviation of repeatability
SR
=
standard deviation of reproducibility
CVr
=
coefficient of variation of repeatability
CVR
=
coefficient of variation of reproducibility.
9.ObservationU.K.
Although the method has not been validated for feeds containing more than 100 mg/kg of olaquindox, it may be possible to obtain satisfactory results by taking a smaller sample weight and/or diluting the extract (5.2) to reach a concentration within the range of the calibration graph (5.3.2).
C.DETERMINATION OF AMPROLIUMU.K.
1-[(4-amino-2-propylpyrimidin-5-yl)methyl]-2-methyl-pyridinium chloride hydrochloride
1.Purpose and ScopeU.K.
This method makes it possible to determine the level of amprolium in feed and premixtures. The detection limit is 1 mg/kg, the limit of quantification is 5 mg/kg.
2.PrincipleU.K.
The sample is extracted with a methanol-water mixture. After dilution with the mobile phase and membrane filtration the content of amprolium is determined by cation exchange high performance liquid chromatography (HPLC) using a UV detector.
3.ReagentsU.K.
3.1.Methanol.U.K.
3.2.Acetonitrile, equivalent to HPLC grade.U.K.
3.3.Water, equivalent to HPLC grade.U.K.
3.4.Sodium dihydrogen phosphate solution, c = 0,1 mol/l.U.K.
Dissolve 13,8 g of sodium dihydrogen phosphate monohydrate in water (3.3) in a 1 000 ml graduated flask, make up to the mark with water (3.3) and mix.
3.5.Sodium perchlorate solution, c = 1,6 mol/l.U.K.
Dissolve 224,74 g of sodium perchlorate monohydrate in water (3.3) in a 1 000 ml graduated flask, make up to the mark with water (3.3) and mix.
3.6.Mobile phase for HPLC (see observation 9.1).U.K.
Mixture of acetonitrile (3.2), sodium dihydrogen phosphate solution (3.4) and sodium perchlorate solution (3.5), 450+450+100 (v+v+v). Prior to use filter through a 0,22 μm membrane filter (4.3) and degas the solution (e.g. in the ultrasonic bath (4.4) for at least 15 minutes).
3.7.Standard substance: pure amprolium, 1-[(4-amino-2-propylpyrimidin-5-yl)methyl]-2-methyl-pyridinium chloride hydrochloride, E 750 (see 9.2).U.K.
3.7.1.Amprolium stock standard solution, 500 μg/mlU.K.
Weigh to the nearest 0,1 mg, 50 mg of amprolium (3.7) in a 100 ml graduated flask, dissolve in 80 ml methanol (3.1) and place the flask for 10 min. in an ultrasonic bath (4.4). After ultrasonic treatment bring the solution to room temperature, make up to the mark with water and mix. At a temperature of ≤ 4 oC the solution is stable for 1 month.
3.7.2.Amprolium intermediate standard solution, 50 μg/mlU.K.
Pipette 5,0 ml of the stock standard solution (3.7.1) into a 50 ml graduated flask, make up to the mark with the extraction solvent (3.8) and mix. At a temperature of ≤ 4 oC the solution is stable for 1 month.
3.7.3.Calibration solutionsU.K.
Transfer 0,5, 1,0 and 2,0 ml of the intermediate standard solution (3.7.2) into a series of 50 ml graduated flasks. Make up to the mark with the mobile phase (3.6) and mix. These solutions correspond to 0,5, 1,0 and 2,0 μg of amprolium per ml respectively. These solutions must be prepared freshly before use.
3.8.Extraction solvent.U.K.
Methanol (3.1)-water mixture 2+1 (v+v).
4.ApparatusU.K.
4.1.HPLC equipment with injection system, suitable for injection volumes of 100 μl.U.K.
4.1.1.Liquid chromatographic column 125 mm × 4 mm, cation exchange Nucleosil 10 SA, 5 or 10 μm packing, or equivalent.U.K.
4.1.2.UV detector with variable wavelength adjustment or diode array detector.U.K.
4.2.Membrane filter, PTFE material, 0,45 μm.U.K.
4.3.Membrane filter, 0,22 μm.U.K.
4.4.Ultrasonic bath.U.K.
4.5.Mechanical shaker or magnetic stirrer.U.K.
5.ProcedureU.K.
5.1.GeneralU.K.
5.1.1.Blank feedU.K.
For the performance of the recovery test (5.1.2) a blank feed shall be analysed to check that neither amprolium nor interfering substances are present. The blank feed shall be similar in type to that of the sample and amprolium or interfering substances must not be detected.
5.1.2.Recovery testU.K.
A recovery test shall be carried out by analysing the blank feed which has been fortified by addition of a quantity of amprolium, similar to that present in the sample. To fortify at a level of 100 mg/kg, transfer 10,0 ml of the stock standard solution (3.7.1) to a 250 ml conical flask and evaporate the solution to approximately 0,5 ml. Add 50 g of the blank feed, mix thoroughly and leave for 10 min. mixing again several times before proceeding with the extraction step (5.2).
Alternatively, if a blank feed similar in type to that of the sample is not available (see 5.1.1), a recovery test can be performed by means of the standard addition method. In this case, the sample to be analysed is fortified with a quantity of amprolium similar to that already present in the sample. This sample is analysed together with the unfortified sample and the recovery can be calculated by subtraction.
5.2.ExtractionU.K.
5.2.1.Premixtures (content < 1 % amprolium) and feedU.K.
Weigh to the nearest 0,01 g, 5-40 g of the sample depending on the amprolium content into a 500 ml conical flask and add 200 ml extraction solvent (3.8). Place the flask in the ultrasonic bath (4.4) and leave for 15 minutes. Remove the flask from the ultrasonic bath and shake it for 1 h on the shaker or stir on the magnetic stirrer (4.5). Dilute an aliquot of the extract with the mobile phase (3.6) to an amprolium content of 0,5-2 μg/ml and mix (see observation 9.3). Filter 5-10 ml of this diluted solution on a membrane filter (4.2). Proceed to the HPLC determination (5.3).
5.2.2.Premixtures (content ≥ 1 % amprolium)U.K.
Weigh to the nearest 0,001 g, 1-4 g of the premixture depending on the amprolium content into a 500 ml conical flask and add 200 ml extraction solvent (3.8). Place the flask in the ultrasonic bath (4.4) and leave for 15 minutes. Remove the flask from the ultrasonic bath and shake it for 1 h on the shaker or stir on the magnetic stirrer (4.5). Dilute an aliquot of the extract with the mobile phase (3.6) to an amprolium content of 0,5-2 μg/ml and mix. Filter 5-10 ml of this diluted solution on a membrane filter (4.2). Proceed to the HPLC determination (5.3).
5.3.HPLC determinationU.K.
5.3.1.Parameters:U.K.
The following conditions are offered for guidance, other conditions may be used provided that they give equivalent results.
| Liquid chromatographic | |
| column (4.1.1): | 125 mm × 4 mm, cation exchange Nucleosil 10 SA, 5 or 10 μm packing, or equivalent |
| Mobile phase (3.6): | Mixture of acetonitrile (3.2), sodium dihydrogen phosphate solution (3.4) and sodium perchlorate solution (3.5), 450+450+100 (v+v+v). |
| Flow rate: | 0,7-1 ml/min |
| Detection wavelength: | 264 nm |
| Injection volume: | 100 μl |
Check the stability of the chromatographic system, injecting several times the calibration solution (3.7.3) containing 1,0 μg/ml, until constant peak heights and retention times are achieved.
5.3.2.Calibration graphU.K.
Inject each calibration solution (3.7.3) several times and determine the mean peak heights (areas) for each concentration. Plot a calibration graph using the mean peak heights (areas) of the calibration solutions as the ordinates and the corresponding concentrations in μg/ml as the abscissae.
5.3.3.Sample solutionU.K.
Inject the sample extract (5.2) several times using the same volume as taken for the calibration solutions and determine the mean peak height (area) of the amprolium peaks.
6.Calculation of the resultsU.K.
From the mean height (area) of the amprolium peaks of the sample solution determine the concentration of the sample solution in μg/ml by reference to the calibration graph (5.3.2).
The amprolium content w in mg/kg of the sample is given by the following formula:

in which:
V
=
volume of the extraction solvent (3.8) in ml according to 5.2 (i.e. 200 ml)
c
=
amprolium concentration of the sample extract (5.2) in μg/ml
f
=
dilution factor according to 5.2
m
=
weight of the test portion in g.
7.Validation of the resultsU.K.
7.1.IdentityU.K.
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract (5.2) and the calibration solution (3.7.3) containing 2,0 μg/ml are compared.
7.1.1.Co-chromatographyU.K.
A sample extract (5.2) is fortified by addition of an appropriate amount of calibration solution (3.7.3). The amount of added amprolium must be similar to the amount of amprolium found in the sample extract.
Only the height of the amprolium peak shall be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its height, must be within ± 10 % of the original width of the amprolium peak of the unfortified sample extract.
7.1.2.Diode array detectionU.K.
The results are evaluated according to the following criteria:
(a)
The wavelength of maximum absorption of the sample and of the standard spectra, recorded at the peak apex on the chromatogram, must be the same within a margin determined by the resolving power of the detection system. For diode-array detection this is typically within ± 2 nm.
(b)
Between 210 and 320 nm, the sample and standard spectra recorded at the peak apex of the chromatogram, must not be different for those parts of the spectrum within the range 10 %-100 % of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15 % of the absorbance of the standard analyte.
(c)
Between 210 and 320 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 %-100 % of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15 % of the absorbance of the spectrum of the peak apex.
If one of these criteria is not met, the presence of the analyte has not been confirmed.
7.2.RepeatabilityU.K.
The difference between the results of two parallel determinations carried out on the same sample must not exceed:
15 % relative to the higher value for amprolium contents from 25 mg/kg to 500 mg/kg,
75 mg/kg for amprolium contents between 500 mg/kg and 1 000 mg/kg,
7,5 % relative to the higher value for amprolium contents of more than 1 000 mg/kg.
7.3.RecoveryU.K.
For a fortified (blank) sample the recovery shall be at least 90 %.
8.Results of a collaborative studyU.K.
A collaborative study was arranged in which three poultry feeds (sample 1-3), one mineral feed (sample 4) and one premix (sample 5) were analysed. The results are given in the following table:
| Sample 1 (blank feed) | Sample 2 | Sample 3 | Sample 4 | Sample 5 | |
|---|---|---|---|---|---|
| L | 14 | 14 | 14 | 14 | 15 |
| n | 56 | 56 | 56 | 56 | 60 |
| mean [mg/kg] | — | 45,5 | 188 | 5 129 | 25 140 |
| sr [mg/kg] | — | 2,26 | 3,57 | 178 | 550 |
| CVr [%] | — | 4,95 | 1,9 | 3,46 | 2,2 |
| sR [mg/kg] | — | 2,95 | 11,8 | 266 | 760 |
| CVR [%] | — | 6,47 | 6,27 | 5,19 | 3,0 |
| nominal content [mg/kg] | — | 50 | 200 | 5 000 | 25 000 |
L
=
number of laboratories
n
=
number of single values
sr
=
standard deviation of repeatability
CVr
=
coefficient of variation of repeatability
sR
=
standard deviation of reproducibility
CVR
=
coefficient of variation of reproducibility.
9.ObservationsU.K.
9.1.If the sample contains thiamine, the thiamine peak in the chromatogram appears shortly before the amprolium peak. Following this method amprolium and thiamine must be separated. If the amprolium and thiamine are not separated by the column (4.1.1) used in this method, replace up to 50 % of the acetonitrile portion of the mobile phase (3.6) by methanol.U.K.
9.2.According to the British Pharmacopoeia, the spectrum of an amprolium solution (c = 0,02 mol/l) in hydrochloric acid (c = 0,1 mol/l) shows maxima at 246 nm and 262 nm. The absorbance shall amount to 0,84 at 246 nm and 0,8 at 262 nm.U.K.
9.3.The extract must always be diluted with the mobile phase, because otherwise the retention time of the amprolium peak may shift significantly, due to changes in the ionic strength.U.K.
D.DETERMINATION OF CARBADOXU.K.
Methyl 3-(2-quinoxalinylmethylene)carbazate N1,N4-dioxide
1.Purpose and scopeU.K.
This method makes it possible to determine the level of carbadox in feed, premixtures and preparations. The detection limit is 1 mg/kg. The limit of quantification is 5 mg/kg.
2.PrincipleU.K.
The sample is equilibrated with water and extracted with methanol-acetonitrile. For feed, an aliquot portion of the filtered extract is subjected to clean-up on an aluminium oxide column. For premixtures and preparations an aliquot portion of the filtered extract is diluted to an appropriate concentration with water, methanol and acetonitrile. The content of carbadox is determined by reversed-phase high-performance liquid chromatography (HPLC) using a UV detector.
3.ReagentsU.K.
3.1.Methanol.U.K.
3.2.Acetonitrile, equivalent to HPLC grade.U.K.
3.3.Acetic acid, w = 100 %.U.K.
3.4.Aluminium oxide: neutral, activity grade I.U.K.
3.5.Methanol-acetonitrile 1 + 1 (v + v).U.K.
Mix 500 ml of methanol (3.1) with 500 ml of acetonitrile (3.2).
3.6.Acetic acid, σ = 10 %.U.K.
Dilute 10 ml acetic acid (3.3) to 100 ml with water.
3.7.Sodium acetate.U.K.
3.8.Water, equivalent to HPLC grade.U.K.
3.9.Acetate buffer solution, c = 0,01 mol/l, pH = 6,0.U.K.
Dissolve 0,82 g of sodium acetate (3.7) in 700 ml of water (3.8) and adjust the pH to 6,0 with acetic acid (3.6). Transfer to a 1 000 ml graduated flask, make up to the mark with water (3.8) and mix.
3.10.Mobile phase for HPLC.U.K.
Mix 825 ml of acetate buffer solution (3.9) with 175 ml of acetonitrile (3.2).
Filter through a 0,22 μm filter (4.5) and degas the solution (e.g. by ultrasonification for 10 minutes).
3.11.Standard substance.U.K.
Pure carbadox: Methyl 3-(2-quinoxalinylmethylene)carbazate N1,N4-dioxide, E 850.
3.11.1.Carbadox stock standard solution, 100 μg/ml (see Note 5. Procedure):U.K.
Weigh to the nearest 0,1 mg, 25 mg of carbadox standard substance (3.11) into a 250 ml graduated flask. Dissolve in methanol-acetonitrile (3.5) by ultrasonification (4.7). After ultrasonic treatment bring the solution to room temperature, make up to the mark with methanol-acetonitrile (3.5) and mix. Wrap the flask with aluminium foil or use amber glassware and store in a refrigerator. At a temperature of ≤ 4 oC the solution is stable for 1 month.
3.11.2.Calibration solutionsU.K.
Transfer 2,0, 5,0, 10,0, and 20,0 ml of the stock standard solution (3.11.1) into a series of 100 ml calibrated flasks. Add 30 ml of water, make up to the mark with methanol-acetonitrile (3.5) and mix. Wrap the flasks with aluminium foil. These solutions correspond to 2,0, 5,0, 10,0 and 20,0 μg/ml of carbadox respectively.
Calibration solutions must be freshly prepared before use.
Note:
For the determination of carbadox in feed containing less than 10 mg/kg, calibration solutions with a concentration below 2,0 μg/ml must be prepared.
3.12.Water-[methanol-acetonitrile] (3.5) mixture, 300 + 700 (v + v).U.K.
Mix 300 ml of water with 700 ml of the mixture of methanol-acetonitrile (3.5).
4.ApparatusU.K.
4.1.Laboratory shaker or magnetic stirrer.U.K.
4.2.Glass fibre filter paper (Whatman GF/A or equivalent).U.K.
4.3.Glass column (length 300 to 400 mm, internal diameter approximately 10 mm) with sintered glass frit and draw-off valve.U.K.
Note:
a glass column fitted with a stopcock or a glass column with a tapered end may also be used; in this case, a small glass-wool plug is inserted into the lower end and it is tamped down using a glass rod.
4.4.HPLC equipment with injection system, suitable for injection volumes of 20 μl.U.K.
4.4.1.Liquid chromatographic column: 300 mm x 4 mm, C18, 10 μm packing or equivalent.U.K.
4.4.2.UV detector with variable wavelength adjustment or diode array detector operating in the range of 225 to 400 nm.U.K.
4.5.Membrane filter, 0,22 μm.U.K.
4.6.Membrane filter, 0,45 μm.U.K.
4.7.Ultrasonic bath.U.K.
5.ProcedureU.K.
Note:
Carbadox is light-sensitive. Carry out all procedures under subdued light or use amber glassware or glassware wrapped in aluminium foil.
5.1.GeneralU.K.
5.1.1.Blank feedU.K.
For the performance of the recovery test (5.1.2) a blank feed shall be analysed to check that neither carbadox nor interfering substances are present. The blank feed shall be similar in type to that of the sample and on analysis carbadox or interfering substances must not be detected.
5.1.2.Recovery testU.K.
A recovery test shall be carried out by analysing the blank feed (5.1.1) which has been fortified by the addition of a quantity of carbadox, similar to that present in the sample. To fortify at a level of 50 mg/kg, transfer 5,0 ml of the stock standard solution (3.11.1) to a 200 ml conical flask. Evaporate the solution to approximately 0,5 ml in a stream of nitrogen. Add 10 g of the blank feed, mix and wait for 10 minutes before proceeding with the extraction step (5.2).
Alternatively, if a blank feed similar in type to that of the sample is not available (see 5.1.1), a recovery test can be performed by means of the standard addition method. In this case, the sample is fortified with a quantity of carbadox, similar to that already present in the sample. This sample is analysed, together with the unfortified sample and the recovery can be calculated by subtraction.
5.2.ExtractionU.K.
5.2.1.FeedU.K.
Weigh to the nearest 0,01 g, 10 g of the sample and transfer to a 200 ml conical flask. Add 15,0 ml of water, mix, and equilibrate for 5 min. Add 35,0 ml of methanol-acetonitrile (3.5), stopper and shake for 30 min. on the shaker or stir on the magnetic stirrer (4.1). Filter the solution through a glass fibre filter paper (4.2). Retain this solution for the purification step (5.3).
5.2.2.Premixtures (0,1 %-2,0 %)U.K.
Weigh to the nearest 0,001 g, 1 g of the unground sample and transfer to a 200 ml conical flask. Add 15,0 ml of water, mix, and equilibrate for 5 min. Add 35,0 ml of methanol-acetonitrile (3.5), stopper and shake for 30 min. on the shaker or stir on the magnetic stirrer (4.1). Filter the solution through a glass fibre filter paper (4.2).
Pipet an aliquot of filtrate into a 50 ml calibrated flask. Add 15,0 ml of water, make up to the mark with methanol-acetonitrile (3.5) and mix. The carbadox concentration of the final solution shall be approximately 10 μg/ml. An aliquot is filtered through a 0,45 μm filter (4.6).
Proceed to the HPLC determination (5.4).
5.2.3.Preparations (> 2 %)U.K.
Weigh to the nearest 0,001 g, 0,2 g of the unground sample and transfer to a 250 ml conical flask. Add 45,0 ml of water, mix, and equilibrate for 5 min. Add 105,0 ml of methanol-acetonitrile (3.5), stopper and homogenise. Sonicate (4.7) the sample for 15 min. followed by shaking or stirring for 15 min. (4.1). Filter the solution through a glass fibre filter paper (4.2).
Dilute an aliquot of filtrate with the mixture of water-methanol-acetonitrile (3.12) to a final carbadox concentration of 10-15 μg/ml (for a 10 % preparation, the dilution factor is 10). An aliquot is filtered through a 0,45 μm filter (4.6).
Proceed to the HPLC determination (5.4).
5.3.PurificationU.K.
5.3.1.Preparation of the aluminium oxide columnU.K.
Weigh 4 g of aluminium oxide (3.4) and transfer it to the glass column (4.3).
5.3.2.Sample purificationU.K.
Apply 15 ml of the filtered extract (5.2.1) to the aluminium oxide column and discard the first 2 ml of eluate. Collect the next 5 ml and filter an aliquot through a 0,45 μm filter (4.6).
Proceed to the HPLC determination (5.4).
5.4.HPLC determinationU.K.
5.4.1.ParametersU.K.
The following conditions are offered for guidance, other conditions may be used provided they yield equivalent results:
| Liquid chromatographic | |
| column (4.4.1): | 300 mm × 4 mm, C18, 10 μmpacking or equivalent |
| Mobile phase (3.10): | Mixture of acetate buffer solution (3.9) and acetonitrile (3.2), 825 + 175 (v+v) |
| Flow rate: | 1,5-2 ml/min. |
| Detection wavelength: | 365 nm |
| Injection volume: | 20 μl |
Check the stability of the chromatographic system, injecting the calibration solution (3.11.2) containing 5,0 μg/ml several times, until constant peak heights (areas) and retention times are achieved.
5.4.2.Calibration graphU.K.
Inject each calibration solution (3.11.2) several times and measure the peak heights (areas) for each concentration. Plot a calibration curve using the mean peak heights or areas of the calibration solutions as the ordinates and corresponding concentrations in μg/ml as the abscissae.
5.4.3.Sample solutionU.K.
Inject the sample extract [(5.3.2) for feed, (5.2.2) for premixtures and (5.2.3) for preparations] several times and determine the mean peak height (area) of the carbadox peaks.
6.Calculation of the resultsU.K.
From the mean height (area) of the carbadox peaks of the sample solution determine the carbadox concentration of the sample solution in μg/ml by reference to the calibration graph (5.4.2).
6.1.FeedU.K.
The content of carbadox w (mg/kg) in the sample is given by the following formula:

in which:
c
=
carbadox concentration of the sample extract (5.3.2) in μg/ml
V1
=
extraction volume in ml (i.e. 50)
m
=
weight of the test portion in g.
6.2.Premixtures and preparationsU.K.
The content of carbadox w (mg/kg) in the sample is given by the following formula:

in which:
c
=
carbadox concentration of the sample extract (5.2.2 or 5.2.3) in μg/ml
V2
=
extraction volume in ml (i.è. 50 for premixtures; 150 for preparations)
f
=
dilution factor according to 5.2.2 (premixtures) or 5.2.3 (preparations)
m
=
weight of the test portion in g.
7.Validation of the resultsU.K.
7.1.IdentityU.K.
The identity of the analyte can be confirmed by co-chromatography, or by using a diode-array detector by which the spectra of the sample extract and the calibration solution (3.11.2) containing 10,0 μg/ml are compared.
7.1.1.Co-chromatographyU.K.
A sample extract is fortified by addition of an appropriate amount of calibration solution (3.11.2). The amount of added carbadox must be similar to the estimated amount of carbadox found in the sample extract.
Only the height of the carbadox peak shall be enhanced after taking into account both the amount added and the dilution of the extract. The peak width, at half of its maximum height, must be within approximately 10 % of the original width.
7.1.2.Diode-array detectionU.K.
The results are evaluated according to the following criteria:
(a)
the wavelength of maximum absorption of the sample and of the standard spectra, recorded at the peak apex on the chromatogram, must be the same within a margin determined by the resolving power of the detection system. For diode-array detection, this is typically within + 2 nm;
(b)
between 225 and 400 nm, the sample and standard spectra recorded at the peak apex on the chromatogram, must not be different for those parts of the spectrum within the range 10 % to 100 % of relative absorbance. This criterion is met when the same maxima are present and at no observed point the deviation between the two spectra exceeds 15 % of the absorbance of the standard analyte;
(c)
between 225 and 400 nm, the spectra of the upslope, apex and downslope of the peak produced by the sample extract must not be different from each other for those parts of the spectrum within the range 10 % to 100 % of relative absorbance. This criterion is met when the same maxima are present and when at all observed points the deviation between the spectra does not exceed 15 % of the absorbance of the spectrum of the apex.
If one of these criteria is not met the presence of the analyte has not been confirmed.
7.2.RepeatabilityU.K.
For contents of 10 mg/kg and higher, the difference between the results of two parallel determinations carried out on the same sample must not exceed 15 % relative to the higher result.
7.3.RecoveryU.K.
For a fortified (blank) sample the recovery shall be at least 90 %.
8.Results of a collaborative studyU.K.
A collaborative study was arranged in which 6 feed, 4 premixtures and 3 preparations were analysed by 8 laboratories. Duplicate analyses were performed on each sample. (More detailed information on this collaborative study can be found in the Journal of the AOAC, Volume 71, 1988, p. 484-490). The results (excluding outliers) are shown below:
Table 1
Results of the collaborative study for feed
| Sample 1 | Sample 2 | Sample 3 | Sample 4 | Sample 5 | Sample 6 | |
|---|---|---|---|---|---|---|
| L | 8 | 8 | 8 | 8 | 8 | 8 |
| n | 15 | 14 | 15 | 15 | 15 | 15 |
| Mean (mg/kg) | 50,0 | 47,6 | 48,2 | 49,7 | 46,9 | 49,7 |
| Sr (mg/kg) | 2,9 | 2,69 | 1,38 | 1,55 | 1,52 | 2,12 |
| CVr (%) | 5,8 | 5,6 | 2,9 | 3,1 | 3,2 | 4,3 |
| SR (mg/kg) | 3,92 | 4,13 | 2,23 | 2,58 | 2,26 | 2,44 |
| CVR (%) | 7,8 | 8,7 | 4,6 | 5,2 | 4,8 | 4,9 |
| Nominal content (mg/kg) | 50,0 | 50,0 | 50,0 | 50,0 | 50,0 | 50,0 |
Table 2
Results of the collaborative study for premixtures and preparations
| Premixtures | Preparations | ||||||
|---|---|---|---|---|---|---|---|
| A | B | C | D | A | B | C | |
| L | 7 | 7 | 7 | 7 | 8 | 8 | 8 |
| n | 14 | 14 | 14 | 14 | 16 | 16 | 16 |
| Mean (g/kg) | 8,89 | 9,29 | 9,21 | 8,76 | 94,6 | 98,1 | 104 |
| Sr (g/kg) | 0,37 | 0,28 | 0,28 | 0,44 | 4,1 | 5,1 | 7,7 |
| CVr (%) | 4,2 | 3,0 | 3,0 | 5,0 | 4,3 | 5,2 | 7,4 |
| SR (g/kg) | 0,37 | 0,28 | 0,4 | 0,55 | 5,4 | 6,4 | 7,7 |
| CVR (%) | 4,2 | 3,0 | 4,3 | 6,3 | 5,7 | 6,5 | 7,4 |
| Nominal content (g/kg) | 10,0 | 10,0 | 10,0 | 10,0 | 100 | 100 | 100 |
L
=
number of laboratories
n
=
number of single values
Sr
=
standard deviation of repeatability
CVr
=
coefficient of variation of repeatability
SR
=
standard deviation of reproducibility
CVR
=
coefficient of variation of reproducibility.
ANNEX IXU.K.CORRELATION TABLES REFERRED TO IN ARTICLE 6
1.Directive 71/250/EECU.K.
| Directive 71/250/EEC | This Regulation |
|---|---|
| Article 1 first subparagraph | Article 3 |
| Article 1 second subparagraph | Article 2 |
| Article 2 | — |
| Article 3 | — |
| Annex, part 1 | Annex II |
| Annex, part 2 | — |
| Annex, part 3 | — |
| Annex, part 4 | Annex III, part O |
| Annex, part 5 | Annex III, part M |
| Annex, part 6 | Annex III, part N |
| Annex, part 7 | Annex III, part Q |
| Annex, part 9 | Annex III, part K |
| Annex, part 10 | — |
| Annex, part 11 | — |
| Annex, part 12 | Annex III, part J |
| Annex, part 14 | Annex III, part D |
| Annex, part 16 | — |
2.Directive 71/393/EECU.K.
| Directive 71/393/EEC | This Regulation |
|---|---|
| Article 1 | Article 3 |
| Article 2 | — |
| Article 3 | — |
| Annex, part I | Annex III, part A |
| Annex, part II | Annex III, part E |
| Annex, part III | Annex III, part P |
| Annex, part IV | Annex III, part H |
3.Directive 72/199/EECU.K.
| Directive 72/199/EEC | This Regulation |
|---|---|
| Article 1 | Article 3 |
| Article 2 | — |
| Article 3 | — |
| Article 4 | — |
| Annex I, part 1 | Annex III, part L |
| Annex I, part 2 | Annex III, part C |
| Annex I, part 3 | — |
| Annex I, part 4 | — |
| Annex I, part 5 | Annex V, part A |
| Annex II | — |
4.Directive 73/46/EECU.K.
| Directive 73/46/EEC | This Regulation |
|---|---|
| Article 1 | Article 3 |
| Article 3 | — |
| Article 4 | — |
| Annex I, part 1 | Annex III, part B |
| Annex I, part 2 | — |
| Annex I, part 3 | Annex III, part I |
5.Directive 76/371/EECU.K.
| Directive 76/371/EEC | This Regulation |
|---|---|
| Article 1 | Article 1 |
| Article 2 | — |
| Article 3 | — |
| Annex | Annex I |
6.Directive 76/372/EECU.K.
| Directive 76/372/EEC | This Regulation |
|---|---|
| Article 1 | — |
| Article 2 | — |
| Article 3 | — |
| Annex | — |
7.Directive 78/633/EECU.K.
| Directive 78/633/EEC | This Regulation |
|---|---|
| Article 1 | Article 3 |
| Article 2 | — |
| Article 3 | — |
| Annex, part 1 | — |
| Annex, part 2 | — |
| Annex, part 3 | Annex IV, part C |
8.Directive 81/715/EECU.K.
| Directive 81/715/EEC | This Regulation |
|---|---|
| Article 1 | — |
| Article 2 | — |
| Article 3 | — |
| Annex | — |
9.Directive 84/425/EECU.K.
| Directive 84/425/EEC | This Regulation |
|---|---|
| Article 1 | — |
| Article 2 | — |
| Article 3 | — |
| Annex | — |
10.Directive 86/174/EECU.K.
| Directive 86/174/EEC | This Regulation |
|---|---|
| Article 1 | Article 4 |
| Article 2 | — |
| Article 3 | — |
| Annex | Annex VII |
11.Directive 93/70/EECU.K.
| Directive 93/70/EEC | This Regulation |
|---|---|
| Article 1 | Article 3 |
| Article 2 | — |
| Article 3 | — |
| Annex | Annex IV, part D |
12.Directive 93/117/ECU.K.
| Directive 93/117/EC | This Regulation |
|---|---|
| Article 1 | Articles 3 and 5 |
| Article 2 | — |
| Article 3 | — |
| Annex, part 1 | Annex IV, part E |
| Annex, part 2 | Annex VIII, part A |
13.Directive 98/64/ECU.K.
| Directive 98/64/EC | This Regulation |
|---|---|
| Article 1 | Articles 3 and 5 |
| Article 2 | — |
| Article 3 | — |
| Article 4 | — |
| Annex, part A | Annex III, part F |
| Annex, part C | Annex VIII, part B |
14.Directive 1999/27/ECU.K.
| Directive 1999/27/EC | This Regulation |
|---|---|
| Article 1 | Articles 3 and 5 |
| Article 2 | — |
| Article 3 | — |
| Article 4 | — |
| Article 5 | — |
| Article 6 | — |
| Article 7 | — |
| Annex, part A | Annex VIII, part C |
| Annex, part B | Annex IV, part F |
| Annex, part C | Annex VIII, part D |
15.Directive 1999/76/ECU.K.
| Directive 1999/76/EC | This Regulation |
|---|---|
| Article 1 | Article 3 |
| Article 2 | — |
| Article 3 | — |
| Article 4 | — |
| Annex | Annex IV, part G |
16.Directive 2000/45/ECU.K.
| Directive 2000/45/EC | This Regulation |
|---|---|
| Article 1 | Article 3 |
| Article 2 | — |
| Article 3 | — |
| Article 4 | — |
| Annex, part A | Annex IV, part A |
| Annex, part B | Annex IV, part B |
| Annex, part C | Annex III, part G |
17.Directive 2002/70/ECU.K.
| Directive 2002/70/EC | This Regulation |
|---|---|
| Article 1 | Article 1 |
| Article 2 | Articles 2 and 3 |
| Article 3 | — |
| Article 4 | — |
| Article 5 | — |
| Annex I | Annex I and Annex V part B(I) |
| Annex II | Annex II and Annex V part B(II) |
18.Directive 2003/126/ECU.K.
| Directive 2003/126/EC | This Regulation |
|---|---|
| Article 1 | Article 3 |
| Article 2 | — |
| Article 3 | — |
| Article 4 | — |
| Article 5 | — |
| Article 6 | — |
| Annex | Annex VI |
(1)
OJ L 165, 30.4.2004, p. 1, corrected by OJ L 191, 28.5.2004, p. 1.
(20)
(21)
(22)
(23)
[F1Any lumps shall be broken up (if necessary by separating them out and returning them to the sample).]
(24)
[F1Except in the case of roughage or forage with low specific gravity.]
(25)
[F1Except in the case of roughage or forage with low specific gravity.]
(26)
(27)
For the drying of cereals, flour, groats and meal, the oven must have a thermal capacity such that, when pre-set at 131 oC, it will return to that temperature in less than 45 minutes after the maximum number of test samples have been placed inside to dry simultaneously. Ventilation must be such that, when as many samples of common wheat as it can contain are dried for two hours, the results differ from those obtained after four hours of drying by less than 0,15 %.
(28)
Where the oil or fat has to undergo subsequent quality tests, replace the fragments of pumice stone by glass beads.
(30)
Conducted by the Feed Working Group of Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(31)
Conducted by the Feed Working Group of Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(32)
Other methods of digestion may be used provided they have been demonstrated to have similar results (such as microwave pressure digestion).
(33)
Green fodder (fresh or dried) is liable to contain large amounts of vegetable silica, which may retain trace elements and must be removed. For samples of these feed, therefore, the following modified procedure must be followed. Carry out operation 5.1.1.1. as far as the filtration. Wash the filter paper containing the insoluble residue twice with boiling water and place it in a quartz or platinum crucible. Ignite in the muffle furnace (4.1) at a temperature below 550 oC until all carbonaceous material has completely disappeared. Allow to cool, add a few drops of water followed by 10 to 15 ml of hydrofluoric acid (3.4) and evaporate to dryness at about 150 oC. If any silica remains in the residue, redissolve it in a few millilitres of hydrofluoric acid (3.4) and evaporate to dryness. Add five drops of sulphuric acid (3.5) and heat until no more white fumes are given off. After the addition of 5 ml of 6 mol/litre hydrochloric acid (3.2) and about 30 ml of water, heat, filter the solution into the 250 ml volumetric flask and make up to the mark with water (HCl concentration about 0,5 mol/l). Proceed then with the determination from point 5.1.2.
(34)
The Analyst 108, 1983, pp. 1252 to 1256.
(35)
Analyst, 1995, 120, 2175-2180.
(36)
[F17Table of TEF (= toxic equivalency factors) for PCDDs, PCDFs and dioxin-like PCBs: WHO-TEFs for human risk assessment based on the conclusions of the World Health Organization (WHO) — International Programme on Chemical Safety (IPCS) expert meeting which was held in Geneva in June 2005 (Martin van den Berg et al., The 2005 World Health Organization Re-evaluation of Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-like Compounds. Toxicological Sciences 93(2), 223–241 (2006)).
| Congener | TEF value | Congener | TEF value |
|---|---|---|---|
| Dibenzo-p-dioxins (‘PCDDs’) and Dibenzo-p-furans '(‘PCDFs’) | ‘Dioxin-like’ PCBs Non-ortho PCBs + Mono-ortho PCBs | ||
| 2,3,7,8-TCDD | 1 | ||
| 1,2,3,7,8-PeCDD | 1 | Non-ortho PCBs | |
| 1,2,3,4,7,8-HxCDD | 0,1 | PCB 77 | 0,0001 |
| 1,2,3,6,7,8-HxCDD | 0,1 | PCB 81 | 0,0003 |
| 1,2,3,7,8,9-HxCDD | 0,1 | PCB 126 | 0,1 |
| 1,2,3,4,6,7,8-HpCDD | 0,01 | PCB 169 | 0,03 |
| OCDD | 0,0003 | Mono-ortho PCBs | |
| 2,3,7,8-TCDF | 0,1 | PCB 105 | 0,00003 |
| 1,2,3,7,8-PeCDF | 0,03 | PCB 114 | 0,00003 |
| 2,3,4,7,8-PeCDF | 0,3 | PCB 118 | 0,00003 |
| 1,2,3,4,7,8-HxCDF | 0,1 | PCB 123 | 0,00003 |
| 1,2,3,6,7,8-HxCDF | 0,1 | PCB 156 | 0,00003 |
| 1,2,3,7,8,9-HxCDF | 0,1 | PCB 157 | 0,00003 |
| 2,3,4,6,7,8-HxCDF | 0,1 | PCB 167 | 0,00003 |
| 1,2,3,4,6,7,8-HpCDF | 0,01 | PCB 189 | 0,00003 |
| 1,2,3,4,7,8,9-HpCDF | 0,01 | ||
| OCDF | 0,0003 | ||
Abbreviations used: ‘ T ’ = tetra; ‘ Pe ’ = penta; ‘ Hx ’ = hexa; ‘ Hp ’ = hepta; ‘ O ’ = octa; ‘ CDD ’ = chlorodibenzodioxin; ‘ CDF ’ = chlorodibenzofuran; ‘ CB ’ = chlorobiphenyl.]
(37)
[F17Commission Decision 2002/657/EC of 14 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and interpretation of results ( OJ L 221, 17.8.2002, p. 8 ).]
(38)
[F17The principles described in the ‘ Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry ’ (http://ec.europa.eu/food/safety/animal-feed_en) shall be followed when applicable.]
(39)
[F17The concept of ‘ upper-bound ’ requires using the limit of quantification for the contribution of each non-quantified congener. The concept of ‘ lower-bound ’ requires using zero for the contribution of each non-quantified congener. The concept of ‘ medium-bound ’ requires using half of the limit of quantification calculating the contribution of each non-quantified congener.]
(40)
[F17Duplicate analysis: Separate analysis of the analytes of interest using a second aliquot of the same homogenized sample. In general, the requirements for duplicate analysis as provided for in Annex II, Chapter C, point 3 apply. However, for methods with the use of 13 C-labelled internal standard for the relevant analytes, the duplicate analysis is only necessary if the result of the first determination is not compliant. The duplicate analysis is necessary to exclude the possibility of internal cross-contamination or an accidental mix-up of samples. In case the analysis is performed in the course of a contamination incident, confirmation by duplicate analysis may be omitted in case the samples selected for analysis are through traceability linked to the contamination incident and the level found is significantly above the maximum level.]
(41)
[F17The concept of ‘ upper-bound ’ requires using the limit of quantification for the contribution of each non-quantified congener to the Toxic Equivalent (TEQ). The concept of ‘ lower-bound ’ requires using zero for the contribution of each non-quantified congener to the TEQ. The concept of ‘ medium-bound ’ requires using half of the limit of quantification calculating the contribution of each non-quantified congener to the TEQ.]
(42)
[F17In general, the requirements for duplicate analysis as provided for in Annex II, Chapter C, point 2 apply. However, for confirmatory methods with the use of 13 C-labelled internal standard for the relevant analytes, the duplicate analysis is only necessary if the result of the first determination is not compliant. The duplicate analysis is necessary to exclude the possibility of internal cross-contamination or an accidental mix-up of samples. In case the analysis is performed in the course of a contamination incident, confirmation by duplicate analysis may be omitted in case the samples selected for analysis are through traceability linked to the contamination incident and the level found is significantly above the maximum level.]
(43)
[F17Identical explanation and requirements for duplicate analysis for control of action thresholds as in footnote 2 above for maximum levels.]
(44)
[F17Regulation (EC) No 183/2005 of the European Parliament and of the Council of 12 January 2005 laying down requirements for feed hygiene ( OJ L 35, 8.2.2005, p. 1 ).]
(45)
[F17Bioanalytical methods are not specific to those congeners included in the TEF-scheme. Other structurally related AhR-active compounds may be present in the sample extract which contribute to the overall response. Therefore, bioanalytical results cannot be an estimate but rather an indication of the TEQ level in the sample.]
(46)
[F17‘ Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry ’ (http://ec.europa.eu/food/safety/animal-feed_en), ‘ Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food ’ (http://ec.europa.eu/food/safety/animal-feed_en).]
(47)
[F17Current requirements are based on the TEFs published in: M. Van den Berg et al, Toxicol Sci 93 (2), 223–241 (2006).]
(48)
[F17Congeners often found to co-elute are for example PCB 28/31, PCB 52/69 and PCB 138/163/164. For GC-MS also possible interferences from fragments of higher chlorinated congeners shall be considered.]
(49)
[F17The principles as described in the ‘ Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food ’ (http://ec.europa.eu/food/safety/animal-feed_en) shall be followed when applicable.]
(50)
[F17It is highly recommendable to have a lower contribution of the reagent blank level to the level of a contaminant in a sample. It is in the responsibility of the laboratory to control the variation of blank levels, in particular, if the blank levels are subtracted.]
(51)
[F17Current requirements are based on the TEFs published in: M. Van den Berg et al, Toxicol Sci 93(2), 223–241 (2006).]
(52)
[F21http://eurl.craw.eu/]
(54)
[F21The list of these primers and probes for each animal species targeted by the assay is available on the EURL-AP website.]
(55)
[F21Examples of Master Mixes that are functional are available on the EURL-AP website.]
Textual Amendments
F1Substituted by Commission Regulation (EU) No 691/2013 of 19 July 2013 amending Regulation (EC) No 152/2009 as regards methods of sampling and analysis (Text with EEA relevance).
F17Substituted by Commission Regulation (EU) 2017/771 of 3 May 2017 amending Regulation (EC) No 152/2009 as regards the methods for the determination of the levels of dioxins and polychlorinated biphenyls (Text with EEA relevance).
Options/Help
Print Options
PrintThe Whole Regulation
You have chosen to open the Whole Regulation
The Whole Regulation you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
Y Rhestrau you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Mae deddfwriaeth ar gael mewn fersiynau gwahanol:
Y Diweddaraf sydd Ar Gael (diwygiedig):Y fersiwn ddiweddaraf sydd ar gael o’r ddeddfwriaeth yn cynnwys newidiadau a wnaed gan ddeddfwriaeth ddilynol ac wedi eu gweithredu gan ein tîm golygyddol. Gellir gweld y newidiadau nad ydym wedi eu gweithredu i’r testun eto yn yr ardal ‘Newidiadau i Ddeddfwriaeth’.
Gwreiddiol (Fel y’i mabwysiadwyd gan yr UE): Mae'r wreiddiol version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Pwynt Penodol mewn Amser: This becomes available after navigating to view revised legislation as it stood at a certain point in time via Advanced Features > Show Timeline of Changes or via a point in time advanced search.
Gweler y wybodaeth ychwanegol ochr yn ochr â’r cynnwys
Rhychwant ddaearyddol: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Dangos Llinell Amser Newidiadau: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Dewisiadau Agor
Dewisiadau gwahanol i agor deddfwriaeth er mwyn gweld rhagor o gynnwys ar y sgrin ar yr un pryd
Rhagor o Adnoddau
Gallwch wneud defnydd o ddogfennau atodol hanfodol a gwybodaeth ar gyfer yr eitem ddeddfwriaeth o’r tab hwn. Yn ddibynnol ar yr eitem ddeddfwriaeth sydd i’w gweld, gallai hyn gynnwys:
- y PDF print gwreiddiol y fel adopted version that was used for the EU Official Journal
- rhestr o newidiadau a wnaed gan a/neu yn effeithio ar yr eitem hon o ddeddfwriaeth
- pob fformat o’r holl ddogfennau cysylltiedig
- slipiau cywiro
- dolenni i ddeddfwriaeth gysylltiedig ac adnoddau gwybodaeth eraill
Llinell Amser Newidiadau
Mae’r llinell amser yma yn dangos y fersiynau gwahanol a gymerwyd o EUR-Lex yn ogystal ag unrhyw fersiynau dilynol a grëwyd ar ôl y diwrnod ymadael o ganlyniad i newidiadau a wnaed gan ddeddfwriaeth y Deyrnas Unedig.
Cymerir dyddiadau fersiynau’r UE o ddyddiadau’r dogfennau ar EUR-Lex ac efallai na fyddant yn cyfateb â’r adeg pan ddaeth y newidiadau i rym ar gyfer y ddogfen.
Ar gyfer unrhyw fersiynau a grëwyd ar ôl y diwrnod ymadael o ganlyniad i newidiadau a wnaed gan ddeddfwriaeth y Deyrnas Unedig, bydd y dyddiad yn cyd-fynd â’r dyddiad cynharaf y daeth y newid (e.e. ychwanegiad, diddymiad neu gyfnewidiad) a weithredwyd i rym. Am ragor o wybodaeth gweler ein canllaw i ddeddfwriaeth ddiwygiedig ar Ddeall Deddfwriaeth.
Rhagor o Adnoddau
Defnyddiwch y ddewislen hon i agor dogfennau hanfodol sy’n cyd-fynd â’r ddeddfwriaeth a gwybodaeth am yr eitem hon o ddeddfwriaeth. Gan ddibynnu ar yr eitem o ddeddfwriaeth sy’n cael ei gweld gall hyn gynnwys:
- y PDF print gwreiddiol y fel adopted fersiwn a ddefnyddiwyd am y copi print
- slipiau cywiro
liciwch ‘Gweld Mwy’ neu ddewis ‘Rhagor o Adnoddau’ am wybodaeth ychwanegol gan gynnwys
- rhestr o newidiadau a wnaed gan a/neu yn effeithio ar yr eitem hon o ddeddfwriaeth
- manylion rhoi grym a newid cyffredinol
- pob fformat o’r holl ddogfennau cysylltiedig
- dolenni i ddeddfwriaeth gysylltiedig ac adnoddau gwybodaeth eraill
The data on this page is available in the alternative data formats listed:
- HTML5 alternative version
- Akoma Ntoso alternative version
- PDF alternative version
- PDF (Original PDF) alternative version
- PDF (Revised PDF) alternative version
- PDF (Revised PDF) alternative version
- PDF (Revised PDF) alternative version
- PDF (Revised PDF) alternative version
- PDF (Revised PDF) alternative version
- PDF (Revised PDF) alternative version
- PDF (Revised PDF) alternative version
- RDF/XML alternative version
- HTML snippet alternative version
- XML alternative version
- CSV alternative version
- HTML RDFa alternative version
 Mae’r holl gynnwys ar gael dan Drwydded Llywodraeth Agored v3.0 ac eithrio ble nodir yn wahanol. Yn ychwanegol mae’r safle hwn â chynnwys sy’n deillio o EUR-Lex, a ailddefnyddiwyd dan delerau Penderfyniad y Comisiwn 2011/833/EU ar ailddefnyddio dogfennau o sefydliadau’r UE. Am ragor o wybodaeth gweler ddatganiad cyhoeddus Swyddfa Gyhoeddiadau’r UE ar ailddefnyddio.
Mae’r holl gynnwys ar gael dan Drwydded Llywodraeth Agored v3.0 ac eithrio ble nodir yn wahanol. Yn ychwanegol mae’r safle hwn â chynnwys sy’n deillio o EUR-Lex, a ailddefnyddiwyd dan delerau Penderfyniad y Comisiwn 2011/833/EU ar ailddefnyddio dogfennau o sefydliadau’r UE. Am ragor o wybodaeth gweler ddatganiad cyhoeddus Swyddfa Gyhoeddiadau’r UE ar ailddefnyddio.