- Latest available (Revised)
- Original (As made)
The Veterinary Medicines Regulations 2013
You are here:
- UK Statutory Instruments
- 2013 No. 2033
- Schedules only

What Version
Advanced Features
- Show Geographical Extent(e.g. England, Wales, Scotland and Northern Ireland)
- Show Timeline of Changes
More Resources
Changes to legislation:
There are currently no known outstanding effects for The Veterinary Medicines Regulations 2013.

Changes to Legislation
Revised legislation carried on this site may not be fully up to date. At the current time any known changes or effects made by subsequent legislation have been applied to the text of the legislation you are viewing by the editorial team. Please see ‘Frequently Asked Questions’ for details regarding the timescales for which new effects are identified and recorded on this site.
Regulation 4(3)
SCHEDULE 1U.K.Marketing authorisations [F1in Great Britain] [F2in Northern Ireland]
Textual Amendments
F1Words in Sch. 1 heading inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(7)(a)
F2Words in Sch. 1 heading inserted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(a)
PART 1U.K.Application for a marketing authorisation
Application for a marketing authorisationU.K.
1. An application under these Regulations for a marketing authorisation for a veterinary medicinal product must be made to the Secretary of State.
[F3Information with the application: generalE+W+S
2.—(1) An application must include the matters mentioned in sub-paragraph (2) and—
(a)where the veterinary medicinal product is an antimicrobial, the matters mentioned in sub-paragraph (4);
(b)subject to sub-paragraph (6), where the product is to be administered to a food-producing animal and is a product containing pharmacologically active substances that are not permitted under Regulation (EC) No 470/2009 of the European Parliament and of the Council, the matter mentioned in sub-paragraph (5);
(c)where the product contains or consists of genetically modified organisms, the matters mentioned in sub-paragraph (7).
(2) For the purposes of sub-paragraph (1) the matters are—
(a)the name of the person who will hold the marketing authorisation and that person’s address or registered place of business;
(b)the name and the address or registered place of business of—
(i)the manufacturer of the finished product;
(ii)any importer of the finished product;
(iii)the manufacturer of any active substances involved at each stage of the manufacture;
(c)the name and address of the sites where—
(i)each stage of the manufacture is carried out;
(ii)any imported products are held; or
(iii)any control or batch release is carried out;
(d)the nature of the marketing authorisation being applied for, and the provisions in Part 2 of Schedule 1 which are relevant to the application;
(e)in relation to the veterinary medicinal product—
(i)the name and the ATCvet code;
(ii)a description of the active substances within the product and, if applicable, a description of any diluent;
(iii)the strength of the product, or, in the case of an immunological veterinary medicinal product or a biological veterinary medicinal product that is not immunological, the biological activity, potency or titre;
(iv)the pharmaceutical form of the product;
(v)the route of administration;
(vi)a description of the target species;
(f)a document showing that the manufacturer is authorised to produce veterinary medicinal products or a certificate of good manufacturing practice issued by the Secretary of State or equivalent certification issued by an authority recognised by the Secretary of State for that purpose, together with a description of the manufacturing process for the active substances and finished product which falls within scope of that authorisation or certificate;
(g)the reference number and a summary of the pharmacovigilance system master file in relation to the product and, where appropriate, the risk management plan that the applicant will put in place;
(h)the proposed summary of product characteristics;
(i)a description of the final presentation, the packaging and labelling of the product;
(j)the proposed text of the information to be included on the immediate packaging, the outer packaging and the information leaflet accompanying the product;
(k)details of any country where—
(i)a marketing authorisation has been granted or revoked in relation to the product;
(ii)a marketing authorisation has been submitted or refused;
(l)a summary of product characteristics included in the terms of any marketing authorisation granted by another country;
(m)technical documentation demonstrating the quality, safety and efficacy of the product;
(n)a report (a “critical expert report”) on the quality, safety and efficacy of the product.
(3) For the purposes of sub-paragraph (2)(n), each critical expert report must—
(a)be prepared with regard to the state of scientific knowledge at the time of the application;
(b)include an evaluation of each test and trial referred to in the application, addressing all aspects relevant to quality, safety and efficacy, with detailed results and precise bibliographic references (including copies of the referenced material);
(c)where technical documentation within sub-paragraph (2)(m) is referenced, include precise cross-references;
(d)be signed and dated by the author, and include details of the author’s educational background, training and occupational experience, and the author’s professional relationship with the applicant.
(4) For the purposes of sub-paragraph (1)(a), the matters are—
(a)information on the direct or indirect risks to public or animal health or to the environment arising from use of the antimicrobial product in animals;
(b)information about the methods of mitigating the development of antimicrobial resistance as a result of the use of the product.
(5) For the purposes of sub-paragraph (1)(b), the matter is a document certifying that a valid application for the establishment of maximum residue levels has been submitted to the Secretary of State.
(6) Sub-paragraph (1)(b) does not apply in respect of a veterinary medicinal product which—
(a)is for administration to a horse that has been declared on its horse passport as not intended for slaughter for human consumption, and
(b)includes an active substance that has been classified under Article 14 of Regulation (EC) No 470/2009 of the European Parliament and of the Council as prohibited for use in food-producing animals.
(7) For the purposes of sub-paragraph (1)(c) the matters are—
(a)a copy of the written consent to the deliberate release into the environment of the genetically modified organisms for research and development purposes issued under the GMO Deliberate Release Regulations;
(b)the complete technical file containing the information provided in respect of the application for that consent under the GMO Deliberate Release Regulations;
(c)the environmental risk assessment provided in respect of the application for that consent under the GMO Deliberate Release Regulations;
(d)the results of any investigations performed for the purposes of research or development.
(8) In assembling the application for an authorisation under this Schedule, the applicant must—
(a)take into account the most up-to-date veterinary medicinal knowledge and scientific guidelines relating to the quality, safety and efficacy of veterinary medicinal products (including relevant monographs of the European Pharmacopoeia and British Pharmacopoeia);
(b)include in the application all information which is relevant to the evaluation of the veterinary medicinal product to which it relates, whether favourable or unfavourable to the product (including information relating to any incomplete or abandoned study or trial);
(c)ensure that the application supports, by reference to specific studies and trials, each claim made by the applicant with regard to the properties, effects and uses of the veterinary medicinal product to which it relates;
(d)otherwise ensure the accuracy of the information in the application.
(9) For the purposes of sub-paragraph (8)(c), pharmacological, toxicological, residue and pre-clinical studies and clinical trials must be carried out in conformity with the principles of good laboratory practice, where applicable.]
Extent Information
E1This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F3Sch. 1 paras. 2-2C substituted for Sch. 1 para. 2 (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 28
Information with the applicationN.I.
2.—(1) An application must include all necessary administrative information, and all scientific documentation necessary for demonstrating the safety, quality and efficacy of the product.
(2) In particular, the applicant must provide all the data required in Annex I to Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products(1), generated in accordance with that Annex.
(3) The application must contain the following information—
(a)the name of the person who will hold the marketing authorisation, that person’s address and, if different, the name and address of all the manufacturers involved in each stage of the manufacture, and the sites where the manufacture will take place;
(b)the name of the veterinary medicinal product, which may be either—
(i)an invented name provided that this is not liable to be confused with the common name of the product or the international non-proprietary name (INN) recommended by the World Health Organization; or
(ii)a common or scientific name accompanied by a trademark or the name of the marketing authorisation holder;
(c)the qualitative and quantitative particulars of all the constituents of the veterinary medicinal product, including its INN recommended by the World Health Organization, where an INN exists, or its chemical name;
(d)a description of the method of manufacture;
(e)all therapeutic indications, contra-indications and adverse reactions;
(f)the dosage for each species of animal for which the veterinary medicinal product is intended, its pharmaceutical form, method and route of administration and proposed shelf life;
(g)any proposed precautionary and safety measures to be taken when storing the veterinary medicinal product, administering it to animals or disposing of waste, together with an indication of potential risks that the veterinary medicinal product might pose to the environment, to human or animal health or to plants, together with the reasons;
(h)in the case of medicinal products intended for food‑producing species, the proposed withdrawal period necessary to ensure that the maximum residue limits specified in Regulation (EC) No 470/2009 of the European Parliament and of the Council are not exceeded;
(i)a description of the testing methods to be used during manufacture;
(j)the results of—
(i)pharmaceutical (physico-chemical, biological or microbiological) tests;
(ii)safety tests and residue tests;
(iii)pre-clinical and clinical trials;
(iv)tests assessing the potential risks to the environment from the product;
(k)a detailed description of the pharmacovigilance system and, where appropriate, the risk management system that the applicant will put in place;
(l)a summary of the product characteristics, mock-ups of all proposed packaging and the proposed package leaflet, if any;
(m)a document showing that the manufacturer is authorised to produce veterinary medicinal products;
(n)copies (which must be updated if there are any changes while the application is being considered) of—
(i)any marketing authorisation obtained in [F565a] member State or in a third country for the relevant veterinary medicinal product, and a list of any F566... member States in which an application for authorisation of the product has been submitted;
(ii)if the product is already authorised outside the United Kingdom, the summary of product characteristics for each authorisation;
(iii)any decision to refuse authorisation, whether in the Community or a third country and the reasons for that decision;
(o)proof that the applicant has the services of a qualified person responsible for pharmacovigilance (referred to in these Regulations as a qualified person (pharmacovigilance)) and has the necessary means for the notification of any adverse reaction suspected of occurring either in the Community or in a third country;
(p)if the veterinary medicinal product is intended for food-producing species and contains one or more pharmacologically active substances not yet included for the species in question in Commission Regulation (EU) No 37/2010, a document certifying that a valid application for the establishment of maximum residue limits has been submitted to the Agency in accordance with Regulation (EC) No 470/2009 of the European Parliament and of the Council.
(4) All documents relating to the results of tests or trials must be accompanied by a detailed and critical expert report that has been drafted and signed by a person with the requisite technical or professional qualifications and that has a brief curriculum vitae of the person signing the report attached to it.
(5) In the case of immunological products, the applicant must submit a description of the methods used to establish that the manufacturing process will consistently produce a veterinary medicinal product that is in accordance with the marketing authorisation.
Extent Information
E154This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F565Word in Sch. 1 para. 2(3)(n)(i) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(b)
F566Word in Sch. 1 para. 2(3)(n)(i) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(b)
[F3Information with the application: formatE+W+S
2A.—(1) An application must be submitted electronically.
(2) Subject to sub-paragraph (3), the application must be structured as a single dossier in four parts—
(a)Part 1 (administrative information);
(b)Part 2 (pharmaceutical quality (physicochemical, biological or microbiological) data);
(c)Part 3 (safety documentation, including safety and residue tests);
(d)Part 4 (efficacy documentation, including pre-clinical studies and clinical trials).
(3) An application concerning the release of GMOs must set out the environmental risk assessment in respect of that release as a separate document, and that assessment must be presented in accordance with the following provision of the GMO Deliberate Release Regulations—
(a)as regards England or Scotland, regulation 6;
(b)as regards Wales, regulation 7.
Textual Amendments
F3Sch. 1 paras. 2-2C substituted for Sch. 1 para. 2 (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 28
Information with the application: animal testingE+W+S
2B.—(1) Where information to be included in an application under paragraph 2(8) includes information concerning experiments on animals, this paragraph applies in respect of that information.
(2) The application must state whether the information was obtained from an experiment conducted in accordance with the requirements in sub-paragraph (4).
(3) The Secretary of State must, in assessing the application, disregard any information to which this paragraph applies which was not obtained from an experiment conducted in accordance with the requirements in sub-paragraph (4).
(4) The requirements are—
(a)the experiment was conducted in accordance with a detailed written protocol prepared in advance;
(b)the experiment was designed to use the minimum number of animals and cause the least pain, suffering or lasting harm, and there was no satisfactory alternative in vitro test available to be used which would have reduced these impacts;
(c)informed consent to the experiment and its consequences (including as regards disposal of treated animals and the taking of produce from treated animals) was obtained in writing from the owner of the animal before the animal was first treated under the experiment;
(d)the welfare of the animals was subject to veterinary supervision throughout the experiment.
Textual Amendments
F3Sch. 1 paras. 2-2C substituted for Sch. 1 para. 2 (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 28
Information with the application: POM-VPS, NFA-VPS and AVM-GSLE+W+S
2C.—(1) Where an applicant proposes, under paragraph 2(2)(d), that a marketing authorisation be granted on the basis that the veterinary medicinal product is classified as POM-VPS, NFA-VPS or AVM-GSL, the requirements in this paragraph apply.
(2) The application must include a document which sets out a detailed justification for the suitability of such classification, having regard to—
(a)animal safety (both as regards treated animals and other animals);
(b)public health; and
(c)environmental safety.]
Textual Amendments
F3Sch. 1 paras. 2-2C substituted for Sch. 1 para. 2 (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 28
[F4Summary of product characteristicsE+W+S
3.—(1) Subject to sub-paragraph (2), the summary of product characteristics required under paragraph 2(2)(h) must include the following information in the order indicated below—
| 1. | Name of the veterinary medicinal product, followed by its strength and pharmaceutical form. | |
| 2. | Qualitative and quantitative composition of the active substances and qualitative composition of excipients and other constituents stating their common name or their chemical description and their quantitative composition, if that information is essential for proper administration of the veterinary medicinal product. | |
| 3. | Clinical information as regards— | |
| 3.1 | target species; | |
| 3.2 | indications for use for each target species; | |
| 3.3 | contra-indications; | |
| 3.4 | special warnings; | |
| 3.5 | special precautions for use, including in particular special precautions for safe use in the target species, special precautions to be taken by the person administering the veterinary medicinal product to the animals and special precautions for the protection of the environment; | |
| 3.6 | frequency and seriousness of adverse events; | |
| 3.7 | use during pregnancy, lactation or lay; | |
| 3.8 | interaction with other medicinal products and other forms of interaction; | |
| 3.9 | administration route and dosage; | |
| 3.10 | symptoms of overdose and, where applicable, emergency procedures and antidotes in the event of overdose; | |
| 3.11 | special restrictions for use; | |
| 3.12 | special conditions for use, including restrictions on the use of antimicrobial and antiparasitic veterinary medicinal products in order to limit the risk of development of resistance; | |
| 3.13 | if applicable, withdrawal periods, even if such periods are zero. | |
| 4. | Pharmacological information as regards— | |
| 4.1 | the ATCvet Code; | |
| 4.2 | pharmacodynamics; | |
| 4.3 | pharmacokinetics. | |
| 5. | Pharmaceutical particulars as regards— | |
| 5.1 | major incompatibilities; | |
| 5.2 | shelf-life, where applicable after reconstitution of the medicinal product or after the immediate packaging has been opened for the first time; | |
| 5.3 | special precautions for storage; | |
| 5.4 | nature and composition of immediate packaging; | |
| 5.5 | requirement to use take-back schemes for veterinary medicinal products for the disposal of unused veterinary medicinal products or waste materials derived from the use of such products and, if appropriate, additional precautions regarding hazardous waste disposal of unused veterinary medicinal products or waste materials derived from the use of such products. | |
| 6. | Name of the holder of the marketing authorisation. | |
| 7. | Marketing authorisation number or numbers. | |
| 8. | Date of the first marketing authorisation. | |
| 9. | Date of the last revision of the summary of product characteristics. | |
| 10. | If applicable, the statement— | |
| 10.1 | “marketing authorisation granted for a limited market and therefore assessment based on customised requirements for documentation”; or | |
| 10.2 | “marketing authorisation in exceptional circumstances and therefore assessment based on customised requirements for documentation”. | |
| 11. | Information on the take-back schemes referred to in point 5.5 applicable to the veterinary medicinal product concerned. | |
| 12. | Classification of the veterinary medicinal product. | |
(2) In the case of an immunological veterinary medicinal product or a biological veterinary medicinal product that is not immunological, in place of the information at points 4, 4.1, 4.2 and 4.3, the summary of product characteristics must include immunological information.]
Textual Amendments
F4Sch. 1 para. 3 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 29
Summary of product characteristicsN.I.
3. The summary of product characteristics required under the preceding paragraph must include the following information, set out in the same format—
Summary of product characteristics | ||
| 1 | Name of the veterinary medicinal product, followed by its strength and pharmaceutical form. | |
| 2 | The name and proportion of each active substance, and of any excipient if knowledge of the excipient is needed for safety reasons. | |
| 3 | Pharmaceutical form. | |
| 4 | Clinical particulars— | |
| 4.1 | target species; | |
| 4.2 | indications for use, specifying the target species; | |
| 4.3 | contra-indications; | |
| 4.4 | special warnings for each target species; | |
| 4.5 | special precautions for use, including special precautions to be taken by the person administering the medicinal product to the animals; | |
| 4.6 | adverse reactions (frequency and seriousness); | |
| 4.7 | use during pregnancy, lactation or lay; | |
| 4.8 | interaction with other medicinal products and other forms of interaction; | |
| 4.9 | amounts to be administered and administration route; | |
| 4.10 | overdose (symptoms, emergency procedures, antidotes) if necessary; | |
| 4.11 | withdrawal periods for the various foodstuffs, including those for which the withdrawal period is zero. | |
| 5 | Pharmacological properties— | |
| 5.1 | pharmacodynamic properties; | |
| 5.2 | pharmacokinetic particulars; | |
| 6 | Pharmaceutical particulars— | |
| 6.1 | list of excipients; | |
| 6.2 | major incompatibilities; | |
| 6.3 | shelf life, when necessary after reconstitution of the medicinal product or when the immediate packaging is opened for the first time; | |
| 6.4 | special precautions for storage; | |
| 6.5 | nature and contents of immediate packaging; | |
| 6.6 | special precautions for the disposal of unused veterinary medicinal products or waste materials derived from the use of such products, if appropriate; | |
| 7 | Marketing authorisation holder; | |
| 8 | Marketing authorisation number; | |
| 9 | Date of the first authorisation or date of renewal of the authorisation; | |
| 10 | Date of any revision of the text; | |
| 11 | Any other information required by the Secretary of State. | |
Supply of a copy of the summary of product characteristicsU.K.
4. A holder of a marketing authorisation must supply a copy of the summary of product characteristics to any person on demand.
Time limits for applications for products for use in food-producing animalsE+W+S
F55. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E2This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F5Sch. 1 para. 5 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 30
Time limits for applications for products for use in food-producing animalsN.I.
5. In the case of a veterinary medicinal product for food-producing animals, a marketing authorisation may not be applied for until at least six months after a valid application has been made for the establishment of a maximum residue limit in accordance with Regulation (EC) No 470/2009 of the European Parliament and of the Council.
Extent Information
E155This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
PART 2U.K.Derogations from some of the requirements in Part 1
ScopeU.K.
6. This Part provides for applications for marketing authorisations in which not all the information required in Part 1 is required, but for the avoidance of doubt any applicant may apply for a marketing authorisation using Part 1 if the applicant wishes to do so.
Bibliographic applicationE+W+S
7.—(1) An applicant for a marketing authorisation need not provide the results of safety tests, residue tests, pre-clinical trials or clinical trials [F6if the applicant demonstrates that the active substances of the veterinary medicinal product have been in well-established veterinary use for at least 10 years, that their efficacy is documented and that they provide an acceptable level of safety].
[F7(1A) Sub-paragraph (1) does not apply to applications for—
(a)biological (including immunological) veterinary medicinal products;
(b)novel therapies.]
(2) The applicant may use any publicly available document.
(3) If an applicant makes use of scientific literature to obtain authorisation for a food-producing species, and submits, in respect of the same medicinal product and with a view to obtaining authorisation for another food-producing species, new residue studies, together with further clinical trials, a third party may not use those studies or trials in an application for a pharmacologically equivalent product for a period of three years from the grant of the authorisation for the additional species.
Extent Information
E3This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F6Words in Sch. 1 para. 7(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 31(a)
F7Sch. 1 para. 7(1A) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 31(b)
Bibliographic applicationN.I.
7.—(1) An applicant for a marketing authorisation need not provide the results of safety tests, residue tests, pre-clinical trials or clinical trials if the active substance of the veterinary medicinal product has been in an authorised veterinary medicinal product for that species in the Community for at least ten years, and the applicant provides appropriate scientific literature to demonstrate this.
(2) The applicant may use any publicly available document.
(3) If an applicant makes use of scientific literature to obtain authorisation for a food-producing species, and submits, in respect of the same medicinal product and with a view to obtaining authorisation for another food-producing species, new residue studies, together with further clinical trials, a third party may not use those studies or trials in an application for a pharmacologically equivalent product for a period of three years from the grant of the authorisation for the additional species.
Extent Information
E156This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for a product using a new combination of active substancesU.K.
8.—[F8(1)] If an application is for a veterinary medicinal product containing active substances already used in an authorised veterinary medicinal product but not previously used in that combination in a veterinary medicinal product, the applicant need not provide the safety and efficacy data for the individual active substances.
[F9(2) Notwithstanding sub-paragraph (1) the applicant must provide a sound scientific justification based on valid therapeutic principles for the combination of active substances, including clinical data, which demonstrates the need for and contribution of all active substances at the moment of treatment.]
Textual Amendments
F8Sch. 1 para. 8 renumbered as Sch. 1 para. 8(1) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 32(a)
F9Sch. 1 para. 8(2) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 32(b)
Application using existing dataU.K.
9. If the Secretary of State has granted a marketing authorisation, the Secretary of State may, with the permission of the holder, use the data submitted in support of that marketing authorisation when assessing an application for another marketing authorisation.
Application for a [F10generic veterinary] medicinal productE+W+S
10.—(1) [F11Subject to sub-paragraphs (2A), (9) and (10) and paragraph 10A,] an applicant need not provide the results of safety tests, residue tests, pre-clinical trials or clinical trials if the applicant can demonstrate that the veterinary medicinal product is [F12a generic of a] veterinary medicinal product already authorised in the [F13United Kingdom] [F14(“the reference veterinary medicinal product”), provided that the applicant provides data demonstrating the matters referred to in sub-paragraph (2)].
(2) For the purposes of this paragraph a product is [F15a generic of] an existing product if—
(a)it has the same qualitative and quantitative composition in active substances;
(b)it has the same pharmaceutical form [F16as the reference product]; and
[F17(c)bioequivalence with the reference product has been demonstrated]
[F18(2A) Sub-paragraph (1) does not apply to applications for biological (including immunological) veterinary medicinal products.]
(3) For the purposes of this paragraph—
(a)the different salts, esters, ethers, isomers, mixtures of isomers, complexes or derivatives of an active substance are considered to be the same active substance, unless they differ significantly in properties with regard to safety or efficacy; and
(b)if they do differ significantly in properties with regard to efficacy or safety, additional information intended to provide proof of the safety or efficacy of the various salts, esters or derivatives of an authorised active substance must be supplied by the applicant.
(4) Different immediate-release oral pharmaceutical forms are regarded as the same pharmaceutical form.
(5) Bioavailability studies are not required if the bioequivalence guidelines produced by the [F19European Medicines] Agency [F20or the Secretary of State] exempt the product.
F21(6) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
[F22(7) For the purposes of these Regulations, subject to sub-paragraph (8), the summary of product characteristics of a generic veterinary medicinal product must be essentially similar to the summary of product characteristics for the reference product.
(8) The requirement in sub-paragraph (7) does not apply in relation to those parts of the summary of product characteristics of the reference product that refer to indications or pharmaceutical forms which are covered by patents at the time when the generic veterinary medicinal product is authorised.
(9) Notwithstanding sub-paragraph (1), in respect of generic veterinary medicinal products intended to be administered by intramuscular, subcutaneous or transdermal routes, the applicant must provide—
(a)administration site target animal tolerance data;
(b)in respect of products intended for administration to food-producing species only, residues depletion data from the site of administration.
(10) Notwithstanding sub-paragraph (1), in respect of generic veterinary medicinal products containing antimicrobial or antiparasitic substances, the applicant must provide all available data (including published data) on the current level of resistance, together with a review of that data as it relates to target pathogens to the active substances concerned.
(11) An applicant must provide an environmental risk assessment for a generic veterinary medicinal product where—
(a)the marketing authorisation for the reference veterinary medicinal product was granted before 1st October 2005, and
(b)no marketing authorisation has been granted since 1st October 2005 in respect of a veterinary medicinal product which has the same active substance and pharmaceutical form as the reference veterinary medicinal product, and which is indicated for use in the same target species when administered at the same or a higher total dose,
unless the Secretary of State holds an environmental risk assessment for the reference veterinary medicinal product and has confirmed this to the applicant.]
Extent Information
E4This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F10Words in Sch. 1 para. 10 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(g)
F11Words in Sch. 1 para. 10(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(a)(i)
F12Words in Sch. 1 para. 10(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(a)(ii)
F13Words in Sch. 1 para. 10(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(13)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F14Words in Sch. 1 para. 10(1) added (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(a)(iii)
F15Words in Sch. 1 para. 10(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(b)(i)
F16Words in Sch. 1 para. 10(2)(b) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(b)(ii)
F17Sch. 1 para. 10(2)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(b)(iii)
F18Sch. 1 para. 10(2A) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(c)
F19Words in Sch. 1 para. 10(5) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(13)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F20Words in Sch. 1 para. 10(5) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(d)
F21Sch. 1 para. 10(6) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(e)
F22Sch. 1 para. 10(7)-(11) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 33(f)
Application for a pharmacologically equivalent medicinal productN.I.
10.—(1) An applicant need not provide the results of safety tests, residue tests, pre-clinical trials or clinical trials if the applicant can demonstrate that the veterinary medicinal product is pharmacologically equivalent to a veterinary medicinal product already authorised in the Community.
(2) For the purposes of this paragraph a product is pharmacologically equivalent to an existing product if—
(a)it has the same qualitative and quantitative composition in active substances;
(b)it has the same pharmaceutical form; and
(c)bioequivalence has been demonstrated by means of appropriate bioavailability studies.
(3) For the purposes of this paragraph—
(a)the different salts, esters, ethers, isomers, mixtures of isomers, complexes or derivatives of an active substance are considered to be the same active substance, unless they differ significantly in properties with regard to safety or efficacy; and
(b)if they do differ significantly in properties with regard to efficacy or safety, additional information intended to provide proof of the safety or efficacy of the various salts, esters or derivatives of an authorised active substance must be supplied by the applicant.
(4) Different immediate-release oral pharmaceutical forms are regarded as the same pharmaceutical form.
(5) Bioavailability studies are not required if the bioequivalence guidelines produced by the Agency exempt the product.
(6) In the case of a reference product authorised in [F567a] member State but not in [F568Northern Ireland], the Secretary of State must be satisfied that the risk-benefit balance of the original product is appropriate for the product to be placed on the market in the United Kingdom, and if the data provided under Article 13, third paragraph of Directive 2001/82/EC by the member State in which the product is authorised are insufficient for the Secretary of State to be satisfied of this, the Secretary of State may notify the applicant and require the applicant to provide further data.
Extent Information
E157This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F567Word in Sch. 1 para. 10(6) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(c)(i)
F568Words in Sch. 1 para. 10(6) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(c)(ii)
[F23Hybrid veterinary medicinal productsE+W+S
10A. An applicant for a marketing authorisation must provide the results of relevant pre-clinical studies or clinical trials where—
(a)bioavailability studies are not capable of demonstrating bioequivalence between the veterinary medicinal product for which the authorisation is sought and a reference veterinary medicinal product for the purposes of paragraph 10; or
(b)the veterinary medicinal product for which the authorisation is sought is not pharmacologically equivalent to a reference veterinary medicinal product for the purposes of paragraph 10 as a result of a difference in relation to—
(i)the active substance or substances contained in the product;
(ii)the strength of the product;
(iii)the indications for use of the product;
(iv)the pharmaceutical form of the product;
(v)the route of administration of the product;
(vi)the withdrawal period for the product.]
Textual Amendments
F23Sch. 1 para. 10A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 34
Time limits for marketing authorisations granted under the procedure for a [F24generic veterinary medicinal] productE+W+S
11.—(1) This paragraph establishes the time limits relating to granting a marketing authorisation under the procedure for a [F25generic veterinary medicinal] product.
(2) An application for a marketing authorisation cannot be made until two years before the product may be placed on the market in accordance with this paragraph.
[F26(3) The product may not be placed on the market until the end of the longest of the following periods which is relevant—
(a)subject to sub-paragraph (3A), 10 years in the case of a veterinary medicinal product authorised for major species;
(b)18 years in the case of a veterinary medicinal product authorised for bees; and
(c)14 years for a veterinary medicinal product authorised for all other species.
(3A) Where the product—
(a)is intended for administration to a major species; and
(b)contains an active substance which is an antimicrobial which has not been an active substance in a veterinary medicinal product previously subject to a marketing authorisation in Great Britain,
the period mentioned in sub-paragraph (3)(a) is 14 years.
(3B) Where a patent in relation to a reference product has lapsed, the summary of product characteristics of the relevant generic product must be updated in order to include the protected information.
(3C) Where, as a result of a variation to an existing marketing authorisation a product is accorded a new marketing authorisation number any relevant protection period applies in relation to that product.
(3D) In this regulation “major species” means cattle, sheep (for meat production), pigs, chickens, dogs and cats.]
(4) Time limits in this paragraph are calculated from the first grant of the marketing authorisation for the reference product.
Textual Amendments
F24Words in Sch. 1 para. 11 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 35(c)
F25Words in Sch. 1 para. 11(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 35(a)
F26Sch. 1 para. 11(3)-(3D) substituted for Sch. 1 para. 11(3) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 35(b)
Time limits for marketing authorisations granted under the procedure for a pharmacologically equivalent productN.I.
11.—(1) This paragraph establishes the time limits relating to granting a marketing authorisation under the procedure for a pharmacologically equivalent product.
(2) An application for a marketing authorisation cannot be made until two years before the product may be placed on the market in accordance with this paragraph.
(3) The product may not be placed on the market until ten years (or, in the case of medicinal products for fish or bees where the application for a marketing authorisation was submitted after 30th October 2005, thirteen years) have elapsed from the initial authorisation of the reference product.
(4) Time limits in this paragraph are calculated from the first grant of the marketing authorisation for the reference product.
Extension of time limitsE+W+S
12.—F27(1) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
[F28(2) Subject to sub-paragraph (2B), if a person submits an application for a marketing authorisation or for a variation to a marketing authorisation for a product, and within five years of the original marketing authorisation being granted, the marketing authorisation is extended to include an additional major species or a new antimicrobial product, the relevant protection period is extended by one year for each additional major species added to the marketing authorisation.
(2A) Subject to sub-paragraph (2B), if a person submits an application for a marketing authorisation mentioned in sub-paragraph (2) and the marketing authorisation is extended to include an additional minor species, the relevant protection period is extended by four years.
(2B) Sub-paragraphs (2) and (2A) do not apply where the application to extend the marketing authorisation is made fewer than three years before the expiration of the protection period.]
(3) The total period may not exceed [F2918] years.
F30(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Textual Amendments
F27Sch. 1 para. 12(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 36(a)
F28Sch. 1 para. 12(2)-(2B) substituted for Sch. 1 para. 12(2) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 36(b)
F29Word in Sch. 1 para. 12(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 36(c)
F30Sch. 1 para. 12(4) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 36(d)
Extension of time limitsN.I.
12.—(1) This paragraph applies in relation to veterinary medicinal products that—
(a)are intended for administration to food-producing species; and
(b)contain a new active substance that was not authorised in the Community by 30th April 2004.
(2) If a person submitted an application for a marketing authorisation for a product on or after 30th October 2005, and within 5 years of the original marketing authorisation being granted, the marketing authorisation is extended to include additional food-producing species, the ten-year protection period is extended by one year for each additional food-producing species added to the marketing authorisation.
(3) The total period may not exceed 13 years.
(4) The extension applies only if the marketing authorisation holder originally applied for determination of the maximum residue limits for the active substance.
[F31Time limits – supplementaryE+W+S
12A.—(1) Subject to sub-paragraph (3), a study, residue test or pre-clinical study in relation to the establishment of residue limits submitted by an applicant in relation to an application for a marketing authorisation or a variation of a marketing authorisation may not be used for any other such application or variation until the period of five years from that submission has elapsed.
(2) Subject to sub-paragraph (3), a study, residue test or preclinical study submitted by an applicant for a marketing authorisation or a variation in a marketing authorisation which demonstrates a reduction in antimicrobial resistance in relation to a reference product may not be used for any other such application until a period of four years in addition to the relevant protection period has elapsed.
(3) Sub-paragraphs (1) and (2) do not apply where an applicant has obtained a written authorisation to access a study, residue test or pre-clinical study mentioned in the relevant sub-paragraph.]
Textual Amendments
F31Sch. 1 para. 12A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 37
Parallel importsE+W+S
F3213. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E5This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F32Sch. 1 para. 13 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 38
Parallel importsN.I.
13.—(1) The Secretary of State may grant a marketing authorisation in relation to a veterinary medicinal product authorised in [F569a] member State and imported into [F570Northern Ireland] from that member State in accordance with this paragraph without the data required in Part 1 if the applicant can demonstrate compliance with this paragraph.
(2) If the product is for a food-producing species it must be identical to a product authorised in [F571Northern Ireland].
(3) Other products must be therapeutically the same as a product authorised in [F572Northern Ireland] unless the importer can justify any differences.
(4) The member State from which it is imported must have authorised the product in accordance with Directive 2001/82/EC.
(5) The applicant must be established within the Community.
(6) The applicant must hold (or have a contract with the holder of) a wholesale dealer’s authorisation in [F573Northern Ireland] appropriate to the type of product to be imported.
(7) If re-labelling is to take place in [F574Northern Ireland] the applicant must also be (or have a contract with) the holder of a suitable manufacturing authorisation in [F574Northern Ireland].
Extent Information
E158This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F569Word in Sch. 1 para. 13(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(i)
F570Words in Sch. 1 para. 13(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
F571Words in Sch. 1 para. 13(2) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
F572Words in Sch. 1 para. 13(3) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
F573Words in Sch. 1 para. 13(6) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
F574Words in Sch. 1 para. 13(7) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(d)(ii)
Specific batch control schemeE+W+S
14.—(1) Where a veterinary medicinal product (other than a biological veterinary medicinal product) has been granted a marketing authorisation or an animal test certificate, and any starting material (active substance, excipient or packaging) or any batch of the product does not fully meet the requirements of the authorisation or animal test certificate, the holder may apply to the Secretary of State to place one or more batches on the market notwithstanding this.
(2) The Secretary of State may authorise the placing on the market on being satisfied that the safety, quality and efficacy of the product are not compromised, and that in all the circumstances of the case the product should be placed on the market.
F33(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F34(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E6This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F33Sch. 1 para. 14(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(16) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F34Sch. 1 para. 14(4) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 39
Specific batch control schemeN.I.
14.—(1) Where a veterinary medicinal product (other than a biological veterinary medicinal product) has been granted a marketing authorisation or an animal test certificate, and any starting material (active substance, excipient or packaging) or any batch of the product does not fully meet the requirements of the authorisation or animal test certificate, the holder may apply to the Secretary of State to place one or more batches on the market notwithstanding this.
(2) The Secretary of State may authorise the placing on the market on being satisfied that the safety, quality and efficacy of the product are not compromised, and that in all the circumstances of the case the product should be placed on the market.
(3) This paragraph does not apply in relation to a product recognised in more than one member State.
(4) In this paragraph a biological veterinary medicinal product is a veterinary medicinal product, the active substance of which is a biological substance; and a biological substance is a substance that is produced by or extracted from a biological source and for which a combination of physico-chemical-biological testing and the production process and its control is needed for its characterisation and the determination of its quality.
Extent Information
E159This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Similar immunological productsU.K.
[F3515. Where an immunological veterinary medicinal product is pharmacologically equivalent to a reference product other than differences in raw materials or in the manufacturing process, the results of the appropriate pre-clinical tests or clinical trials must be provided, but the applicant need not provide the results of safety tests or residue tests.]
Textual Amendments
F35Sch. 1 para. 15 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 40
Marketing a product authorised in another countryE+W+S
16. Where the health situation so requires, the Secretary of State may authorise the placing on the market of a veterinary medicinal product that has been authorised [F36in another] country.
Extent Information
E7This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F36Words in Sch. 1 para. 16 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(17) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Marketing a product authorised in another countryN.I.
16. Where the health situation so requires, the Secretary of State may authorise the placing on the market of a veterinary medicinal product that has been authorised by [F575a] member State or, if there is no such authorised product, authorised in a third country.
Extent Information
E160This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F575Word in Sch. 1 para. 16 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(e)
PART 3U.K.Grant of a marketing authorisation
Time limitsU.K.
17.—[F37(1)] The Secretary of State must ensure that the procedure for granting an authorisation for a veterinary medicinal product is completed within a maximum of 210 days after the submission of the application.
[F38(2) Sub-paragraph (1) does not apply where a simultaneous assessment of the application is being conducted by the Secretary of State and the relevant authority in another country.]
Textual Amendments
F37Sch. 1 para. 17 renumbered as Sch. 1 para. 17(1) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 41(a)
F38Sch. 1 para. 17(2) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 41(b)
[F39Place of establishment of applicantE+W+S
18. Only an applicant established in the United Kingdom or in a country which the Secretary of State considers to have demonstrated equivalent standards to those in the United Kingdom may be granted (or hold) a marketing authorisation or a veterinary homeopathic registration.]
Extent Information
E8This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F39Sch. 1 para. 18 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 42
Place of establishment of applicantN.I.
18. Only an applicant established in [F576the United Kingdom or] a member State may be granted a marketing authorisation.
Extent Information
E161This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F576Words in Sch. 1 para. 18 inserted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(i)
ProcedureU.K.
19. The Secretary of State may require the applicant to provide additional information or to generate additional data, including laboratory testing, or may require the applicant to provide samples of any medicinal product, its starting materials and intermediate products or other constituent materials for testing in a laboratory.
Products authorised in another member StateE+W+S
F4020. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E9This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F40Sch. 1 para. 20 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(19) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Products authorised in another member StateN.I.
20. Where the Secretary of State is informed or discovers that [F577a] member State has authorised a veterinary medicinal product that is the subject of an application for authorisation by the Secretary of State, the Secretary of State must reject the application unless it was submitted in accordance with the mutual recognition procedure or the decentralised procedure in Part 6.
Extent Information
E162This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F577Word in Sch. 1 para. 20 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(ii)
Assessment reportsU.K.
21. The Secretary of State must produce an assessment of the dossier, consisting of an evaluation of the results of the pharmaceutical, safety and residue tests and the pre-clinical and clinical trials of the veterinary medicinal product concerned, and any additional related information.
Grant of a marketing authorisationU.K.
22.—[F41(1) The Secretary of State must, before granting a marketing authorisation—
(a)verify that the data submitted complies with the requirements set out in these Regulations;
(b)assess the application and data submitted in respect of the veterinary medicinal product; and
(c)reach a conclusion in relation to the benefit-risk balance of granting a marketing authorisation in respect of the veterinary medicinal product.]
[F42(2)] When granting a marketing authorisation, the Secretary of State must inform the applicant of the summary of product characteristics that has been approved, and the distribution category of the product.
[F43(3) The Secretary of State must set out any terms and conditions in connection with placing the product on the market when granting a marketing authorisation.
(4) Where the marketing authorisation relates to a veterinary medicinal product that contains an antimicrobial the Secretary of State may require the holder of the marketing authorisation to conduct post-authorisation studies in order to ensure that the benefit-risk balance remains positive in relation to the development of antimicrobial resistance.]
Textual Amendments
F41Sch. 1 para. 22(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 43(b)
F42Sch. 1 para. 22 renumbered as Sch. 1 para. 22(2) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 43(a)
F43Sch. 1 para. 22(3)(4) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 43(c)
[F44Withdrawal of application for marketing authorisationE+W+S
22A.—(1) Where an applicant for a marketing authorisation withdraws the application before the Secretary of State has produced an assessment of the dossier under paragraph 21 the applicant must give written reasons for so doing.
(2) Where an applicant withdraws an application for a marketing authorisation in the circumstances mentioned in sub-paragraph (1) the Secretary of State must publish—
(a)the fact that the application has been withdrawn; and
(b)a summary of the reasons for withdrawal.]
Textual Amendments
F44Sch. 1 para. 22A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 44
Marketing authorisations for food-producing speciesE+W+S
23.—(1) The Secretary of State must not grant a marketing authorisation for a veterinary medicinal product for food-producing species unless [F45maximum residue limits have been established under Regulation (EC) No 470/2009 of the European Parliament and of the Council in respect of ] all its pharmacologically active substances F46....
(2) This does not apply in the case of a marketing authorisation for a veterinary medicinal product for administration to a horse that has been declared on its horse passport as not intended for slaughter for human consumption; but in this case the product must not include an active substance that [F47has been classified under Article 14 of Regulation (EC) No 470/2009 of the European Parliament and of the Council as prohibited for use in food producing animals.]
Extent Information
E10This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F45Words in Sch. 1 para. 23(1) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(20)(a)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F46Words in Sch. 1 para. 23(1) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(20)(a)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F47Words in Sch. 1 para. 23(2) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(20)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Marketing authorisations for food-producing speciesN.I.
23.—(1) The Secretary of State must not grant a marketing authorisation for a veterinary medicinal product for food-producing species unless all its pharmacologically active substances appear in Table 1 in the Annex to Commission Regulation (EU) No 37/2010.
(2) This does not apply in the case of a marketing authorisation for a veterinary medicinal product for administration to a horse that has been declared on its horse passport as not intended for slaughter for human consumption; but in this case the product must not include an active substance that appears in Table 2 in the Annex to Commission Regulation (EU) No 37/2010 and must not be intended for the treatment of a condition for which a veterinary medicinal product is already authorised for horses.
Extent Information
E163This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Refusal of a marketing authorisationE+W+S
24.—(1) The Secretary of State must refuse to grant a marketing authorisation if the application does not comply with these Regulations.
(2) In addition, the Secretary of State must refuse to grant it if—
(a)the data submitted with the application are inadequate;
(b)the [F48benefit-risk balance] of the veterinary medicinal product is unfavourable;
[F49(c)the applicant has not provided sufficient evidence of the efficacy of the product in relation to the target species;]
(d)the withdrawal period proposed by the applicant is not long enough to ensure [F50food safety], or is insufficiently substantiated;
(e)the veterinary medicinal product is for a prohibited use;
(f)the way that the product will be used will have an unnecessarily undesirable effect on the environment.
[F51(g)the veterinary medicinal product is a veterinary medicinal product which contains an antimicrobial which is presented for use in order to promote the growth of treated animals or to increase yields from treated animals;
(h)the risk for public health in case of development of antimicrobial resistance or antiparasitic resistance outweighs the benefits of the veterinary medicinal product to animal health;
(i)the risks to public or animal health or to the environment are not sufficiently addressed;
(j)the qualitative or quantitative composition of the veterinary medicinal product is not as stated in the application;
(k)the active substance within the veterinary medicinal product meets the criteria for being considered persistent, bio-accumulative and toxic and the veterinary medicinal product is intended to be used in food-producing animals (except where it is demonstrated that the active substance is essential to prevent or control a serious risk to animal health).]
(3) The Secretary of State may refuse to grant a marketing authorisation—
(a)if there is F52... legislation pending that is incompatible with the requested authorisation; or
(b)if additional data have been requested and those data are not provided within such time limit as may be stipulated.
(4) If the Secretary of State, on the grounds of safety, quality or efficacy, intends to refuse an application, or proposes to grant a marketing authorisation that is different from the one applied for, the Secretary of State must notify the applicant accordingly, and the applicant may appeal to the Veterinary Products Committee.
Extent Information
E11This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F48Words in Sch. 1 para. 24(2)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 45(a)
F49Sch. 1 para. 24(2)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 45(b)
F50Words in Sch. 1 para. 24(2)(d) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(21)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F51Sch. 1 para. 24(2)(g)-(k) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 45(c)
F52Word in Sch. 1 para. 24(3)(a) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(21)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Refusal of a marketing authorisationN.I.
24.—(1) The Secretary of State must refuse to grant a marketing authorisation if the application does not comply with these Regulations.
(2) In addition, the Secretary of State must refuse to grant it if—
(a)the data submitted with the application are inadequate;
(b)the risk-benefit balance of the veterinary medicinal product is unfavourable;
(c)the product has insufficient therapeutic effect;
(d)the withdrawal period proposed by the applicant is not long enough to ensure that Regulation (EC) No 470/2009 of the European Parliament and of the Council is complied with, or is insufficiently substantiated;
(e)the veterinary medicinal product is for a prohibited use;
(f)the way that the product will be used will have an unnecessarily undesirable effect on the environment.
(3) The Secretary of State may refuse to grant a marketing authorisation—
(a)if there is Community legislation pending that is incompatible with the requested authorisation; or
(b)if additional data have been requested and those data are not provided within such time limit as may be stipulated.
(4) If the Secretary of State, on the grounds of safety, quality or efficacy, intends to refuse an application, or proposes to grant a marketing authorisation that is different from the one applied for, the Secretary of State must notify the applicant accordingly, and the applicant may appeal to the Veterinary Products Committee.
Extent Information
E164This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Publication following the grant [F53, refusal, suspension, variation or revocation] of a marketing authorisationU.K.
25.—(1) On granting a marketing authorisation the Secretary of State must publish—
(a)the notice granting the marketing authorisation;
(b)the summary of the product characteristics;
(c)the assessment report that has already been prepared but with any commercially confidential or personal information deleted.
(2) The Secretary of State must update the assessment report whenever new information that is of importance and relates to the quality, safety or efficacy of the veterinary medicinal product becomes available.
(3) The Secretary of State must send a copy of the assessment report, and any update, to the holder of the marketing authorisation before publication to enable the holder to make representations concerning any confidential or personal information that may be in it, and may specify a date by which representations must be made.
[F54(4) Where the Secretary of State refuses to grant a marketing authorisation or suspends or revokes an authorisation the Secretary of State must publish that fact.
(5) Where the Secretary of State varies a marketing authorisation in relation to the summary of product characteristics the Secretary of State must publish the terms of the variation.]
Textual Amendments
F53Words in Sch. 1 para. 25 heading inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 46(b)
F54Sch. 1 para. 25(4)(5) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 46(a)
Marketing authorisations in exceptional circumstancesU.K.
26.—(1) In exceptional circumstances, and if there is no other product with a full marketing authorisation for the indicated condition in the target species, the Secretary of State may grant an exceptional marketing authorisation consisting of—
(a)a provisional marketing authorisation subject to a requirement for the applicant to provide further data [F55, taking into account the benefit of the immediate availability on the market of the veterinary medicinal product in comparison to the risks]; or
(b)a limited marketing authorisation for a product with a limited market [F56, taking into account the benefit in relation to public or animal health of the availability of the product on the market in comparison to the risks.].
[F57(1A) An exceptional marketing authorisation may be granted subject to such further conditions, including any restrictions, as the Secretary of State considers appropriate.]
(2) The Secretary of State must reassess each provisional or limited marketing authorisation annually.
Textual Amendments
F55Words in Sch. 1 para. 26(1)(a) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 47(a)
F56Words in Sch. 1 para. 26(1)(b) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 47(b)
F57Sch. 1 para. 26(1A) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 47(c)
Provisions of samples and expertiseU.K.
27.—(1) The Secretary of State may require a marketing authorisation holder to provide, at any time and at any stage of the manufacturing process, samples of starting materials or the veterinary medicinal product for testing [F58and to provide the results of any control tests carried out in relation to such materials or the finished product in accordance with the methods to be used under the terms of the marketing authorisation].
(2) At the request of the Secretary of State, the marketing authorisation holder must provide technical expertise to facilitate any analysis of the product.
[F59(3) The Secretary of State may require an applicant for a marketing authorisation to provide samples of a veterinary medicinal product for testing.
(4) The samples mentioned in sub-paragraph (3) may be used—
(a)to test the veterinary medicinal product and its constituents at any stage of development of the product in order to ensure that the control methods used by the manufacturer are satisfactory; and
(b)to verify that, where a veterinary medicinal product is intended for administration to a food-producing animal, the means used for residue detection in relation to pharmacologically active substances are satisfactory.]
Textual Amendments
F58Words in Sch. 1 para. 27(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 48(a)
F59Sch. 1 para. 27(3)(4) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 48(b)
[F60Records and] supply of informationE+W+S
28.—(1) A marketing authorisation holder must immediately inform the Secretary of State on receipt of any new information that might adversely affect the [F61benefit-risk balance] of the veterinary medicinal product.
(2) The holder must immediately inform the Secretary of State of any prohibition or restriction imposed by the competent authorities of any country in which the veterinary medicinal product is authorised.
(3) The Secretary of State may at any time require the marketing authorisation holder to provide data relating to the [F61benefit-risk balance].
[F62(4) A marketing authorisation holder must retain all of the original documents from every clinical trial from which data was derived in support of the application for authorisation under this Schedule, and in support of any variation of the authorisation (whether granted or otherwise), for at least five years from the date on which the authorisation ceases.]
Textual Amendments
F60Words in Sch. 1 para. 28 heading inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 49(c)
F61Words in Sch. 1 para. 28 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 49(a)
F62Sch. 1 para. 28(4) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 49(b)
Supply of informationN.I.
28.—(1) A marketing authorisation holder must immediately inform the Secretary of State on receipt of any new information that might adversely affect the risk-benefit balance of the veterinary medicinal product.
(2) The holder must immediately inform the Secretary of State of any prohibition or restriction imposed by the competent authorities of any country in which the veterinary medicinal product is authorised.
(3) The Secretary of State may at any time require the marketing authorisation holder to provide data relating to the risk-benefit balance.
Duties on the holder of a marketing authorisation relating to an immunological productE+W+S
29.—[F63(1) Before placing an immunological product on the market the holder of the marketing authorisation must notify the Secretary of State asking for written approval to do so.]
(2) If notified under sub-paragraph [F64(1)] the Secretary of State must give or refuse a written approval as soon as is reasonably practicable.
(3) No person may place an immunological product on the market without a written approval issued by the Secretary of State F65....
Extent Information
E12This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F63Sch. 1 para. 29(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(22)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F64Word in Sch. 1 para. 29(2) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(22)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F65Words in Sch. 1 para. 29(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(22)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Duties on the holder of a marketing authorisation relating to an immunological productN.I.
29.—(1) Before placing an immunological product on the market the holder of the marketing authorisation must either—
(a)notify the Secretary of State asking for written approval to do so; or
(b)if the holder has already received written approval from [F578a] member State permitting the release of the product, send a copy of that approval to the Secretary of State.
(2) If notified under sub-paragraph (1)(a) the Secretary of State must give or refuse a written approval as soon as is reasonably practicable.
(3) No person may place an immunological product on the market without a written approval issued by the Secretary of State or (if the approval was issued by [F579a] member State) without sending a copy of that approval to the Secretary of State.
Extent Information
E165This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F578Word in Sch. 1 para. 29(1)(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(iii)
F579Word in Sch. 1 para. 29(3) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(iii)
Control testsU.K.
[F6630. The holder of a marketing authorisation must give to the Secretary of State on demand evidence that the holder has carried out all control tests required under the marketing authorisation, and the results of those tests.]
Textual Amendments
F66Sch. 1 para. 30 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 50
Placing on the marketE+W+S
31.—(1) A holder of a marketing authorisation must notify the Secretary of State when the veterinary medicinal product is first placed on the market in the United Kingdom, and the date on which it was placed on the market.
(2) A holder of a marketing authorisation who removes the veterinary medicinal product from the market in the United Kingdom must notify the Secretary of State at least two months (or a shorter period in exceptional circumstances) before doing so.
[F67(2A) A holder of a marketing authorisation who identifies a shortage of the veterinary medicinal product must notify the Secretary of State as soon as is reasonably practicable.
(2B) For the purposes of sub-paragraph (2A) a shortage of a veterinary medicinal product occurs when supply does not meet demand at a national level within the United Kingdom.]
(3) Upon request by the Secretary of State, the marketing authorisation holder must provide—
(a)all data relating to the volume of sales of the veterinary medicinal product by the holder; and
(b)any data in the holder’s possession relating to the number of prescriptions written for the product and the total volume supplied under those prescriptions.
Extent Information
E13This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F67Sch. 1 para. 31(2A)(2B) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 51
Placing on the marketN.I.
31.—(1) A holder of a marketing authorisation must notify the Secretary of State when the veterinary medicinal product is first placed on the market in [F580Northern Ireland], and the date on which it was placed on the market.
(2) A holder of a marketing authorisation who removes the veterinary medicinal product from the market in [F581Northern Ireland] must notify the Secretary of State at least two months (or a shorter period in exceptional circumstances) before doing so.
(3) Upon request by the Secretary of State, the marketing authorisation holder must provide—
(a)all data relating to the volume of sales of the veterinary medicinal product by the holder; and
(b)any data in the holder’s possession relating to the number of prescriptions written for the product and the total volume supplied under those prescriptions.
Extent Information
E166This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F580Words in Sch. 1 para. 31(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(iv)
F581Words in Sch. 1 para. 31(2) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(iv)
[F68Duration and validity of marketing authorisationE+W+S
32. Subject to any power of revocation provided under these Regulations a marketing authorisation is valid indefinitely.]
Extent Information
E14This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F68Sch. 1 para. 32 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 52
Duration and validity of a marketing authorisationN.I.
32.—(1) A marketing authorisation is initially valid for five years.
(2) The authorisation may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance.
(3) An application for renewal must be made at least six months, and not more than nine months, before the marketing authorisation ceases to be valid.
(4) An applicant who applies for the renewal of the marketing authorisation must enclose a list of all documents concerning the product that the applicant has submitted to the Secretary of State since the marketing authorisation was granted.
(5) The Secretary of State may require the applicant to provide a copy of any of the listed documents at any time.
(6) Once renewed, the marketing authorisation is valid indefinitely unless, within five years of the renewal, the Secretary of State notifies the holder, on justified grounds relating to pharmacovigilance, that the authorisation will cease to be valid five years from the first renewal unless the holder applies for a further renewal.
(7) The further renewal is not time-limited.
(8) Any marketing authorisation granted under these Regulations that is not followed within three years of its granting by the actual placing on the market of the authorised veterinary medicinal product in [F582Northern Ireland] ceases to be valid.
(9) When a veterinary medicinal product authorised under these Regulations and previously placed on the market in [F583Northern Ireland] is not present on the market in [F583Northern Ireland] for a period of three consecutive years, its marketing authorisation ceases to be valid.
(10) The Secretary of State may, on human or animal health grounds, grant exemptions from sub-paragraphs (8) and (9).
Extent Information
E167This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F582Words in Sch. 1 para. 32(8) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(v)
F583Words in Sch. 1 para. 32(9) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(f)(v)
PART 4U.K.Variations of marketing authorisations on the application of the holder
Variation of a marketing authorisationE+W+S
33.—F69(1) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(2) The holder of a marketing authorisation may apply to the Secretary of State for a variation of that marketing authorisation.
[F70(3) An application for a variation under paragraph (2) may only relate to—
(a)a single variation, which may relate to one or more marketing authorisations, or
(b)one or more variations to a single marketing authorisation.]
(4) The Secretary of State, when granting a variation of a veterinary medicinal product, may (unless there are exceptional circumstances necessary to protect human or animal health or the environment) specify transitional measures to enable products produced in accordance with the previous authorisation to continue to be marketed for the transitional period.
Textual Amendments
F69Sch. 1 para. 33(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 53(a)
F70Sch. 1 para. 33(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 53(b)
Variation of a marketing authorisationN.I.
33.—(1) The Secretary of State is the competent authority for the purposes of Commission Regulation (EC) No 1234/2008(2).
(2) The holder of a marketing authorisation may apply to the Secretary of State for a variation of that marketing authorisation.
(3) An application for a variation under paragraph (2) may only relate to a “single variation” unless the application is submitted in accordance with—
(a)Article 7 of Commission Regulation (EC) No 1234/2008 (“grouped variations”), or
(b)Article 20 of Commission Regulation (EC) No 1234/2008 (“workshare variations”).
(4) The Secretary of State, when granting a variation of a veterinary medicinal product, may (unless there are exceptional circumstances necessary to protect human or animal health or the environment) specify transitional measures to enable products produced in accordance with the previous authorisation to continue to be marketed for the transitional period.
Extent Information
E168This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F71Variation procedureE+W+S
33A.—(1) Subject to sub-paragraphs (2) and (6), an application for a variation must be submitted to the Secretary of State electronically.
(2) Sub-paragraph (1) does not apply where the application is an emergency application.
(3) The application must contain—
(a)a description of the proposed variation;
(b)information in relation to any of the matters referred to in paragraph 2 which are relevant to the proposed variation;
(c)details of any marketing authorisation which may be affected by the proposed variation; and
(d)where the proposed variation requires consequential variations to the terms of the marketing authorisation, a description of those variations.
(4) The Secretary of State must produce an assessment of the application.
(5) The Secretary of State may require the applicant to provide additional information during the assessment process.
(6) Where the Secretary of State is satisfied that it is not necessary for the application to contain certain information for the purposes of conducting an assessment, having regard to the risks involved with the proposed variation, the Secretary of State may waive the requirement to provide that information under sub-paragraph (3) (and the requirement in sub-paragraph (4) does not apply in respect of that information).
(7) The Secretary of State must send a copy of the assessment mentioned in sub-paragraph (4) to the applicant.
(8) Having assessed the application, the Secretary of State must—
(a)amend the authorisation to correspond with the proposed variation; or
(b)reject the proposed variation.
(9) Where the Secretary of State amends the authorisation in accordance with sub-paragraph (8)(a) the Secretary of State must notify the applicant in writing.
(10) The Secretary of State must ensure that the determination of an application for a variation of a marketing authorisation is completed within a maximum of 180 days after the submission of the application.]
Textual Amendments
F71Sch. 1 para. 33A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 54
Refusal of a variation of a marketing authorisationE+W+S
34.—F72(1) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(2) The grounds on which the Secretary of State may refuse an application for a variation of a marketing authorisation are those set out in paragraph 24 of this Schedule (refusal of a marketing authorisation).
(3) The Secretary of State must give written reasons for refusing to grant a variation; and if—
(a)those reasons are on the grounds of safety, quality or efficacy; and
[F73(b)the Secretary of State produced an assessment in respect of the variation under paragraph 33A(4),]
the applicant may appeal to the Veterinary Products Committee.
Textual Amendments
F72Sch. 1 para. 34(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 55(a)
F73Sch. 1 para. 34(3)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 55(b)
Refusal of a variation of a marketing authorisationN.I.
34.—(1) This paragraph applies in relation to the refusal by the Secretary of State of an application for a variation unless the procedure following the refusal of a variation is one of those set out in Article 13 of Regulation 1234/2008.
(2) The grounds on which the Secretary of State may refuse an application for a variation of a marketing authorisation are those set out in paragraph 24 of this Schedule (refusal of a marketing authorisation).
(3) The Secretary of State must give written reasons for refusing to grant a variation; and if—
(a)those reasons are on the grounds of safety, quality or efficacy; and
(b)the variation is Type II or an extension application (whether or not in each case as part of an application for a worksharing or grouped application),
the applicant may appeal to the Veterinary Products Committee.
Extent Information
E169This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Administrative variationsU.K.
[F7435.—(1) The holder of a marketing authorisation may apply for a minor change in a marketing authorisation to be made without the Secretary of State considering any scientific data (an “administrative variation”).
(2) If the Secretary of State grants an administrative variation, and subsequently establishes that this should have been a variation requiring consideration of scientific data, the Secretary of State may notify the marketing authorisation holder, require the holder to submit an application for a variation enabling data to be assessed and revoke the administrative variation.]
Textual Amendments
F74Sch. 1 para. 35 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 56
Changes after a marketing authorisation has been issuedU.K.
36. After a marketing authorisation has been issued, the holder must take account of scientific and technical progress in manufacturing and control methods, and apply to the Secretary of State for any variation in the marketing authorisation that may be required to enable that veterinary medicinal product to be manufactured and checked by means of generally accepted scientific methods.
Compulsory variationU.K.
37.—(1) If the Secretary of State decides, for any of the reasons for suspending a marketing authorisation specified in paragraph 38, or because the classification of a veterinary medicinal product should be changed, that a variation to a marketing authorisation is necessary, the Secretary of State must by a notification in writing to the holder of the marketing authorisation require that person to apply for a variation of the marketing authorisation, giving reasons for requiring the application to be made.
(2) The notification may specify a time limit within which the marketing authorisation holder must apply for the variation.
(3) If the variation is on the grounds of safety, quality or efficacy, the applicant may, within 28 days of the notification, appeal to the Veterinary Products Committee.
(4) If the marketing authorisation holder fails to apply for the variation within that time limit the Secretary of State may suspend or revoke the marketing authorisation.
PART 5U.K.Suspension, etc. of a marketing authorisation
Suspension [F75, revocation, etc] of a marketing authorisation: groundsE+W+S
38.—[F76(1) If the Secretary of State is satisfied at any time that the benefit-risk balance of a veterinary medicinal product is not positive or is insufficient to ensure food safety, the Secretary of State may—
(a)suspend the marketing authorisation;
(b)require the holder of the marketing authorisation to submit an application for its variation;
(c)revoke the marketing authorisation.]
(2) The Secretary of State may also suspend a marketing authorisation on being satisfied that a marketing authorisation holder has failed to make an application for a variation to take account of scientific and technical progress in manufacturing and control methods to enable the veterinary medicinal product to be manufactured and checked by means of generally accepted scientific methods.
[F77(3) The Secretary of State may take the steps set out in sub-paragraph (1)(a), (b) and (c) on being satisfied at any time that—
(a)information given in the application documents is incorrect;
(b)any control tests required have not been carried out;
(c)changes have been made to the manufacturing process without the authority of the Secretary of State;
(d)any information required to be supplied to the Secretary of State has not been so supplied;
(e)the holder of the marketing authorisation has failed to comply with the requirements of these Regulations;
(f)the pharmacovigilance system in relation to a veterinary medicinal product is inadequate;
(g)in the case of a generic authorisation, the reference product is updated to show a reduction in antimicrobial resistance;
(h)the qualified person (pharmacovigilance) has failed to comply with the requirements of these Regulations]
Extent Information
E15This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F75Words in Sch. 1 para. 38 heading inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 57(c)
F76Sch. 1 para. 38(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 57(a)
F77Sch. 1 para. 38(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 57(b)
Suspension of a marketing authorisation: groundsN.I.
38.—(1) The Secretary of State may suspend a marketing authorisation at any time on being satisfied that —
(a)this is necessary for the protection of animal or public health or the environment;
(b)the terms of the marketing authorisation have not been complied with; or
(c)the veterinary medicinal product has insufficient therapeutic effect.
(2) The Secretary of State may also suspend a marketing authorisation on being satisfied that a marketing authorisation holder has failed to make an application for a variation to take account of scientific and technical progress in manufacturing and control methods to enable the veterinary medicinal product to be manufactured and checked by means of generally accepted scientific methods.
(3) The Secretary of State must suspend a marketing authorisation on being satisfied that—
(a)the risk-benefit balance is unfavourable;
(b)the withdrawal period does not ensure that residues in foodstuffs obtained from the treated animal comply with Regulation (EC) No 470/2009 of the European Parliament and of the Council;
(c)information given in the application documents is incorrect;
(d)any control tests required have not been carried out;
(e)changes have been made to the manufacturing process without the authority of the Secretary of State; or
(f)any information required to be supplied to the Secretary of State has not been so supplied.
Extent Information
E170This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Suspension of a marketing authorisation: procedureE+W+S
39.—(1) If a marketing authorisation is suspended the Secretary of State must notify the holder immediately, and, unless the Secretary of State directs otherwise, the suspension has immediate effect, and continues in effect unless the marketing authorisation is reinstated.
(2) If the suspension is on the grounds of safety, quality or efficacy, the holder may, within 28 days of the notification, appeal to the Veterinary Products Committee.
F78(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F79(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E16This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F78Sch. 1 para. 39(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(23) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F79Sch. 1 para. 39(4) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 58
Suspension of a marketing authorisation: procedureN.I.
39.—(1) If a marketing authorisation is suspended the Secretary of State must notify the holder immediately, and, unless the Secretary of State directs otherwise, the suspension has immediate effect, and continues in effect unless the marketing authorisation is reinstated.
(2) If the suspension is on the grounds of safety, quality or efficacy, the holder may, within 28 days of the notification, appeal to the Veterinary Products Committee.
(3) If the veterinary medicinal product is authorised in more than one member State, the Secretary of State—
(a)must immediately refer the matter to the Agency, and must comply with a decision of the Commission within 30 days of the decision; and
(b)may suspend the marketing and the use of the veterinary medicinal product in [F584Northern Ireland] pending a decision of the Agency, but must inform the Commission and the F585... member States no later than the following working day of the reasons for the action.
(4) When a marketing authorisation is suspended, the Secretary of State may in addition prohibit the supply of the veterinary medicinal product, and if necessary require the marketing authorisation holder to recall the product.
Extent Information
E171This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F584Words in Sch. 1 para. 39(3)(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(g)(i)
F585Word in Sch. 1 para. 39(3)(b) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(g)(i)
RevocationU.K.
40. The Secretary of State may revoke any marketing authorisation that has been suspended for more than 28 days unless there is a current appeal to the Veterinary Products Committee, and may publicise a revocation in such manner as the Secretary of State sees fit.
Prohibiting the supply of veterinary medicinal productsE+W+S
41.—[F80(1) The Secretary of State may prohibit the supply of a veterinary medicinal product or require the recall of the product at any time on being satisfied that—
(a)the benefit-risk balance of the veterinary medicinal product is not positive;
(b)the qualitative or quantitative composition of the veterinary medicinal product is not as stated in the summary of product characteristics;
(c)the recommended withdrawal period is insufficient to ensure food safety;
(d)the required control tests have not been carried out; or
(e)the incorrect labelling of the product might lead to a serious risk to human or animal health]
(2) The prohibition on supply and the requirement for recall may be confined to specific production batches.
F81(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E17This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F80Sch. 1 para. 41(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 59
F81Sch. 1 para. 41(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(24) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Prohibiting the supply of veterinary medicinal productsN.I.
41.—(1) In addition to the powers to suspend a marketing authorisation, the Secretary of State, on being satisfied that a product has not been manufactured in accordance with the marketing authorisation, may prohibit the supply of a veterinary medicinal product, and if necessary require the marketing authorisation holder to recall it.
(2) The prohibition on supply and the requirement for recall may be confined to specific production batches.
(3) In the case of an immunological veterinary medicinal product manufactured outside the United Kingdom, if a batch has had all the tests that were originally carried out by the manufacturer repeated by the competent authority of [F586a] member State, the Secretary of State may not prohibit the release of that batch if all the results have been submitted to the Secretary of State and the results demonstrate that the product is within the terms of the authorisation.
Extent Information
E172This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F586Word in Sch. 1 para. 41(3) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(g)(ii)
[F82Temporary restrictionsE+W+S
41A. Where urgent action is necessary for protecting human or animal health or the environment, the Secretary of State may, on a temporary basis—
(a)restrict the supply of a veterinary medicinal product;
(b)restrict the use of a veterinary medicinal product;
(c)suspend the authorisation of a veterinary medicinal product;
(d)require the holder of a marketing authorisation for a veterinary medicinal product to submit an application for variation of the authorisation.]
Textual Amendments
F82Sch. 1 paras. 41A, 41B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 60
[F82Restrictions in relation to immunological veterinary medicinesU.K.
41B. The Secretary of State may prohibit the manufacture, importation, distribution, supply or use of immunological veterinary medicines in any part of Great Britain where—
(a)the administration of the product to an animal interferes with the implementation of a programme for the diagnosis, control or eradication of animal disease;
(b)the administration of the product to an animal causes difficulty in relation to the certifying of absence of disease in live animals or contamination of foodstuffs or other products from treated animals; or
(c)the strains of disease agents in relation to which the product is intended to confer immunity are largely absent from the territory concerned.]
Textual Amendments
F82Sch. 1 paras. 41A, 41B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 60
[F83PART 6U.K.Mutual recognition and multiple applications
Textual Amendments
F83Sch. 1 Pt. 6 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(25) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Application for a marketing authorisation where one already exists in [F84a] member StateU.K.
42.—(1) If a veterinary medicinal product has already received a marketing authorisation in [F85a] member State at the time of application, and the holder of the marketing authorisation applies for a marketing authorisation in [F86Northern Ireland], the following procedure (“the mutual recognition procedure”) applies.
(2) The applicant must submit to the Secretary of State a dossier identical to the one submitted to the competent authority of the member State in which the veterinary medicinal product has been authorised (“the reference member State”).
(3) If there is a marketing authorisation current in more than one member State the applicant must identify which member State is acting as the reference member State.
(4) An applicant applying in more than one member State must supply the Secretary of State with a list of all the States in which the applicant is applying.
(5) The Secretary of State must obtain an assessment report from the reference member State and, where appropriate, an explanation of any extension of the period of data protection.
(6) Within 90 days after receipt of the assessment report, the Secretary of State must, subject to the following provisions, either—
(a)approve the assessment report, the summary of product characteristics, the labelling and the package leaflet, and inform the reference member State accordingly; or
(b)notify the reference member State that they have not been approved, and provide the reference member State with a detailed statement of the reasons.
(7) The Secretary of State may only refuse an application on the grounds of serious risk to human or animal health or the environment.
(8) If the assessment report, the summary of product characteristics, the labelling and the package leaflet are approved, the Secretary of State must ensure that a decision whether or not to grant a marketing authorisation can be made within 30 days of the approval.
(9) If the Secretary of State is notified by the reference member State that—
(a)not all member States concerned have within 90 days approved the assessment report, summary of product characteristics, labelling or package leaflet; and
(b)the reference member State has sent a detailed statement of the reasons to the other member States involved in the application, the applicant and the coordination group for action in accordance with Article 33(3) of Directive 2001/82/EC,
the Secretary of State must within 30 days comply with the decision of the coordination group or, if the coordination group refers the matter to the Agency, the decision of the Commission.
(10) The Secretary of State may grant the marketing authorisation even though not all member States have agreed to grant it, but must revoke or vary the authorisation if this is necessary to comply with the decision of the Commission when it is received.
Textual Amendments
F84Word in Sch. 1 para. 42 heading substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(i)(aa)
F85Word in Sch. 1 para. 42(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(i)(aa)
F86Words in Sch. 1 para. 42(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(i)(bb)
Application in [F87a] member StateU.K.
43.—(1) When the Secretary of State has granted a marketing authorisation for a veterinary medicinal product and is notified by the marketing authorisation holder that the marketing authorisation holder has applied to have that veterinary medicinal product authorised in [F88a] member State, the Secretary of State must prepare an assessment report for the product within 90 days of the notification and send it to the member State or States concerned.
(2) If the other member State (or, if there is more than one, all of them) agrees with the assessment report, the summary of product characteristics, the labelling and the package leaflet the Secretary of State need take no further action.
(3) If not all the other member States concerned so agree within a further 90 days the Secretary of State must send a detailed statement setting out why they have disagreed to the other member States, the applicant and the coordination group for action in accordance with Article 33(3) of Directive 2001/82/EC.
(4) The Secretary of State must within 30 days comply with the decision of the coordination group or, if the coordination group refers the matter to the Agency, the decision of the Commission.
Textual Amendments
F87Word in Sch. 1 para. 43 heading substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(ii)(aa)
F88Word in Sch. 1 para. 43(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(h)(ii)(bb)
Application for a marketing authorisation in multiple member States where a marketing authorisation does not exist in any member StateU.K.
44.—(1) If an applicant wishes to apply for a marketing authorisation in more than one member State, and a marketing authorisation does not exist in any member State for the product (“the decentralised procedure”), the applicant must—
(a)apply simultaneously in all the relevant member States;
(b)submit a dossier to the Secretary of State that is identical to the dossier being submitted to all the other member States;
(c)include a list of all member States in which applications have been made; and
(d)nominate one of them to act as the reference member State to prepare a draft assessment report and drafts of the summary of product characteristics, labelling and package leaflet for consideration by the other member States (“the concerned member States”).
(2) If the United Kingdom is the reference member State, the Secretary of State must prepare a draft assessment report and drafts of the summary of product characteristics, labelling and package leaflet within 120 days of the receipt of a valid application and must send them to the other concerned member States and to the applicant.
(3) If the United Kingdom is not the reference member State, within 90 days after receipt of the assessment report and drafts of the summary of product characteristics, labelling and package leaflet from the reference member State, the Secretary of State must, subject to the following provisions, either—
(a)approve the assessment report, the summary of product characteristics, the labelling and the package leaflet, and inform the reference member State accordingly; or
(b)notify the reference member State that the Secretary of State will not approve it, and provide the reference member State with a detailed statement of the reasons.
(4) The Secretary of State may only refuse an application on the grounds of serious risk to human or animal health or the environment.
(5) If all the member States involved agree the assessment report, the summary of product characteristics, the labelling and the package leaflet within 90 days, the Secretary of State must ensure that a decision whether or not to grant a marketing authorisation can be made within 30 days.
(6) If, within 90 days, not all the member States have agreed the assessment report, summary of product characteristics, labelling and package leaflet on grounds of a potential serious risk to human or animal health or to the environment, the Secretary of State (if the United Kingdom is the reference member State) must send a detailed statement of the reasons to the other member States involved in the application, the applicant, and the coordination group to act in accordance with Article 33(3) of Directive 2001/82/EC.
(7) If reference has been made to the coordination group by any member State, the Secretary of State must within 30 days comply with the decision of the coordination group or, if the coordination group refers the matter to the Agency, the decision of the Commission.
(8) If the Secretary of State wishes to do so, the Secretary of State may grant the marketing authorisation even though not all member States have agreed to grant it, but must revoke or vary the authorisation if this is necessary to comply with the decision of the Commission when it is received.]
PART 7U.K.Labelling and package leaflets
Approval by the Secretary of StateU.K.
45. The Secretary of State, when issuing a marketing authorisation, must approve all containers, packaging, labels and package leaflets.
Reference to being authorisedE+W+S
46. A label and package leaflet of an authorised veterinary medicinal product may contain in legible characters the words “UK authorised veterinary medicinal product” or, if the marketing authorisation provides, other wording specified in the authorisation indicating that the product is authorised in the United Kingdom.
Extent Information
E18This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Reference to being authorisedN.I.
46. A label and package leaflet of an authorised veterinary medicinal product may contain in legible characters the words “[F587UK(NI)] authorised veterinary medicinal product” or, if the marketing authorisation provides, other wording specified in the authorisation indicating that the product is authorised in [F588Northern Ireland].
Extent Information
E173This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F587Word in Sch. 1 para. 46 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(i)(i)
F588Words in Sch. 1 para. 46 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(i)(ii)
LanguageU.K.
47.—(1) All labels and package leaflets must be in English, but may contain other languages provided that the information given is identical in all the languages.
(2) This requirement does not apply in the case of a product imported by a veterinary surgeon and administered by or under the responsibility of that same veterinary surgeon.
[F89Labelling of immediate packaging of veterinary medicinal productsE+W+S
48.—(1) Subject to paragraph 50, the following information must be provided on the immediate packaging of a veterinary medicinal product—
(a)the name of the product, followed by its strength and pharmaceutical form;
(b)a statement of the active substances expressed qualitatively and quantitatively per unit or according to the form of administration for a particular volume or weight, using their common names;
(c)the batch number, preceded by the word “Lot”;
(d)the name or company name or logo of the marketing authorisation holder;
(e)the target species;
(f)the expiry date, in the format ‘mm/yyyy’, preceded by the abbreviation “Exp.”;
(g)special storage precautions, if any;
(h)the route of administration;
(i)if applicable, the withdrawal period, even if such period is zero.
(2) Where there is no outer packaging for the product, the information set out in paragraph 49 must be included on the immediate packaging of the veterinary medicinal product.
(3) The information referred to in paragraph (1) must appear in easily legible and clearly comprehensible characters, or in abbreviations or pictograms.]
Extent Information
E19This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F89Sch. 1 para. 48 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 61 (with reg. 200)
Labelling with all the information on the immediate packagingN.I.
48.—(1) If it is reasonably practicable to do so, the following must be provided on the immediate packaging, in legible characters—
(a)the name, strength and pharmaceutical form of the veterinary medicinal product;
(b)the name and strength of each active substance, and of any excipient if this is required under paragraph 2 of the summary of product characteristics;
(c)the route of administration (if not immediately apparent);
(d)the batch number;
(e)the expiry date;
(f)the words “For animal treatment only” and, if appropriate, “To be supplied only on veterinary prescription”;
(g)the contents by weight, volume or number of dose units;
(h)the marketing authorisation number;
(i)the name and address of the marketing authorisation holder or, if there is a distributor authorised in the marketing authorisation, that distributor;
(j)a suitably labelled space to record discard date (if relevant);
(k)the target species;
(l)the distribution category;
(m)the words “Keep out of reach of children”;
(n)storage instructions;
(o)the in-use shelf-life (if appropriate);
(p)for food-producing species, the withdrawal period for each species or animal product concerned;
(q)any warning specified in the marketing authorisation;
(r)disposal advice;
(s)full indications;
(t)dosage instructions;
(u)contra-indications;
(v)further information required in the marketing authorisation;
(w)if the product is one that requires a dose to be specified for the animal being treated, a space for this.
(2) If all this is on the immediate packaging, there is no need for any outer packaging or a package leaflet.
Extent Information
E174This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F90Labelling of the outer packaging of veterinary medicinal productsE+W+S
49.—(1) The following information must be provided on any outer packaging of a veterinary medicinal product—
(a)the information referred to in paragraph 48(1);
(b)the contents by weight, volume or number of the immediate packaging units of the veterinary medicinal product;
(c)a warning that the veterinary medicinal product must be kept out of the sight and reach of children;
(d)a warning that the veterinary medicinal product is “for animal treatment only”;
(e)a recommendation to read the package leaflet, if there is one;
(f)in the case of a veterinary medicinal product not subject to a veterinary prescription, the indication for use;
(g)the marketing authorisation number.
(2) The information referred to in sub-paragraph (1) must appear in easily legible and clearly comprehensible characters, or in abbreviations or pictograms.]
Extent Information
E20This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F90Sch. 1 para. 49 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 62 (with reg. 200)
Products with immediate and outer packagingN.I.
49.—(1) If it is not reasonably practicable to have all the required information on the immediate packaging then this paragraph applies.
(2) The immediate packaging must have at least the following information—
(a)the name of the veterinary medicinal product, including its strength and pharmaceutical form;
(b)the name and proportion of each active substance, and of any excipient if knowledge of the excipient is needed for safety reasons;
(c)the route of administration (if not immediately apparent);
(d)the batch number;
(e)the expiry date;
(f)the words “For animal treatment only” and if appropriate, “To be supplied only on veterinary prescription”;
(g)the words “Keep the container in the outer carton”.
(3) In addition, the immediate packaging must have as much of the required information as is reasonably practicable.
(4) The outer packaging must contain all the required information if it is reasonably practicable to do this, and if it is not reasonably practicable to do this a package leaflet must be supplied with the product in accordance with the following paragraph.
Extent Information
E175This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F91Labelling of small immediate packaging units of veterinary medicinal productsE+W+S
50.—(1) Where the immediate packaging units of a veterinary medicinal product are too small to include in a legible form all of the information set out in paragraph 48, the immediate packaging must instead provide the following information—
(a)the name of the veterinary medicinal product;
(b)the quantitative particulars of the active substances contained in the product;
(c)the batch number, preceded by the word “Lot”;
(d)the expiry date, in the form ‘mm/yyyy’, preceded by the abbreviation “Exp”.
(2) The immediate packaging units mentioned in sub-paragraph (1) must be packed within outer packaging which provides the information required by paragraph 49.]
Extent Information
E21This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F91Sch. 1 para. 50 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 63 (with reg. 200)
Package leafletsN.I.
50.—(1) If it is not reasonably practicable to have all the required information on the immediate packaging or all of this information on the outer packaging, there must be a package leaflet supplied with the product, containing all the required information except for the batch number and the expiry date, and including the name of both the marketing authorisation holder and, if different, the name of the distributor named in the marketing authorisation.
(2) If there is a package leaflet, the immediate packaging and the outer packaging must both refer the user to it.
(3) A package leaflet must relate solely to the veterinary medicinal product with which it is included.
(4) It must be written in plain English.
(5) Only a package leaflet approved in the marketing authorisation may be included with the veterinary medicinal product.
Extent Information
E176This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F92Package leaflet of veterinary medicinal productsE+W+S
51.—(1) Subject to sub-paragraphs (5) and (7), a package leaflet must be supplied with each veterinary medicinal product.
(2) The package leaflet must provide the following information—
(a)the name and address of the marketing authorisation holder and of the manufacturer and, where applicable, the distributor;
(b)the name of the veterinary medicinal product, followed by its strength and pharmaceutical form;
(c)the qualitative and quantitative composition of any active substance;
(d)the target species, the dosage for each species, the method and route of administration and if necessary, advice on the correct administration;
(e)the indications for use;
(f)the contra-indications and adverse events;
(g)if applicable, the withdrawal period for each species, even if such a period is zero;
(h)special storage precautions, if any;
(i)information essential for safety or health protection, including any special precautions relating to use and any other warnings;
(j)the words “use take-back schemes for the disposal of any unused veterinary medicinal product or associated waste materials in accordance with local requirements and with any applicable national collection schemes”;
(k)the marketing authorisation number;
(l)contact details for the marketing authorisation holder or its representative, as appropriate, for the reporting of suspected adverse events;
(m)classification of the veterinary medicinal product as referred to in the summary of product characteristics.
(3) Providing that it complies with the marketing authorisation, the package leaflet may include additional information concerning distribution, possession or any necessary precaution required, provided that this information is not promotional in character.
(4) The package leaflet must be in legible form and designed to be clear and understandable, in terms that are comprehensible to the general public.
(5) Only a package leaflet approved in the marketing authorisation may be published or included with the veterinary medicinal product.
(6) The Secretary of State may require the information set out in sub-paragraph (2) to be made available in written form or electronically, or both.
(7) Where the Secretary of State requires the leaflet to be made available electronically—
(a)an electronic package information leaflet which includes the information required by this paragraph must be provided in place of a leaflet in written form;
(b)the packaging of the veterinary medicinal product must include—
(i)a statement that the information which must be included on a package leaflet is provided electronically;
(ii)any necessary electronic link in order to access the relevant part of the website where the electronic package information leaflet is to be found;
(iii)a statement that a copy of the information in written form may be obtained on request; and
(iv)instructions on how to obtain such a copy.
(8) Any information required by this paragraph to be provided on a package leaflet in written form may be otherwise provided on the packaging of the veterinary medicinal product.]
Extent Information
E22This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F92Sch. 1 para. 51 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 64 (with reg. 200)
AmpoulesN.I.
51.—(1) In the case of ampoules or other unit dose forms, where the container cannot bear legibly the required information, only the following information must be shown on the immediate packaging—
(a)the name of the veterinary medicinal product;
(b)the name and strength of the active ingredient;
(c)the route of administration (if not immediately apparent);
(d)the batch number;
(e)the expiry date;
(f)the words “For animal treatment only” and if appropriate, “To be supplied only on veterinary prescription”.
(2) The outer packaging must contain all the required information if it is reasonably practicable to do this, and if it is not reasonably practicable to do this a package leaflet must be supplied with the product, except that the ampoule need not refer to the package leaflet.
Extent Information
E177This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Small containers other than ampoulesU.K.
[F9352. As regards small immediate packaging containing a single dose, other than ampoules, on which it is impossible to give the required information, all the required information must appear on the outer packaging or outer packaging and package leaflet, but the immediate packaging must be labelled with the batch number and the expiry date and, if there is room, the other information in the preceding paragraph.]
Textual Amendments
F93Sch. 1 para. 52 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 65 (with reg. 200)
Homeopathic remediesE+W+S
53.—(1) A homeopathic remedy registered under these Regulations must be labelled in accordance with this paragraph.
(2) There must be no specific therapeutic indication on the labelling or in any information relating to it.
(3) The labelling (or labelling and package leaflet) must contain the following and no other information—
(a)the words “homeopathic remedy without approved therapeutic indications for veterinary use”;
(b)the scientific name of the stock or stocks followed by the degree of dilution, using the symbols of the pharmacopoeia used (if the homeopathic remedy is composed of more than one stock, the labelling may mention an invented name in addition to the scientific names of the stocks);
[F94(c)the name or company name and the permanent address or registered place of business of the registration holder and of the manufacturer]
(d)the method and, if necessary, route of administration;
(e)the expiry date;
(f)the pharmaceutical form;
(g)the contents of the pack;
(h)any special storage precautions;
(i)the target species;
(j)any necessary special warnings;
(k)the batch number; F95...
(l)the registration number; [F96and]
[F97(m)the withdrawal period, where applicable.]
Extent Information
E23This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F94Sch. 1 para. 53(3)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 66(a) (with reg. 200)
F95Word in Sch. 1 para. 53(3) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 66(b) (with reg. 200)
F96Word in Sch. 1 para. 53(3) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 66(c) (with reg. 200)
F97Sch. 1 para. 53(3)(m) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 66(d) (with reg. 200)
Homeopathic remediesN.I.
53.—(1) A homeopathic remedy registered under these Regulations must be labelled in accordance with this paragraph.
(2) There must be no specific therapeutic indication on the labelling or in any information relating to it.
(3) The labelling (or labelling and package leaflet) must contain the following and no other information—
(a)the words “homeopathic remedy without approved therapeutic indications for veterinary use”;
(b)the scientific name of the stock or stocks followed by the degree of dilution, using the symbols of the pharmacopoeia used (if the homeopathic remedy is composed of more than one stock, the labelling may mention an invented name in addition to the scientific names of the stocks);
(c)the name and address of the registration holder and (on the package leaflet) of the manufacturer;
(d)the method and, if necessary, route of administration;
(e)the expiry date;
(f)the pharmaceutical form;
(g)the contents of the pack;
(h)any special storage precautions;
(i)the target species;
(j)any necessary special warnings;
(k)the batch number; and
(l)the registration number.
Extent Information
E178This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
VariationsU.K.
54. The Secretary of State may permit variations in the above in any individual marketing authorisation if this is necessary for public or animal health purposes or the protection of the environment.
PART 8U.K.Pharmacovigilance
Qualified persons responsible for pharmacovigilanceE+W+S
F9855. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E24This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F98Sch. 1 para. 55 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 67
Qualified persons responsible for pharmacovigilanceN.I.
55. A marketing authorisation holder must have permanently and continuously the services of an appropriately qualified person responsible for pharmacovigilance (“a qualified person (pharmacovigilance)”) who resides in a member State [F589or the United Kingdom].
Extent Information
E179This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F589Words in Sch. 1 para. 55 inserted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(i)
[F99Duties of marketing authorisation holder in relation to pharmacovigilanceE+W+S
56.—(1) The marketing authorisation holder is responsible for pharmacovigilance in relation to a veterinary medicinal product for which it holds a marketing authorisation and must continuously evaluate, by appropriate means, the benefit-risk balance of this veterinary medicinal product and, if necessary, take appropriate measures to address any risk presented by the product.
(2) A marketing authorisation holder must carry out the signal management process mentioned in paragraph 56C in relation to any veterinary medicinal product for which it holds an authorisation.
(3) A marketing authorisation holder must comply with best practice in good veterinary pharmacovigilance practice.
(4) A marketing authorisation holder must establish and maintain a system for collecting, collating and evaluating information in relation to suspected adverse events in respect of any veterinary medicinal product for which it holds an authorisation.
(5) Subject to sub-paragraph (6), a marketing authorisation holder must establish and maintain one or more pharmacovigilance system master files describing in detail the pharmacovigilance system with respect to its authorised veterinary medicinal products.
(6) For each veterinary medicinal product, the marketing authorisation holder must not establish and maintain more than one pharmacovigilance system master file.
(7) A marketing authorisation holder must establish and maintain an adequate and effective local system for the purpose of receiving reports of suspected adverse events.
(8) The system mentioned in sub-paragraph (7) must be staffed by personnel trained for this purpose who are able to communicate in English.
(9) A marketing authorisation holder must designate not more than one qualified person responsible for pharmacovigilance (a “qualified person (pharmacovigilance)”) in relation to each pharmacovigilance system master file whose services are available permanently and continuously.
(10) Where the pharmacovigilance functions or the functions of the qualified person for pharmacovigilance are performed by a third party, any such arrangement must be specified in detail in the pharmacovigilance system master file and within appropriate pharmacovigilance agreements.
(11) A marketing authorisation holder may introduce urgent safety restrictions where evidence comes to the attention of the holder of a risk posed to human or animal health or to the environment from the use of the product.
(12) Where a marketing authorisation holder takes any action under sub-paragraph (11) the holder must inform the Secretary of State no later than the following working day of the reasons for the action.
(13) A marketing authorisation holder must establish and maintain an adequate and effective quality management system for the performance of its pharmacovigilance activities.
(14) The Secretary of State may at any time by notice require a marketing authorisation holder to provide a copy of the pharmacovigilance system master file.
(15) A marketing authorisation holder who is given notice under sub-paragraph (14) must comply with the requirement within seven days of receipt of the notice.]
Extent Information
E25This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F99Sch. 1 paras. 56-56C substituted for Sch. 1 para. 56 (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 68
Duties relating to the qualified personN.I.
56. The marketing authorisation holder must ensure that the qualified person (pharmacovigilance)—
(a)establishes and maintains a system that ensures that information about all suspected adverse reactions reported to the marketing authorisation holder is collected and collated in order to be accessible at least at one point in a member State;
(b)answers any request from the Secretary of State for the provision of additional information necessary for the evaluation of the benefits and risks afforded by a veterinary medicinal product fully and within any time limit imposed by the Secretary of State when the information was requested, including the volume of sales of the veterinary medicinal product concerned and, if available, details of prescriptions;
(c)provides to the Secretary of State any other information relevant to the evaluation of the benefits and risks afforded by a veterinary medicinal product, including appropriate information on post-marketing surveillance studies; and in this paragraph “post-marketing surveillance studies” means a pharmacoepidemiological study or a clinical trial carried out in accordance with the terms of the marketing authorisation, conducted with the aim of identifying and investigating a safety hazard relating to an authorised veterinary medicinal product.
Extent Information
E180This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F99Duties of marketing authorisation holder in relation to signal management processE+W+S
56A.—(1) A marketing authorisation holder must carry out the signal management process mentioned in paragraph 56C on reports received (whether those reports derive from the United Kingdom or any other country) in relation to any veterinary medicinal product for which it holds an authorisation.
(2) The marketing authorisation holder must record on an annual basis the results of the signal management process mentioned in paragraph 56C in relation to the product.
(3) Where, as a result of the carrying out of the signal management process, a new risk or a change in the benefit-risk balance of the product is identified, the marketing authorisation holder must notify the Secretary of State promptly and in any event within 30 days of such identification.
(4) Where the signal management process identifies the necessity for a variation in an authorisation the marketing authorisation holder must submit an application for such a variation to the Secretary of State promptly.
Textual Amendments
F99Sch. 1 paras. 56-56C substituted for Sch. 1 para. 56 (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 68
Duties of qualified person (pharmacovigilance)E+W+S
56B. A qualified person (pharmacovigilance) must—
(a)establish and maintain a system which ensures that all suspected adverse events which are brought to the attention of the marketing authorisation holder in relation to a veterinary medicinal product are collected and recorded;
(b)monitor the performance of each product which is the subject of a marketing authorisation, apply the signal management process mentioned in paragraph 56C and ensure that any relevant requirements in accordance with the process are carried out;
(c)maintain the pharmacovigilance system master file for each such product;
(d)provide to the Secretary of State any information relevant to detecting a change to the benefit-risk balance of a veterinary medicinal product including the results of any study or clinical trial carried out in relation to the product;
(e)communicate the fact that a regulatory measure has been taken in a country other than the United Kingdom as a consequence of pharmacovigilance data and the nature of such measure to the Secretary of State within 30 days of the receipt of such information, if no equivalent to that regulatory measure has already been taken in the United Kingdom;
(f)answer fully and promptly any request from the Secretary of State for the provision of additional information necessary for the evaluation of the benefit-risk balance of that product;
(g)monitor the pharmacovigilance system and ensure that, if required, an appropriate preventative or corrective action plan is prepared and implemented on behalf of the marketing authorisation holder through the use of audits and routine monitoring;
(h)following any action taken in accordance with paragraph (g), ensure that any relevant amendments are made to the pharmacovigilance system master file;
(i)liaise with the Secretary of State in relation to any pharmacovigilance inspection carried out under paragraph 60A;
(j)ensure that any person employed by the marketing authorisation holder who is engaged in pharmacovigilance receives ongoing training which is relevant to that person’s duties.
Textual Amendments
F99Sch. 1 paras. 56-56C substituted for Sch. 1 para. 56 (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 68
Signal management processE+W+S
56C.—(1) For the purposes of these Regulations, “signal management process” means a process for performing active surveillance of pharmacovigilance data for veterinary medicinal products in order to assess the pharmacovigilance data and determine whether there is any change to the benefit-risk balance of those veterinary medicinal products, with a view to detecting risks to animal or public health or protection of the environment.
(2) A signal management process must consist of tasks of signal detection, validation, confirmation, analysis and prioritisation, assessment and recommendation for action.
(3) A signal management process must be capable of identifying, at a minimum, in relation to a product—
(a)a sudden and unexpected increase in the number of adverse events;
(b)an unexpected increase in the frequency of a known clinical sign;
(c)a new clinical sign;
(d)reports in scientific literature of any of the matters mentioned in paragraphs (a) to (c).]
Textual Amendments
F99Sch. 1 paras. 56-56C substituted for Sch. 1 para. 56 (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 68
[F100Adverse events following administration of a veterinary medicinal product]E+W+S
57.—(1) A marketing authorisation holder must act in accordance with this paragraph on learning of any suspected—
(a)[F101adverse event in respect of an animal];
(b)human [F102adverse event]; F103...
(c)unintended transmission of an infectious agent through a veterinary medicinal product,
[F104(d)occurrence of an adverse environmental event, or
(e)lack of efficacy,]
following the administration of the product F105....
[F106(1A) A marketing authorisation holder must also act in accordance with this paragraph where—
(a)after the end of the withdrawal period a product of animal origin is found to include a pharmacologically active substance or marker residue exceeding the maximum residue limit established in accordance with Regulation (EC) No 470/2009 of the European Parliament and of the Council; or
(b)there is evidence in published scientific literature of an adverse event in connection with the product.]
(2) The holder must make a record of what happened.
(3) The holder must without delay and in any event within [F10730] days report it (electronically if this is practicable) to the Secretary of State.
(4) In addition, the holder must supply to the Secretary of State all relevant veterinary pharmacovigilance information that the holder possesses relating to the [F108event], giving a full description of the incident and a list of all the symptoms using internationally recognised veterinary and medical terminology, either with the report or, if the information becomes available after the report has been sent, as soon after it becomes available as is reasonably practicable.
[F109(4A) The Secretary of State may require the marketing authorisation holder—
(a)to collect specific pharmacovigilance data (in addition to the data mentioned in sub-paragraph (4)) and submit those data to the Secretary of State; and
(b)to carry out specific post-marketing surveillance studies.
(4B) Where the Secretary of State exercises the power mentioned in sub-paragraph (4A), the Secretary of State must—
(a)state the reason for the requirement; and
(b)state the time by which, or the period during which, the requirement must be complied with.]
F110(5) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E26This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F100Sch. 1 para. 57 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(g)
F101Words in Sch. 1 para. 57(1)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(a)(i)
F102Words in Sch. 1 para. 57(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(a)(ii)(aa)
F103Word in Sch. 1 para. 57(1)(b) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(a)(ii)(bb)
F104Sch. 1 para. 57(1)(d)(e) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(a)(iii)
F105Words in Sch. 1 para. 57(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(a)(iv)
F106Sch. 1 para. 57(1A) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(b)
F107Word in Sch. 1 para. 57(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(c)
F108Word in Sch. 1 para. 57(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(d)
F109Sch. 1 para. 57(4A)(4B) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(e)
F110Sch. 1 para. 57(5) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 69(f)
Adverse reactions to a veterinary medicinal product administered in [F590Northern Ireland] N.I.
57.—(1) A marketing authorisation holder must act in accordance with this paragraph on learning of any suspected—
(a)serious adverse reaction;
(b)human adverse reaction; or
(c)unintended transmission of an infectious agent through a veterinary medicinal product,
following the administration of the product in [F591Northern Ireland].
(2) The holder must make a record of what happened.
(3) The holder must without delay and in any event within 15 days report it (electronically if this is practicable) to the Secretary of State.
(4) In addition, the holder must supply to the Secretary of State all relevant veterinary pharmacovigilance information that the holder possesses relating to the reaction, giving a full description of the incident and a list of all the symptoms using internationally recognised veterinary and medical terminology, either with the report or, if the information becomes available after the report has been sent, as soon after it becomes available as is reasonably practicable.
(5) In this and the following paragraph—
“human adverse reaction” means a reaction that is noxious and unintended and that occurs in a human being following exposure to a veterinary medicine;
“serious adverse reaction” means an adverse reaction that results in death, is life-threatening, results in significant disability or incapacity, is a congenital anomaly or birth defect, or that results in permanent or prolonged signs in the animals treated.
Extent Information
E181This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F590Words in Sch. 1 para. 57 heading substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(ii)
F591Words in Sch. 1 para. 57(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(ii)
Adverse reactions to a veterinary medicinal product administered in [F111another ] countryE+W+S
F11258. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E27This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F111Word in Sch. 1 para. 58 heading substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(28)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F112Sch. 1 para. 58 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 70
Adverse reactions to a veterinary medicinal product administered in a third countryN.I.
58.—(1) A marketing authorisation holder for a veterinary medicinal product authorised in [F592Northern Ireland] must act in accordance with this paragraph on learning of any suspected—
(a)serious, unexpected adverse reaction (for these purposes a reaction is unexpected if its nature, severity or outcome is not consistent with the summary of the product characteristics);
(b)human adverse reaction; or
(c)unintended transmission of an infectious agent through a veterinary medicinal product,
following the administration of the product in a third country.
(2) The holder must make a record of what happened.
(3) The holder must without delay and in any event within 15 days report the suspected reaction or transmission (electronically if this is practicable) to the Secretary of State, the competent authorities of all member States in which the product is authorised, and the Agency.
(4) In addition to the report, the holder must supply to the Secretary of State, the competent authorities of all F593... member States where the product is authorised and the Agency, all relevant veterinary pharmacovigilance information in the holder’s possession relating to the reaction as in the preceding paragraph.
Extent Information
E182This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F592Words in Sch. 1 para. 58(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(iii)(aa)
F593Word in Sch. 1 para. 58(4) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(iii)(bb)
[F113Annual benefit-risk] reportsE+W+S
59.—(1) The marketing authorisation holder must submit to the Secretary of State [F114a summary of pharmacovigilance activity] in the form of [F115an annual benefit-risk report] for each marketing authorisation in accordance with this paragraph F116....
F117(2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(3) Following the placing on the market in [F118Great Britain], the marketing authorisation holder must submit a [F119benefit-risk report] to the Secretary of State immediately upon [F120request and, in any event, once in the course of every year during the period of validity of the authorisation].
(4) Following the granting of a marketing authorisation, the marketing authorisation holder may apply to the Secretary of State to change the [F121submission dates for the annual benefit-risk reports].
[F122(5) The report must include a statement regarding the benefit-risk balance of the veterinary medicinal product.]
[F123(6) The annual benefit-risk report must include—
(a)the volume of the product sold in the United Kingdom and in other countries in the period covered by the report, with the volume of the product sold in the United Kingdom in each calendar year identified;
(b)the notification of signals detected during the reporting period following pharmacovigilance activity in the United Kingdom or a country other than the United Kingdom for which further regulatory actions are required (including a summary of the regular review of adverse events carried out during the year); and
(c)where it appears from the observed data that there is cause for concern in relation to the safety of the product, recommendations on the need for further intervention by the Secretary of State.]
F124(7) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F125(8) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E28This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F113Words in Sch. 1 para. 59 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(h)
F114Words in Sch. 1 para. 59(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(a)(i)
F115Words in Sch. 1 para. 59(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(a)(ii)
F116Words in Sch. 1 para. 59(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(a)(iii)
F117Sch. 1 para. 59(2) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(b)
F118Words in Sch. 1 para. 59(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(c)(i)
F119Words in Sch. 1 para. 59(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(c)(ii)
F120Words in Sch. 1 para. 59(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(c)(iii)
F121Words in Sch. 1 para. 59(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(d)
F122Sch. 1 para. 59(5) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(e)
F123Sch. 1 para. 59(6) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(f)
F124Sch. 1 para. 59(7) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(g)
F125Sch. 1 para. 59(8) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 71(g)
Periodic safety update reportsN.I.
59.—(1) The marketing authorisation holder must submit to the Secretary of State records of all adverse reactions (including nil reports) in the form of a periodic safety update report for each marketing authorisation in accordance with this paragraph, including a summary of each incident and a list of all the symptoms using internationally recognised veterinary and medical terminology.
(2) A marketing authorisation holder who has not yet placed a product on the market in [F594Northern Ireland] must submit a periodic safety update report immediately upon request of the Secretary of State and at least every six months after authorisation.
(3) Following the placing on the market in [F595Northern Ireland], the marketing authorisation holder must submit a periodic safety update report to the Secretary of State immediately upon request and—
(a)at least every six months during the first two years following the initial placing on the market;
(b)once a year for the following two years; and
(c)thereafter, at three-yearly intervals.
(4) Following the granting of a marketing authorisation, the marketing authorisation holder may apply to the Secretary of State to change the periods of notification.
(5) The periodic safety update report must include a scientific evaluation of the risk-benefit balance of the veterinary medicinal product.
(6) The periodic safety update report must include—
(a)the volume of the product sold in each year covered by the report, calculated on an annual basis beginning 1st January;
(b)the number of adverse reactions for each year of the report;
(c)the ratio of adverse reactions to volume of product sold for each year of the report, together with an explanation of the basis of the calculation;
(d)differentiation of data based on—
(i)target species (if the product is authorised for use in more than one species);
(ii)reaction type (such as serious, non-serious, human, suspected lack of efficacy, unauthorised use or other);
(iii)the country of origin of the report.
(7) If the product is indicated for more than one species, the information in sub-paragraph (6)(c) must be based so far as is practicable on the estimated use of the product.
(8) Data relating to different formulations (either different dosage forms or different strengths) must be provided in separate reports.
Extent Information
E183This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F594Words in Sch. 1 para. 59(2) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(iv)
F595Words in Sch. 1 para. 59(3) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(iv)
Release of information by the marketing authorisation holderU.K.
60.—(1) A marketing authorisation holder must not communicate information relating to pharmacovigilance concerns to [F126veterinary surgeons or] the general public in relation to its authorised veterinary medicinal product without giving prior or simultaneous notification to the Secretary of State.
(2) The marketing authorisation holder must ensure that such information is presented objectively and is not misleading.
[F127(3) For the purposes of this paragraph “information” includes any information contained in advertising material.]
Textual Amendments
F126Words in Sch. 1 para. 60(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 72(a)
F127Sch. 1 para. 60(3) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 72(b)
[F128Pharmacovigilance inspections by Secretary of StateE+W+S
60A.—(1) The Secretary of State must, from time to time, inspect the pharmacovigilance systems of marketing authorisation holders for the purpose of verifying compliance with the provisions of this Schedule in relation to pharmacovigilance.
(2) The frequency of inspections under sub-paragraph (1) must be based on the risks associated with each marketing authorisation holder’s history and the nature of the products included in their pharmacovigilance system.
(3) Within 90 days after an inspection, the Secretary of State must issue an inspection report to the holder of the marketing authorisation if the inspection established compliance with best practice in good veterinary pharmacovigilance practice.]
Textual Amendments
F128Sch. 1 paras. 60A, 60B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 73
[F128Powers of Secretary of State in relation to signal management processE+W+S
60B. The Secretary of State may decide to perform a targeted signal management process for a given veterinary medicinal product or a group of veterinary medicinal products.]
Textual Amendments
F128Sch. 1 paras. 60A, 60B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 73
Action taken on account of pharmacovigilanceE+W+S
61.—(1) Where, as a result of the evaluation of veterinary pharmacovigilance data, the Secretary of State considers that a marketing authorisation [F129, or a group of marketing authorisations containing the same active substance,] should be—
(a)suspended;
(b)revoked; or
(c)varied so as to—
(i)restrict the indications;
(ii)change the distribution category;
(iii)amend the dose;
(iv)add a contraindication; F130...
(v)add a new precautionary measure, [F131or
(vi)implement a risk management plan,]
the Secretary of State must forthwith inform F132... and the marketing authorisation holder.
(2) If urgent action is necessary for protecting human or animal health, the Secretary of State may suspend the marketing authorisation of a veterinary medicinal productF133....
F134(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F135(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E29This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F129Words in Sch. 1 para. 61(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 74(a)
F130Word in Sch. 1 para. 61(1)(c) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 74(aa)(b)
F131Sch. 1 para. 61(1)(c)(vi) and word inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 74(b)(bb)
F132Words in Sch. 1 para. 61(1) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(29)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F133Words in Sch. 1 para. 61(2) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(29)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F134Sch. 1 para. 61(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(29)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F135Sch. 1 para. 61(4) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(29)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Action taken on account of pharmacovigilanceN.I.
61.—(1) Where, as a result of the evaluation of veterinary pharmacovigilance data, the Secretary of State considers that a marketing authorisation should be—
(a)suspended;
(b)revoked; or
(c)varied so as to—
(i)restrict the indications;
(ii)change the distribution category;
(iii)amend the dose;
(iv)add a contraindication; or
(v)add a new precautionary measure,
the Secretary of State must forthwith inform the Agency, all F596... member States (irrespective of whether the product is authorised in [F597a] member State) and the marketing authorisation holder.
(2) If urgent action is necessary for protecting human or animal health, the Secretary of State may suspend the marketing authorisation of a veterinary medicinal product, but must inform the Agency, the Commission and the F598... member States within one working day.
(3) If, following the opinion of the Agency, the Commission requests the Secretary of State to suspend, withdraw or vary the marketing authorisation, the Secretary of State must comply with that request immediately on a temporary basis.
(4) The Secretary of State must take final measures in accordance with the Decision of the Commission.
Extent Information
E184This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F596Word in Sch. 1 para. 61(1) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(v)(aa)
F597Word in Sch. 1 para. 61(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(v)(aa)
F598Word in Sch. 1 para. 61(2) omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(j)(v)(bb)
PART 9U.K.Homeopathic remedies
Meaning of “homeopathic remedy”E+W+S
62. For the purposes of these Regulations, a homeopathic remedy is a veterinary medicinal product (which may contain a number of principles) prepared from homeopathic stocks in accordance with a homeopathic manufacturing procedure described in the European Pharmacopoeia(3) or, if it is not described there, in a pharmacopoeia published by the British Pharmacopoeial Commission F136....
Extent Information
E30This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F136Words in Sch. 1 para. 62 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(30) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Meaning of “homeopathic remedy”N.I.
62. For the purposes of these Regulations, a homeopathic remedy is a veterinary medicinal product (which may contain a number of principles) prepared from homeopathic stocks in accordance with a homeopathic manufacturing procedure described in the European Pharmacopoeia(3) or, if it is not described there, in a pharmacopoeia published by the British Pharmacopoeial Commission or by the competent authority of any member State.
Extent Information
E185This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Placing a homeopathic remedy on the market in accordance with a registrationE+W+S
63.—(1) By way of derogation from the provisions of these Regulations requiring a marketing authorisation for a veterinary medicinal product, a homeopathic remedy may be placed on the market in accordance with a registration by the Secretary of State instead of in accordance with a marketing authorisation if it complies with this paragraph.
(2) It must not be an immunological [F137or, subject to sub-paragraph (2A), a biological] product.
[F138(2A) Sub-paragraph (2) does not apply in relation to a homeopathic remedy which is derived from plants.]
(3) The route of administration must be [F139either topical or oral and must be] as described in the European PharmacopoeiaF140....
(4) There must be a sufficient degree of dilution to guarantee the safety of the product, and in any event it must not contain more than one part in 10,000 of the mother tincture.
(5) All other provisions relating to marketing authorisations apply in the same way to registrations of a homeopathic remedy.
Extent Information
E31This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F137Words in Sch. 1 para. 63(2) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 75(a)
F138Sch. 1 para. 63(2A) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 75(b)
F139Words in Sch. 1 para. 63(3) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 75(c)
F140Words in Sch. 1 para. 63(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(31) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Placing a homeopathic remedy on the market in accordance with a registrationN.I.
63.—(1) By way of derogation from the provisions of these Regulations requiring a marketing authorisation for a veterinary medicinal product, a homeopathic remedy may be placed on the market in accordance with a registration by the Secretary of State instead of in accordance with a marketing authorisation if it complies with this paragraph.
(2) It must not be an immunological product.
(3) The route of administration must be as described in the European Pharmacopoeia or, if it is not described there, by a pharmacopoeia currently used officially in any member State.
(4) There must be a sufficient degree of dilution to guarantee the safety of the product, and in any event it must not contain more than one part in 10,000 of the mother tincture.
(5) All other provisions relating to marketing authorisations apply in the same way to registrations of a homeopathic remedy.
Extent Information
E186This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for registrationE+W+S
64.—(1) An applicant for registration must submit the following to the Secretary of State—
(a)the scientific name or other name of the homeopathic stock [F141or stocks] given in a pharmacopoeia, together with a statement of the various routes of administration, pharmaceutical forms and degree of dilution;
(b)a dossier describing how the homeopathic stock is [F142, or stocks are,] obtained and controlled, and justifying [F143their] homeopathic [F144use], on the basis of an adequate bibliography;
(c)in the case of a product containing biological substances, a description of the measures taken to ensure the absence of pathogens;
(d)the manufacturing and control file for each pharmaceutical form and a description of the method of dilution and potentisation;
(e)a copy of the manufacturing authorisation for the product;
(f)copies of any registrations F145... obtained for the same homeopathic remedyF146...;
[F147(g)the text which is to appear on the package leaflet, outer packaging and immediate packaging of the homeopathic remedy;]
[F148(h)any relevant data concerning the stability of the homeopathic remedy;]
(i)the proposed withdrawal period necessary to ensure that the provisions of Regulation (EC) No 470/2009 of the European Parliament and of the Council are complied with together with all necessary justification.
(2) These documents must demonstrate the pharmaceutical quality and the batch-to-batch homogeneity of the products concerned.
(3) In the case of a food-producing animal, if the applicant states in the application that the homeopathic remedy contains an active substance, or has been manufactured using an active substance, that substance must be one [F149for which a maximum residue limit has been established under Regulation (EC) No 470/2009 of the European Parliament and of the Council.]
F150(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E32This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F141Words in Sch. 1 para. 64(1)(a) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 76(a)
F142Words in Sch. 1 para. 64(1)(b) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 76(b)(i)
F143Word in Sch. 1 para. 64(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 76(b)(ii)
F144Word in Sch. 1 para. 64(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 76(b)(iii)
F145Words in Sch. 1 para. 64(1)(f) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 76(c)
F146Words in Sch. 1 para. 64(1)(f) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(32)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F147Sch. 1 para. 64(1)(g) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 76(d)
F148Sch. 1 para. 64(1)(h) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 76(e)
F149Words in Sch. 1 para. 64(3) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(32)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F150Sch. 1 para. 64(4) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(32)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Application for registrationN.I.
64.—(1) An applicant for registration must submit the following to the Secretary of State—
(a)the scientific name or other name of the homeopathic stock given in a pharmacopoeia, together with a statement of the various routes of administration, pharmaceutical forms and degree of dilution;
(b)a dossier describing how the homeopathic stock is obtained and controlled, and justifying its homeopathic nature, on the basis of an adequate bibliography;
(c)in the case of a product containing biological substances, a description of the measures taken to ensure the absence of pathogens;
(d)the manufacturing and control file for each pharmaceutical form and a description of the method of dilution and potentisation;
(e)a copy of the manufacturing authorisation for the product;
(f)copies of any registrations or authorisations obtained for the same homeopathic remedy in other member States;
(g)a mock-up of the outer packaging and immediate packaging;
(h)stability data;
(i)the proposed withdrawal period necessary to ensure that the provisions of Regulation (EC) No 470/2009 of the European Parliament and of the Council are complied with together with all necessary justification.
(2) These documents must demonstrate the pharmaceutical quality and the batch-to-batch homogeneity of the products concerned.
(3) In the case of a food-producing animal, if the applicant states in the application that the homeopathic remedy contains an active substance, or has been manufactured using an active substance, that substance must be one that appears in Table 1 in the Annex to Commission Regulation (EU) No 37/2010 and complies with any requirements in that Table relating to that substance.
(4) If a product is registered in [F599a] member State, the Secretary of State may waive some or all of the requirements of this paragraph on being satisfied that it is reasonable to do so.
Extent Information
E187This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F599Word in Sch. 1 para. 64(4) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(13)(k)
Procedure for registrationU.K.
65.—(1) The procedure for registration is the same as the procedure for granting a marketing authorisation in accordance with Part 3, except—
(a)the applicant is not required to provide proof of efficacy;
(b)the product is not required to have a summary of product characteristics;
(c)the Secretary of State is not required to publish an assessment report.
(2) The procedure for variation, suspension and revocation is the same as for a marketing authorisation.
[F151(3) The Secretary of State must ensure that the procedure for granting a registration in relation to a homeopathic remedy is completed within a maximum of 210 days after the submission of the application.]
Textual Amendments
F151Sch. 1 para. 65(3) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 77
Products on the market before 1994U.K.
66. A homeopathic remedy that was on the market before 1st January 1994 may be placed on the market without being registered.
ClassificationU.K.
67. The registration must specify the classification of the homeopathic remedy, which must be one of the classifications specified for a veterinary medicinal product in Schedule 3.
OffencesE+W+S
68. It is an offence to fail to comply with—
[F152(za)paragraph 22A(1);]
(a)a requirement made under paragraph 27(1);
(b)a request made under paragraph 27(2);
(c)paragraph 28(1) or (2);
(d)a requirement made under paragraph 28(3);
[F153(da)paragraph 28(4);]
(e)paragraph 29(3);
F154(f). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(g)paragraph 31(1) or (2);
(h)a request made under paragraph 31(3);
F155(i). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(j)a prohibition or requirement made under paragraph 41(1);
[F156(ja)a restriction or requirement made under paragraph 41A;
(jb)a prohibition made under paragraph 41B;]
F157(k). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(l)paragraph 56;
[F158(la)paragraph 56A;
(lb)paragraph 56B;
(lc)paragraph 56C;]
(m)paragraph 57;
F159(n). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(o)paragraph 59; or
(p)paragraph 60.
Extent Information
E33This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F152Sch. 1 para. 68(za) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 78(a)
F153Sch. 1 para. 68(da) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 78(b)
F154Sch. 1 para. 68(f) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 78(c)
F155Sch. 1 para. 68(i) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 78(d)
F156Sch. 1 para. 68(ja)(jb) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 78(e)
F157Sch. 1 para. 68(k) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 78(f)
F158Sch. 1 paras. 68(la)-(lc) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 78(g)
F159Sch. 1 para. 68(n) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 78(h)
OffencesN.I.
68. It is an offence to fail to comply with—
(a)a requirement made under paragraph 27(1);
(b)a request made under paragraph 27(2);
(c)paragraph 28(1) or (2);
(d)a requirement made under paragraph 28(3);
(e)paragraph 29(3);
(f)paragraph 30;
(g)paragraph 31(1) or (2);
(h)a request made under paragraph 31(3);
(i)a prohibition or requirement made under paragraph 39(4);
(j)a prohibition or requirement made under paragraph 41(1);
(k)paragraph 55;
(l)paragraph 56;
(m)paragraph 57;
(n)paragraph 58;
(o)paragraph 59; or
(p)paragraph 60.
Extent Information
E188This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Regulation 4(4)
[F160SCHEDULE 1AE+W+SConverted EU marketing authorisations
Textual Amendments
F160Sch. 1A inserted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(3), Sch. 8 Pt. 1; 2020 c. 1, Sch. 5 para. 1(1)
1. In this Schedule—E+W+S
“converted EU marketing authorisation” means an EU marketing authorisation to which paragraph 2 applies;
“EU marketing authorisation” means a marketing authorisation for a veterinary medicinal product granted by the European Commission in accordance with Title 3 of Regulation (EC) No 726/2004 of the European Parliament and of the Council laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency.
2. This paragraph applies to an EU marketing authorisation which—E+W+S
(a)was granted before exit day, and
(b)remains in force immediately before exit day.
3. A converted EU marketing authorisation has effect on and after exit day for the purposes of these regulations as if it were a marketing authorisation granted by the Secretary of State under these Regulations on the date it was originally granted—E+W+S
(a)on the terms which were in force immediately before exit day,
(b)with the benefit of any periods of data marketing exclusivity from which the holder benefited immediately before exit day, and
(c)subject to any suspension or post-authorisation obligations which were in force immediately before exit day.
4. Without prejudice to the generality of paragraph 3—E+W+S
(a)the holder of a converted EU marketing authorisation is subject to the annual fee as set out in paragraph 26 of Schedule 7;
(b)a converted EU marketing authorisation is to be treated as having been granted in accordance with regulation 4(3) and Schedule 1 for the purposes of Regulation (EC) No 469/2009.]
Regulation 4(5)
[F161SCHEDULE 1BE+W+SQualifying Northern Ireland good (QNIG) certificates
Textual Amendments
F161Sch. 1B inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(8)
1. In this Schedule—E+W+S
“QNIG certificate” means a certificate issued under paragraph 3;
“QNIG certificate holder”, in relation to a QNIG certificate, means the person to whom that certificate was issued under paragraph 3;
“qualifying Northern Ireland goods” has the meaning given to it from time to time in regulations made under section 8C(6) of the European Union (Withdrawal) Act 2018;
“Northern Ireland VMRs” means the Veterinary Medicines Regulations 2013 as they have effect in Northern Ireland.
2. This Schedule applies to a veterinary medicinal product which is—E+W+S
(a)a qualifying Northern Ireland good in respect of which there is a marketing authorisation valid in Northern Ireland under the Northern Ireland VMRs,
(b)not a product in respect of which there is a marketing authorisation which is valid in Great Britain (including any marketing authorisation which has effect under paragraph 3 of Schedule 1A),
(c)not a product in respect of which a QNIG certificate issued under this Schedule already applies, and
(d)not a product to which Article 41(1) of the EU withdrawal agreement applies.
3. If the condition in paragraph 4 is met in respect of the veterinary medicinal product, the Secretary of State must issue a QNIG certificate in respect of that product to the person who holds a marketing authorisation in respect of the product which is valid in Northern Ireland under the Northern Ireland VMRs.E+W+S
4. The condition is that the person who holds a marketing authorisation in respect of the product which is valid in Northern Ireland under the Northern Ireland VMRs, who must be a person established in Northern Ireland, has provided the Secretary of State with the following information—E+W+S
(a)the Northern Ireland address of that person;
(b)all necessary administrative information, and all scientific documentation necessary for demonstrating the safety, quality and efficacy of the veterinary medicinal product, equivalent to that which would need to be provided under Schedule 1 if an application for a marketing authorisation was to be made in respect of that product under paragraph 1 of that Schedule (allowing for any relevant derogations provided for in Part 2 of that Schedule);
(c)the name and address of a person who resides in the United Kingdom or in a member State who is to provide in respect of the veterinary medicinal product, permanently and continuously, the services of a qualified person (pharmacovigilance) for the purposes of Part 8 of Schedule 1.
5. A QNIG certificate has effect as if it were a marketing authorisation granted by the Secretary of State under these Regulations subject to the modification that the qualified person (pharmacovigilance) for the purposes of Part 8 of Schedule 1 is the person identified under paragraph 4(c).E+W+S
6. The QNIG certificate holder must provide to the Secretary of State from time to time such further information as is appropriate to ensure that the information provided under paragraph 4 remains accurate and complete.E+W+S
7. Without prejudice to any other power to suspend a marketing authorisation under Schedule 1, if the Secretary of State considers that a QNIG certificate holder is in breach of these Regulations as modified by paragraph 5, or that the information provided in respect of the matters specified in paragraph 4 is no longer accurate or complete, the Secretary of State may by notice suspend the QNIG certificate.E+W+S
8. The Secretary of State must publish any notice given under paragraph 7 in such manner as the Secretary of State considers appropriate from time to time.E+W+S
9. Paragraphs 39 and 40 of Schedule 1 apply to the suspension of a QNIG certificate under paragraph 7 as they would to the suspension of such a certificate under paragraph 38 of that Schedule as read with paragraph 5.]E+W+S
Regulation 5(2)
SCHEDULE 2U.K.The manufacture of veterinary medicinal products
PART 1U.K.Manufacturing authorisations
[F162Manufacturing authorisationE+W+S
1.—(1) No person may carry out any activity mentioned in sub-paragraph (2) otherwise than in accordance with an authorisation granted under this Schedule (a “manufacturing authorisation”).
(2) For the purposes of sub-paragraph (1) the activities are—
(a)the manufacture of veterinary medicinal products (whether for use in Great Britain or another country);
(b)the carrying out of any part of the manufacturing process or of bringing a veterinary medicinal product to its final state, including the processing, assembling, packaging or repackaging, labelling or relabelling, storing, sterilising or releasing for supply of a veterinary medicinal product;
(c)the importation of any veterinary medicinal product for use in Great Britain.]
Extent Information
E34This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F162Sch. 2 para. 1 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 80
ApplicationN.I.
1. An application for a manufacturing authorisation must be made to the Secretary of State.
Extent Information
E189This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F163Application for authorisationE+W+S
2.—(1) An application for a manufacturing authorisation must be submitted to the Secretary of State electronically and must include the matters mentioned in sub-paragraph (2).
(2) For the purposes of sub-paragraph (1) the matters are—
(a)the name of the person who will hold the manufacturing authorisation and that person’s address or registered place of business;
(b)the names and addresses of the sites (including any site where work is undertaken on behalf of the proposed holder under contract) where—
(i)each stage of the manufacturing process or of bringing a veterinary medicinal product to its final state, including processing, assembling, packaging or repackaging, labelling or relabelling, storing or sterilising, is carried out;
(ii)any imported products are held; or
(iii)any control or batch release is carried out;
(c)a description of the veterinary medicinal products or pharmaceutical forms proposed to be manufactured or imported under the authorisation;
(d)the name of the proposed qualified person (manufacture) for the purposes of paragraph 9;
(e)the name of the person proposed to have responsibility for quality control;
(f)the qualifications and a description of the relevant experience of the person proposed to have responsibility for quality control;
(g)the name of the person proposed to have responsibility for production;
(h)the qualifications and a description of the relevant experience of the person proposed to have responsibility for production;
(i)a declaration that the applicant complies with good manufacturing practice and any relevant legislation; and
(j)a declaration that any site mentioned in paragraph (b) is ready for inspection.]
Extent Information
E35This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F163Sch. 2 para. 2 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 81
Time limitsN.I.
2.—(1) The Secretary of State must process an application for a manufacturing authorisation within 90 days of receiving it.
(2) The Secretary of State must process an application for a variation of a manufacturing authorisation within 30 days unless the Secretary of State notifies the applicant in writing that the time has been extended to 90 days.
Extent Information
E190This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F164Procedure for grant of authorisations and time limitsE+W+S
3.—(1) The Secretary of State must process an application mentioned in paragraph 2 within 90 days of validating the application.
(2) The Secretary of State must inspect the sites mentioned in paragraph 2(2)(b) within 90 days of validating the application.
(3) The Secretary of State must grant the manufacturing authorisation if satisfied, following the inspection mentioned in sub-paragraph (2), that—
(a)the sites are suitable for the intended purposes;
(b)the applicant has—
(i)suitable and sufficient staff, technical equipment and facilities for the proposed activities; and
(ii)a documented quality management system in place.
(4) Where the Secretary of State is not satisfied in relation to one or more of the matters mentioned in sub-paragraph (3), the Secretary of State may—
(a)reject the application; or
(b)grant a conditional manufacturing authorisation for a period specified by the Secretary of State until the deficiency has been addressed.
(5) The Secretary of State may extend the period for which a conditional manufacturing authorisation is granted under sub-paragraph (4)(b).
(6) Where a conditional manufacturing authorisation is granted under sub-paragraph (4)(b) and the deficiency is addressed within the specified period to the satisfaction of the Secretary of State, the authorisation continues to have effect without those conditions.]
Extent Information
E36This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F164Sch. 2 para. 3 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 82
Granting the authorisationN.I.
3. The Secretary of State must grant a manufacturing authorisation on being satisfied that the applicant has suitable and sufficient premises, staff, technical equipment and facilities for the manufacture, control and storage of the products, and will comply with these Regulations.
Extent Information
E191This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
The authorisationE+W+S
4.—(1) The manufacturing authorisation must specify—
(a)the types of veterinary medicinal products and pharmaceutical forms that may be manufactured [F165, controlled] or imported;
[F166(b)the name and address of the site where the products are to be manufactured or controlled, or to which they are to be imported;]
(c)the name and address of the person holding the authorisation;
(d)the address of the premises to which it relates;
(e)the names of all qualified persons nominated to act under this Schedule.
(2) It may specify that different activities must be carried out in different premises or parts of premises, and may require the holder of the manufacturing authorisation to restrict access to premises or parts of premises to persons carrying out activities there.
(3) The holder of a manufacturing authorisation must notify the Secretary of State, and if necessary apply for a variation of the authorisation, before making a material alteration to the premises or facilities used under the authorisation, or to the operations for which they are used.
Extent Information
E37This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F165Word in Sch. 2 para. 4(1)(a) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 83(a)
F166Sch. 2 para. 4(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 83(b)
The authorisationN.I.
4.—(1) The manufacturing authorisation must specify—
(a)the types of veterinary medicinal products and pharmaceutical forms that may be manufactured or imported;
(b)the place where they are to be manufactured or controlled;
(c)the name and address of the person holding the authorisation;
(d)the address of the premises to which it relates;
(e)the names of all qualified persons nominated to act under this Schedule.
(2) It may specify that different activities must be carried out in different premises or parts of premises, and may require the holder of the manufacturing authorisation to restrict access to premises or parts of premises to persons carrying out activities there.
(3) The holder of a manufacturing authorisation must notify the Secretary of State, and if necessary apply for a variation of the authorisation, before making a material alteration to the premises or facilities used under the authorisation, or to the operations for which they are used.
Extent Information
E192This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F167Application for variation to the authorisationE+W+S
4A.—(1) The holder of a manufacturing authorisation must notify the Secretary of State, and apply for a variation of the authorisation, before—
(a)making a material alteration to the premises or facilities used under the authorisation, or to the operations for which they are used;
(b)changing the qualified person (manufacture), the person with responsibility for quality control or the person with responsibility for production.
(2) The Secretary of State must process an application under sub-paragraph (1) within 30 days of receiving it unless the Secretary of State notifies the applicant in writing that the time has been extended to 90 days.
(3) The Secretary of State must grant the application under sub-paragraph (1) if satisfied in respect of the matters in paragraph 3(3) as regards the proposed variation.
(4) The Secretary of State may inspect any site to which the manufacturing authorisation or proposed variation relates in connection with the application.
(5) Where the Secretary of State is not satisfied for the purposes of sub-paragraph (3), the Secretary of State may—
(a)reject the application; or
(b)grant a conditional variation to the manufacturing authorisation for a period specified by the Secretary of State until the deficiency has been addressed.
(6) The Secretary of State may extend the period for which a conditional variation to the marketing authorisation is granted under sub-paragraph (5)(b).
(7) Where a conditional variation to the manufacturing authorisation is granted under sub-paragraph (5)(b) and the deficiency is addressed within the specified period to the satisfaction of the Secretary of State, the authorisation continues to have effect as so varied without those conditions.]
Textual Amendments
F167Sch. 2 para. 4A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 84
[F168Suspension, revocation etc] of the authorisationE+W+S
5.—(1) The Secretary of State may suspend, vary or revoke a manufacturing authorisation if the holder—
(a)has not complied with these Regulations;
(b)has manufactured a veterinary medicinal product not authorised by the manufacturing authorisation;
(c)has produced a veterinary medicinal product outside the terms of a marketing authorisation; F169...
(d)no longer has suitable premises or equipment.
[F170(e)has failed to carry out the activity specified in the authorisation for a period of five years or more; or
(f)has not paid any fee required under these Regulations]
[F171(2) The Secretary of State may also suspend, vary or revoke the authorisation on being satisfied that the qualified person (manufacture), the person responsible for quality control or the person with responsibility for production is not fulfilling that person’s duties under these Regulations.
(3) In particular, the Secretary of State may—
(a)suspend the manufacture or import of veterinary medicinal products;
(b)suspend, revoke or vary the manufacturing authorisation for one or more pharmaceutical forms;
(c)suspend, revoke or vary the manufacturing authorisation for one or more activities in one or more manufacturing sites.]
Extent Information
E38This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F168Words in Sch. 2 para. 5 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 85(c)
F169Word in Sch. 2 para. 5(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 85(a)(i)
F170Sch. 2 para. 5(1)(e)(f) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 85(a)(ii)
F171Sch. 2 para. 5(2)(3) substituted for Sch. 2 para. 5(2) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 85(b)
Suspension, variation or revocation of the authorisationN.I.
5.—(1) The Secretary of State may suspend, vary or revoke a manufacturing authorisation if the holder—
(a)has not complied with these Regulations;
(b)has manufactured a veterinary medicinal product not authorised by the manufacturing authorisation;
(c)has produced a veterinary medicinal product outside the terms of a marketing authorisation; or
(d)no longer has suitable premises or equipment.
(2) The Secretary of State may also suspend, vary or revoke it on being satisfied that the qualified person (manufacture) is not fulfilling their duties under these Regulations.
Extent Information
E193This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F172Good manufacturing practice certificates and inspection of sites]E+W+S
6.—(1) The Secretary of State must, from time to time, inspect [F173sites] [F174authorised] under paragraph 3, basing the frequency of the inspection on the risks associated with each [F175site’s history] and the nature of the products handled at the [F173sites].
[F176(2) Within 90 days after an inspection, the Secretary of State must issue a certificate of good manufacturing practice to the manufacturer if the inspection establishes that the manufacturer has complied with the requirements of these Regulations in respect of the site to which the inspection relates.
(2A) Where the Secretary of State does not consider that compliance is established after inspection in accordance with sub-paragraph (2), the Secretary of State must enter that fact in the register mentioned in paragraph 12(a).
(2B) The Secretary of State may carry out an inspection on a site occupied by a manufacturer established in a country other than the United Kingdom notwithstanding any arrangements that may have been entered into between the United Kingdom and that country.
(2C) The importer of a veterinary medicinal product must ensure before importation that the manufacturer of that product has—
(a)a valid certificate of good manufacturing practice issued by the Secretary of State; or
(b)an equivalent certificate issued by a regulatory authority—
(i)with which the Secretary of State has an agreement or arrangement for such purposes; or
(ii)which the Secretary of State considers to have demonstrated equivalent standards to those in the United Kingdom.]
F177(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F177(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F177(5) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E39This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F172Sch. 2 para. 6 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 86(c)
F173Word in Sch. 2 para. 6(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 86(a)(i)
F174Word in Sch. 2 para. 6(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 86(a)(ii)
F175Words in Sch. 2 para. 6(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 86(a)(iii)
F176Sch. 2 para. 6(2)-(2C) substituted for Sch. 2 para. 6(2) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 86(b)
F177Sch. 2 para. 6(3)-(5) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Inspection of premisesN.I.
6.—(1) The Secretary of State must, from time to time, inspect premises registered under paragraph 3, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
(2) Within 90 days after an inspection, the Secretary of State must issue a certificate of good manufacturing practice to the manufacturer if the inspection established compliance with the principles and guidelines on good manufacturing practice in accordance with Commission Directive 91/412/EEC laying down the principles and guidelines of good manufacturing practice for veterinary medicinal products(4).
(3) If an inspection is carried out at the request of the European Pharmacopoeia to establish compliance with a monograph, the Secretary of State must issue a certificate of compliance with the monograph, if appropriate.
(4) The Secretary of State must provide details of each certificate of good manufacturing practice issued to the Agency for entry into a database.
(5) If the outcome of the inspection is that the manufacturer does not comply with the principles and guidelines of good manufacturing practice, the Secretary of State must provide details to the Agency for entry into the database.
Extent Information
E194This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Report following inspectionU.K.
7.—(1) After each inspection of manufacturing premises, the inspector must make a written report to the Secretary of State on whether the principles and guidelines on good manufacturing practice and the conditions of these Regulations are being complied with.
(2) The Secretary of State must inform the inspected manufacturer of the content of such reports.
Duties on the holder of a manufacturing authorisationE+W+S
8.—(1) A holder of a manufacturing authorisation must ensure that the veterinary medicinal product is manufactured in accordance with the marketing authorisation.
[F178(2) The holder must have permanently at the holder’s disposal the services of—
(a)staff complying with any legal requirements in relation to manufacture of veterinary medicinal products; and
(b)at least one qualified person (manufacture).
(2A) The holder must place at the disposal of any qualified person (manufacture) all necessary documents, premises and technical and other facilities in order to enable that person to discharge their duties as the qualified person.
(2B) Where any qualified person (manufacture) ceases to be available to provide services to the holder, the holder must give notice of the fact to the Secretary of State—
(a)at least 30 days in advance of the person’s ceasing to be so available; or
(b)where such notice is not possible, at the earliest opportunity.]
[F179(3) The holder must—
(a)comply with good manufacturing practice and have a valid certificate of good manufacturing practice;
(b)use as starting materials only active substances which have been manufactured in accordance with good manufacturing practice and distributed in accordance with good distribution practice for active substances;
(c)verify that each manufacturer, distributor or importer from whom the holder obtains active substances and to which paragraph 26 applies is registered with the Secretary of State under that paragraph;
(d)carry out audits based on a risk assessment in relation to the manufacturers, distributors and importers from which the holder obtains active substances;
(e)have in place a system of quality assurance and quality control; and
(f)give to the Secretary of State, on request, proof of any control test specified by the Secretary of State which has been carried out on the veterinary medicinal product or the constituents and intermediate products of the manufacturing process in accordance with the data submitted in support of the application for the marketing authorisation.
(3A) The holder of a manufacturing authorisation must inform the Secretary of State and the holder of any relevant marketing authorisation where the holder obtains information that veterinary medicinal products which fall within the scope of its manufacturing authorisation are falsified, or are suspected of being falsified, irrespective of whether those products were distributed within the legal supply chain or by illegal means.]
(4) A holder who makes up a bulk package of veterinary medicinal products must ensure that the package is labelled, in a way that the label is clearly visible and legible, with—
(a)the name of the veterinary medicinal product, its strength as shown in the summary of product characteristics and its pharmaceutical form;
(b)the batch number;
(c)the expiry date;
(d)any storage requirements; and
(e)any other warning necessary for the safe handling of the package.
(5) A holder must keep an adequate number of representative samples of each batch of a veterinary medicinal product in stock at least until the expiry date of the batch, and must submit any such sample to the Secretary of State if required in writing to do so.
[F180(6) A holder must keep detailed records of all veterinary medicinal products which the holder supplies.]
Extent Information
E40This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F178Sch. 2 para. 8(2)-(2B) substituted for Sch. 2 para. 8(2) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 87(a)
F179Sch. 2 para. 8(3)(3A) substituted for Sch. 2 para. 8(3) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 87(b)
F180Sch. 2 para. 8(6) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 87(c)
Duties on the holder of a manufacturing authorisationN.I.
8.—(1) A holder of a manufacturing authorisation must ensure that the veterinary medicinal product is manufactured in accordance with the marketing authorisation.
(2) The holder must have permanently at their disposal the services of at least one qualified person (manufacture) who is on the register of qualified persons (manufacture) maintained by the Secretary of State and must place all necessary facilities at the qualified person’s disposal.
(3) The holder must—
(a)have a current Certificate of Good Manufacturing Practice;
(b)have in place a system of Quality Assurance and Quality Control; and
(c)give to the Secretary of State on request proof of all control tests carried out on the veterinary medicinal product or the constituents and intermediate products of the manufacturing process in accordance with the data submitted in support of the application for the marketing authorisation.
(4) A holder who makes up a bulk package of veterinary medicinal products must ensure that the package is labelled, in a way that the label is clearly visible and legible, with—
(a)the name of the veterinary medicinal product, its strength as shown in the summary of product characteristics and its pharmaceutical form;
(b)the batch number;
(c)the expiry date;
(d)any storage requirements; and
(e)any other warning necessary for the safe handling of the package.
(5) A holder must keep an adequate number of representative samples of each batch of a veterinary medicinal product in stock at least until the expiry date of the batch, and must submit any such sample to the Secretary of State if required in writing to do so.
Extent Information
E195This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F181Good manufacturing practiceE+W+S
8A.—(1) A holder of a manufacturing authorisation must ensure that the veterinary medicinal product is manufactured in accordance with this paragraph, whether the manufacturing is performed by the holder or another person.
(2) The manufacturing operations must be conducted in accordance with a written methodology, to be known as the “pharmaceutical quality system” or “PQS”.
(3) The PQS must be—
(a)clear;
(b)systematically reviewed from time to time in the light of experience; and
(c)capable of consistently manufacturing veterinary medicinal products which are of the required quality and which meet the requirements of the relevant marketing authorisation.
(4) The critical steps of the manufacturing process set out in the PQS must be validated.
(5) Any significant amendments to the PQS must be validated.
(6) The PQS must provide for—
(a)appropriately qualified and trained personnel;
(b)adequate premises and space;
(c)suitable equipment and access to services;
(d)suitable materials, containers and labelling;
(e)relevant procedures and instructions;
(f)suitable storage and transport;
(g)investigation into complaints and defects.
(7) The PQS must provide for any significant deviations from its provisions to be—
(a)fully recorded, and
(b)investigated, with appropriate corrective and preventative action implemented.
(8) The holder of a manufacturing authorisation must ensure that records of the manufacturing process, including distribution, are kept in a comprehensible and accessible form until the later of—
(a)the date which is five years after the date on which the veterinary medicinal product is placed on the market;
(b)the date which is one year after the expiry date of the batch of veterinary medicinal product.
(9) In this paragraph, a process (or part of a process) is “validated” if scientific evidence is assembled which demonstrates that it is capable of consistently delivering expected results.]
Textual Amendments
F181Sch. 2 paras. 8A, 8B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 88
[F181Recalled and counterfeit productsE+W+S
8B.—(1) The holder of a manufacturing authorisation must comply with any requirement by the Secretary of State to recall a veterinary medicinal product and must record the details of the recall operation.
(2) The holder of a manufacturing authorisation must record any veterinary medicinal product which is—
(a)recalled (whether or not the holder physically receives the recalled product); or
(b)discovered to be counterfeit.
(3) Where any veterinary medicinal product is recalled and physically received, the qualified person (manufacture) must assess the recalled product in order to determine whether—
(a)the product has been stored (including during transport) in accordance with the summary of product characteristics;
(b)the product is a genuine product and not counterfeit.
(4) Where the qualified person (manufacture) determines that a recalled veterinary medicinal product does not satisfy sub-paragraph (3)(a) or (b), or where it is not possible for the qualified person (manufacture) to determine whether the product does so, the product may not be re-sold.
(5) The qualified person (manufacture) must record any assessment and determination made under sub-paragraphs (3) and (4).
(6) Any veterinary medicinal products which may not be re-sold must be identified, held separately and destroyed and the holder of a manufacturing authorisation must develop a suitable procedure to set out the steps to be taken in accordance with this sub-paragraph.
(7) The holder of a manufacturing authorisation must keep any information recorded under this paragraph for five years.]
Textual Amendments
F181Sch. 2 paras. 8A, 8B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 88
Qualified persons for manufactureU.K.
9.—(1) The Secretary of State may appoint as a qualified person (manufacture) any person [F182(including the manufacturer of a veterinary medicinal product)] who is—
(a)a member of the Royal Pharmaceutical Society or registered with the Pharmaceutical Society of Northern Ireland;
(b)a Chartered Chemist or a Fellow, Member or Associate Member of the Royal Society of Chemistry; or
(c)a Chartered Biologist or a Fellow, Member or Associate Member of the Society of Biology;
[F183(1A) For the purposes of sub-paragraph (1), a person has sufficient practical experience if they have been engaged in one or more of the activities mentioned in sub-paragraph (1B) for at least two years in the provision of services to the holder of a manufacturing authorisation.
(1B) For the purposes of sub-paragraph (1A) the activities are—
(a)quality assurance of medicinal products;
(b)qualitative analysis of medicinal products;
(c)quantitative analysis of active substances.
(1C) The Secretary of State may treat the reference in sub-paragraph (1A) to two years of practical experience as a reference to—
(a)one year, where the person’s formal course of study lasted for at least five years;
(b)six months, where the person’s formal course of study lasted for at least six years.]
who qualified on the basis of a formal course of study lasting not less than three years full-time or equivalent and who has sufficient practical experience to carry out the duties under this Schedule.
(2) The Secretary of State may exceptionally appoint a person who is not a member of one of those institutions to act as a qualified person (manufacture) on being satisfied that that person has the educational qualifications or practical experience to carry out the duties under this Schedule.
Textual Amendments
F182Words in Sch. 2 para. 9(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 89(a)
F183Sch. 2 para. 9(1A)-(1C) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 89(b)
[F184Refusal, revocation, suspension or variation of appointment]E+W+S
10. The Secretary of State may refuse [F185, revoke, suspend or vary] an appointment if the Secretary of State is not satisfied that a person has fulfilled or will fulfil duties under these Regulations.
Extent Information
E41This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F184Sch. 2 para. 10 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 90(b)
F185Words in Sch. 2 para. 10 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 90(a)
Refusal or revocation of appointmentN.I.
10. The Secretary of State may refuse or revoke an appointment if the Secretary of State is not satisfied that a person has fulfilled or will fulfil duties under these Regulations.
Extent Information
E196This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Duties on a qualified personE+W+S
11.—(1) The qualified person (manufacture) must ensure that each batch of veterinary medicinal product manufactured under that person’s responsibility is manufactured and checked in compliance with these Regulations and in accordance with the data submitted in support of the application for the marketing authorisation.
(2) If a manufacturer imports a veterinary medicinal product from [F186another] country,F187..., the qualified person (manufacture) must ensure that, following importation, each production batch imported is fully tested F188..., including a full qualitative analysis, a quantitative analysis of at least all the active substances and all the other tests or controls necessary to ensure the quality of a veterinary medicinal product is in accordance with the requirements of the marketing authorisation.
(3) Sub-paragraph (2) does not apply [F189where the exporting country has demonstrated equivalent standards to those of the United Kingdom or ] where appropriate arrangements have been made F190...with the exporting country to ensure that the manufacturer of the veterinary medicinal product applies standards of good manufacturing practice at least equivalent to those laid down in Commission Directive 91/412/EEC and to ensure that the controls in sub‑paragraph (2) have been carried out in the exporting country.
(4) At each stage of manufacture, including release for sale, the qualified person (manufacture) must certify in writing that all control tests required under the marketing authorisation have been carried out, and that the production batch complies with the marketing authorisation.
Extent Information
E42This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F186Word in Sch. 2 para. 11(2) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(b)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F187Words in Sch. 2 para. 11(2) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F188Words in Sch. 2 para. 11(2) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(b)(iii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F189Words in Sch. 2 para. 11(3) inserted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(c)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F190Words in Sch. 2 para. 11(3) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(33)(c)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Duties on a qualified personN.I.
11.—(1) The qualified person (manufacture) must ensure that each batch of veterinary medicinal product manufactured under that person’s responsibility is manufactured and checked in compliance with these Regulations and in accordance with the data submitted in support of the application for the marketing authorisation.
(2) If a manufacturer imports a veterinary medicinal product from a third country, including a product manufactured in a member State, the qualified person (manufacture) must ensure that, following importation, each production batch imported is fully tested in a member State, including a full qualitative analysis, a quantitative analysis of at least all the active substances and all the other tests or controls necessary to ensure the quality of a veterinary medicinal product is in accordance with the requirements of the marketing authorisation.
(3) Sub-paragraph (2) does not apply where appropriate arrangements have been made by the European Union with the exporting country to ensure that the manufacturer of the veterinary medicinal product applies standards of good manufacturing practice at least equivalent to those laid down in Commission Directive 91/412/EEC and to ensure that the controls in sub‑paragraph (2) have been carried out in the exporting country.
(4) At each stage of manufacture, including release for sale, the qualified person (manufacture) must certify in writing that all control tests required under the marketing authorisation have been carried out, and that the production batch complies with the marketing authorisation.
Extent Information
E197This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
RegisterU.K.
12. The Secretary of State must maintain and publish a register of—
(a)holders of manufacturing authorisations; and
(b)qualified persons (manufacture) appointed under paragraph 9(2).
Test sitesE+W+S
13.—(1) The Secretary of State may authorise [F191a site] to act as a test site to carry out contract testing for a holder of a manufacturing authorisation.
(2) The [F192site] must have a current certificate of good manufacturing practice.
[F193(2A) The site must be specified in an existing manufacturing authorisation.]
(3) [F194Inspection of the site is] the same as for a manufacturing authorisation.
Extent Information
E43This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F191Words in Sch. 2 para. 13(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 91(a)
F192Word in Sch. 2 para. 13(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 91(b)
F193Sch. 2 para. 13(2A) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 91(c)
F194Words in Sch. 2 para. 13(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 91(d)
Test sitesN.I.
13.—(1) The Secretary of State may authorise premises to act as a test site to carry out contract testing for a holder of a manufacturing authorisation.
(2) The premises must have a current certificate of good manufacturing practice.
(3) Authorisation and inspection of the premises are the same as for a manufacturing authorisation.
Extent Information
E198This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195PART 2U.K.Authorisation of autogenous vaccines, blood-banks, stem cell centres and products manufactured under the cascade]
Textual Amendments
F195Sch. 2 Pt. 2 (paras. 14-25 entitled "Authorisation of autogenous vaccines, blood-banks, stem cell centres and products manufactured under the cascade") substituted for Sch. 2 Pt. 2 (paras. 14-19 entitled "Authorisation of manufacturers of autogenous vaccines"), Pt. 3 (paras. 20-24 entitled "Authorisation of blood banks"), Pt. 4 (paras. 25-29 entitled "Authorisation of manufacturers of products for administration under the cascade") and Pt. 5 (paras. 30-35 entitled "Authorisation of equine stem cell centres") (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 92
[F195Authorisation to manufacture specific veterinary medicinal productsE+W+S
14.—(1) The Secretary of State may authorise a person to—
(a)manufacture—
(i)autogenous vaccines; or
(ii)an unauthorised veterinary medicinal product for administration under the cascade;
(b)collect, store and supply blood in connection with the treatment of non-food animals;
(c)collect, store and supply blood constituents obtained by the physical separation of donor blood into different fractions within a closed bag system, for the treatment of non-food animals; or
(d)collect, process and store stem cells for use as an autologous treatment in non-food animals,
and may authorise sites for the purpose of carrying out those activities by that person.
(2) A single authorisation under sub-paragraph (1) may confer permission to carry out the activities mentioned in both paragraph (b) and (c) of that sub-paragraph.
(3) In this paragraph, a “closed bag system” means a system in which the blood pack assembly is manufactured under clean conditions, sealed to the external environment and sterilised.]
Extent Information
E44This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Authorisation to manufacture autogenous vaccinesN.I.
14.—(1) The Secretary of State may authorise a person to manufacture autogenous vaccines and may authorise premises for the purpose of such manufacture by that person.
(2) In order to be authorised the premises must be under the supervision of—
(a)a veterinary surgeon; or
(b)a person who the Secretary of State is satisfied has sufficient qualifications and experience to manufacture the product safely.
(3) Before authorising the premises, the Secretary of State must be satisfied that the production process will produce a consistent, safe product.
(4) No person may manufacture an autogenous vaccine other than in accordance with an authorisation under sub-paragraph (1).
Extent Information
E199This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195ProhibitionE+W+S
15. No person may carry out any activity mentioned in paragraph 14 otherwise than—
(a)in accordance with an authorisation mentioned in that paragraph; or
(b)pursuant to paragraph 1(2) of Schedule 4 (administration under the cascade).]
Extent Information
E45This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Types of authorisationN.I.
15.—(1) The authorisation must specify the products that may be manufactured.
(2) It may either be for the production of a single batch of product or for ongoing production of the products specified in the authorisation.
(3) If it is for a single batch it must be time limited.
(4) Only the products specified in the authorisation may be manufactured, and in the case of an authorisation for a single batch the product may only be manufactured before the expiry of the authorisation.
Extent Information
E200This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195PersonnelE+W+S
16. In order to be authorised the site mentioned in paragraph 14(1) must be under the supervision of a named person responsible for release (a “PRR”) who in the opinion of the Secretary of State has sufficient qualifications and experience to manufacture the product safely.]
Extent Information
E46This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
LabellingN.I.
16. The operator of the premises must ensure that every container containing autogenous vaccine is labelled with—
(a)the name of the veterinary surgeon who ordered the vaccine;
(b)a precise description of the vaccine;
(c)the date the vaccine was produced;
(d)the name of the authorisation holder and address of the authorised premises;
(e)the expiry date;
(f)any necessary warnings; and
(g)instructions for use.
Extent Information
E201This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195Process of authorisationE+W+S
17.—(1) An applicant for authorisation under paragraph 14 must, at least two months before commencing an activity mentioned in that paragraph, submit the following to the Secretary of State—
(a)the name and address of the proposed holder of the authorisation;
(b)a description of the activity in which the applicant for authorisation proposes to be engaged;
(c)particulars (including the name and address) in relation to the site at which the relevant activity is to be carried out (whether in the occupation of the proposed holder or otherwise) and a description of the technical equipment on the site;
(d)particulars in relation to the qualifications and experience of the proposed PRR who will supervise the activities at the site.
(2) The application must include a declaration that the applicant will comply with the requirements of these Regulations and confirmation that the site is ready for inspection.
(3) Before granting an authorisation in relation to a site, the Secretary of State must be satisfied that the production process carried out there will produce a consistent, safe product and, in the case of a blood bank or a stem cell centre, that the welfare of the animals involved in the processes will be respected.]
Extent Information
E47This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
RecordsN.I.
17. The operator of the premises must, as soon as is reasonably practicable, record—
(a)the name and address of the veterinary surgeon who ordered the vaccine;
(b)the identification of the source animal;
(c)the expiry date;
(d)the date of supply to the veterinary surgeon,
and must keep the records for at least five years.
Extent Information
E202This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195Authorisation in relation to blood banksE+W+S
18.—(1) No person may collect blood for the purposes of a non-food animal blood bank other than a veterinary surgeon or a person acting under the responsibility of a veterinary surgeon.
(2) The holder of an authorisation to carry out an activity under paragraph 14(1)(b) or (c) may only supply blood or blood constituents to a veterinary surgeon.
(3) No person other than a veterinary surgeon or someone acting under a veterinary surgeon’s responsibility may administer blood to a non-food producing animal.
(4) No person may administer blood to a food-producing animal.]
Extent Information
E48This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Adverse reactionsN.I.
18. The authorised person must notify the Secretary of State of any adverse reactions to an autogenous vaccine within 15 days of learning of the reaction.
Extent Information
E203This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195Authorisation in relation to stem cellsE+W+S
19.—(1) No person may collect stem cells for the purposes of treating animals other than a veterinary surgeon or a person acting under the responsibility of a veterinary surgeon.
(2) No person may collect stem cells from embryonic tissues.
(3) No person may administer any product grown from stem cells to a food-producing animal.]
Extent Information
E49This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Inspection of premisesN.I.
19. The Secretary of State must inspect the authorised premises, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
Extent Information
E204This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195Authorisation in relation to products for administration under the cascadeE+W+S
20.—(1) Subject to sub-paragraph (2), no person may manufacture a product for administration under the cascade that is the pharmaceutical equivalent of an authorised veterinary medicinal product.
(2) The Secretary of State may authorise the manufacture of a product notwithstanding sub-paragraph (1) where there is difficulty in relation to the supply of the authorised veterinary medicinal product.
(3) The holder of an authorisation under paragraph 14(1)(a)(ii) may not supply a product manufactured in accordance with that sub-paragraph other than to a veterinary surgeon who has prescribed the product under the cascade.
(4) The holder of an authorisation under paragraph 14(1)(a)(ii) must—
(a)provide a list of products manufactured in accordance with that sub-paragraph to the Secretary of State annually or at the request of the Secretary of State;
(b)provide sales data for products supplied under sub-paragraph (3) at the request of the Secretary of State.
(5) For the purposes of this paragraph, a product is the pharmaceutical equivalent of an authorised veterinary medicinal product if—
(a)it has the same qualitative and quantitative composition in active substances; and
(b)it has the same pharmaceutical form.]
Extent Information
E50This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Authorisation of blood banksN.I.
20.—(1) The Secretary of State may authorise blood banks for—
(a)the collection, storage and supply of blood, or
(b)the storage and supply of blood constituents obtained by the physical separation of donor blood into different fractions within a closed-bag system,
for the treatment of non-food-producing animals.
(2) The authorisation may be for either or both of these activities.
(3) In order to be authorised a blood bank must be under the supervision of—
(a)a veterinary surgeon named in the authorisation; or
(b)a person named in the authorisation who the Secretary of State is satisfied is suitably qualified to operate the blood bank.
(4) Before authorising a blood bank, the Secretary of State must be satisfied—
(a)that the welfare of animals used in the collection of blood will be respected; and
(b)that the production process will produce a consistent, safe product.
(5) The Secretary of State may suspend, vary or revoke an authorisation of a blood bank if—
(a)the holder no longer uses fit and proper processes;
(b)the premises in which the blood bank is being or is to be operated are not suitable;
(c)the equipment is not suitable; or
(d)the holder has not complied with these Regulations.
(6) Blood may only be collected under the responsibility of a veterinary surgeon.
(7) No person may operate a blood bank for treatment of animals other than in accordance with such an authorisation.
Extent Information
E205This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195Suspension, compulsory variation or revocation of authorisationE+W+S
21. The Secretary of State may by notice suspend, vary or revoke an authorisation under paragraph 14 if the Secretary of State is satisfied that—
(a)the holder of the authorisation no longer uses fit and proper processes;
(b)the site at which the activity takes place is not suitable;
(c)the equipment is not suitable;
(d)the PRR has not carried out adequately the PRR’s responsibilities under these Regulations;
(e)in the case of a person authorised under paragraph 14(1), that person has manufactured a veterinary medicinal product pursuant to that authorisation that is not within its scope;
(f)the holder has not conducted an activity relating to the authorisation for five years or more;
(g)the holder has not paid any fee required under these Regulations; or
(h)the holder has not complied with any other provision in these Regulations.]
Extent Information
E51This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Supply and administration of blood from a blood bankN.I.
21.—(1) The operator of a blood bank may only supply blood to a veterinary surgeon.
(2) No person other than a veterinary surgeon or someone acting under a veterinary surgeon’s responsibility may administer blood.
(3) No person may administer blood to a food-producing animal.
Extent Information
E206This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195LabellingE+W+S
22.—(1) The holder of an authorisation under paragraph 14 must ensure that every container used is labelled with—
(a)a precise description of the product;
(b)the date on which the product was produced;
(c)the name and address of the authorisation holder;
(d)the address of the site named under the authorisation and its authorisation number;
(e)the instructions for use;
(f)the expiry date;
(g)any necessary warnings;
(h)in the case of an autogenous vaccine or an unauthorised veterinary medicinal product for administration under the cascade, the name of the veterinary surgeon who ordered the product;
(i)in the case of blood or a stem cell product—
(i)the identification of the donor animal; and
(ii)the date of collection.
(2) In the case of blood or blood constituents there must be no specific therapeutic indication on the label or on any information related to the product.
(3) In the case of an unauthorised veterinary medicinal product for administration under the cascade the words “this veterinary medicinal product does not hold a marketing authorisation” must appear on the label.]
Extent Information
E52This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
LabellingN.I.
22.—(1) The operator of a blood bank must ensure that every container used for the blood is labelled with—
(a)the identification of the donor animal;
(b)the date of collection;
(c)the authorisation number of the blood bank;
(d)any necessary warnings;
(e)the expiry date.
(2) There must be no specific therapeutic indication on the label or on any information relating to the product.
Extent Information
E207This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195RecordsE+W+S
23. The holder of an authorisation under paragraph 14 must, as soon as is reasonably practicable after the product is supplied, in addition to the expiry date of the product, record the following—
(a)in the case of an unauthorised veterinary medicinal product for administration under the cascade—
(i)the name and address of the veterinary surgeon who ordered the veterinary medicinal product;
(ii)a precise description of the product;
(iii)the date of production;
(iv)the date of supply to the veterinary surgeon;
(b)in the case of stem cells or blood—
(i)the identification of the source animal;
(ii)the name of the veterinary surgeon who collected the product (or under whose responsibility it was collected);
(iii)the date of collection of the product;
(iv)the date that the product was used or if the product was supplied to another veterinary surgeon, the name and address of that veterinary surgeon and the date the product was supplied;
(c)in the case of an autogenous vaccine—
(i)the name and address of the veterinary surgeon who ordered the vaccine;
(ii)the identification of the source animal;
(iii)the date of supply to the veterinary surgeon,
and must keep the records for at least five years.]
Extent Information
E53This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
RecordsN.I.
23. The operator of a blood bank must, as soon as is reasonably practicable, record—
(a)the date of collection;
(b)the identification of the donor animal;
(c)the veterinary surgeon who collected it;
(d)the expiry date;
(e)the date the blood was used or, if it was supplied to another veterinary surgeon, the name of that veterinary surgeon and the date it was supplied;
and must keep the records for at least five years.
Extent Information
E208This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195Adverse eventsE+W+S
24. The holder of an authorisation under paragraph 14 must notify the Secretary of State of any adverse event in relation to a product produced by that person under that authorisation within 30 days of learning of the event.]
Extent Information
E54This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Inspection of premisesN.I.
24. The Secretary of State must inspect the authorised premises, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
Extent Information
E209This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F195Inspection of sitesE+W+S
25. The Secretary of State must inspect any site authorised under paragraph 14, basing the frequency of the inspection on the risks associated with each site’s history and the nature of the products handled at the site.]
Extent Information
E55This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Authorisation to manufacture products for administration under the cascadeN.I.
25.—(1) The Secretary of State may authorise a person and premises to manufacture an unauthorised veterinary medicinal product for administration under the cascade.
(2) In order to be authorised the premises must be under the supervision of a person who the Secretary of State is satisfied has sufficient qualifications and experience to manufacture the product safely.
(3) Before authorising the premises, the Secretary of State must be satisfied that the production process will produce a safe product.
(4) The authorisation must specify what types of product it covers.
(5) No person may manufacture an unauthorised veterinary medicinal product other than in accordance with an authorisation under sub-paragraph (1).
Extent Information
E210This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F196PART 2AU.K.Active Substances]
Textual Amendments
F196Sch. 2 Pts. 2A (paras. 26-31), 2B (para. 32) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 93
[F196Prohibition on manufacture, importation or distribution of active substances unless registeredE+W+S
26.—(1) No person may manufacture, import or distribute an active substance unless the person is registered in the register maintained under sub-paragraph (2).
(2) The Secretary of State must establish and maintain a register of manufacturers, importers and distributors of active substances and the sites occupied by them for the purposes of manufacturing or holding active substances.]
Extent Information
E56This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
LabellingN.I.
26. The authorised person must ensure that, before a veterinary medicinal product is supplied, every container is labelled with—
(a)the name of the veterinary surgeon who ordered the veterinary medicinal product;
(b)a precise description of the veterinary medicinal product;
(c)the date of production;
(d)the name of the authorisation holder and the address of the authorised premises;
(e)the expiry date;
(f)any necessary warnings; and
(g)instructions for use.
Extent Information
E211This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F196Application for registrationE+W+S
27.—(1) An applicant for registration under paragraph 26 must, at least two months before commencing an activity mentioned in paragraph 26(1) or, in the case of an existing manufacturer, within two months of the date on which this provision comes into force, submit the following to the Secretary of State—
(a)the name and address of the proposed registration holder;
(b)the name of the relevant active substance;
(c)a description of the activity proposed to be engaged in in relation to the relevant active substance; and
(d)particulars in relation to the site at which the relevant active substance is to be manufactured or held (as the case may be).
(2) Information may be submitted to the Secretary of State pursuant to sub-paragraph (1) prior to the date on which this provision comes into force, and in such a case—
(a)as regards an applicant for registration who is not an existing manufacturer, the relevant period of two months is to be treated as having started on the date of submission;
(b)as regards an applicant for registration who is an existing manufacturer, the information is to be treated as having been submitted within the relevant period of two months.]
Extent Information
E57This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
RecordsN.I.
27. The authorised person must, as soon as is reasonably practicable, record—
(a)the name and address of the veterinary surgeon who ordered the veterinary medicinal product;
(b)a precise description of the veterinary medicinal product;
(c)the date of production;
(d)the expiry date; and
(e)the date of supply to the veterinary surgeon,
and must keep the record for at least five years.
Extent Information
E212This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F196Good manufacturing or distribution practiceE+W+S
28. A manufacturer, importer or distributor of active substances must comply with good manufacturing practice or good distribution practice, as applicable.]
Extent Information
E58This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Adverse reactionsN.I.
28. The authorised person must notify the Secretary of State of any adverse reactions to a product manufactured by that person within 15 days of learning of the reaction.
Extent Information
E213This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F196Supply of informationE+W+S
29.—(1) A person registered under paragraph 26 must immediately inform the Secretary of State on receipt of any new information that might adversely affect the quality and safety of the active substance.
(2) A person registered under paragraph 26 must immediately inform the Secretary of State of any prohibition or restriction in relation to the active substance imposed by the competent authorities of any country other than the United Kingdom in which the active substance is authorised.]
Extent Information
E59This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Inspection of premisesN.I.
29. The Secretary of State must inspect the authorised premises, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
Extent Information
E214This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F196Inspection of sitesE+W+S
30. The Secretary of State may, from time to time, inspect sites registered under paragraph 26, basing the frequency of the inspections on the risks associated with each site’s history and the nature of the substances handled at the site.]
Extent Information
E60This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Authorisation of stem cell centresN.I.
30.—(1) The Secretary of State may authorise equine stem cell centres for the collection, storage, processing, production and administration of equine stem cells for use as an autologous treatment for horses.
(2) In order to be authorised a centre must be under the supervision of—
(a)a veterinary surgeon named in the authorisation; or
(b)a person named in the authorisation who the Secretary of State is satisfied is suitably qualified to operate the centre.
(3) Before authorising a centre, the Secretary of State must be satisfied—
(a)that the welfare of animals used in the collection of equine stem cells will be respected; and
(b)that the production process will produce a consistent, safe product.
(4) Equine stem cells may only be collected under the responsibility of a veterinary surgeon.
(5) The Secretary of State may suspend, vary or revoke an authorisation of an equine stem cell centre if—
(a)the holder no longer uses fit and proper processes;
(b)the premises in which the centre is being or is to be operated are not suitable;
(c)the equipment of the centre is not suitable; or
(d)the holder has not complied with these Regulations.
(6) No person may operate an equine stem cell centre other than in accordance with such an authorisation.
Extent Information
E215This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F196Report following inspectionE+W+S
31.—(1) After each inspection of a site for the purposes of this Part, the inspector must make a written report to the Secretary of State on whether the requirements in this Part are being complied with.
(2) The Secretary of State must inform the inspected registered person of the content of such reports.]
Extent Information
E61This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Supply and administration of stem cellsN.I.
31.—(1) The operator of an equine stem cell centre may only collect equine stem cells.
(2) The operator of an equine stem cell centre may not collect stem cells from embryonic tissues.
(3) No person other than a veterinary surgeon or someone acting under a veterinary surgeon’s responsibility may administer any product grown from collected equine stem cells.
(4) No person may administer any product grown from collected equine stem cells to a food-producing horse.
Extent Information
E216This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F196PART 2BU.K.Schedule 2 Offences]
[F196OffencesE+W+S
32. It is an offence to fail to comply with—
(a)paragraph 1;
(b)paragraph 4(3);
(c)paragraph 8;
(d)paragraph 11;
(e)paragraph 15;
(f)paragraph 18;
(g)paragraph 19;
(h)paragraph 20(1), (3) or (4);
(i)paragraph 22;
(j)paragraph 23;
(k)paragraph 24;
(l)paragraph 26;
(m)paragraph 28;
(n)paragraph 29.]
Extent Information
E62This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
LabellingN.I.
32.—(1) The operator of an equine stem cell centre must ensure that every container used for the stem cell product is labelled with—
(a)the identification of the donor animal;
(b)the date of collection;
(c)the authorisation number of the equine stem cell centre;
(d)any necessary warnings;
(e)the expiry date.
(2) The operator of an equine stem cell centre must ensure that no specific therapeutic indication is included on the label or on any information relating to the product.
Extent Information
E217This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
RecordsE+W+S
33. F195. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E63This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F195Sch. 2 Pt. 2 (paras. 14-25 entitled "Authorisation of autogenous vaccines, blood-banks, stem cell centres and products manufactured under the cascade") substituted for Sch. 2 Pt. 2 (paras. 14-19 entitled "Authorisation of manufacturers of autogenous vaccines"), Pt. 3 (paras. 20-24 entitled "Authorisation of blood banks"), Pt. 4 (paras. 25-29 entitled "Authorisation of manufacturers of products for administration under the cascade") and Pt. 5 (paras. 30-35 entitled "Authorisation of equine stem cell centres") (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 92
RecordsN.I.
33. The operator of an equine stem cell centre must, as soon as is reasonably practicable, record for each stem cell product—
(a)the identification of the donor animal;
(b)the date of collection;
(c)the veterinary surgeon under whose responsibility the stem cells were collected;
(d)the expiry date;
(e)the date the product was used or, if it was supplied to another veterinary surgeon, the name of that veterinary surgeon and the date it was supplied,
and must keep the records for at least five years.
Extent Information
E218This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Inspection of premisesE+W+S
34. F195. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E64This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F195Sch. 2 Pt. 2 (paras. 14-25 entitled "Authorisation of autogenous vaccines, blood-banks, stem cell centres and products manufactured under the cascade") substituted for Sch. 2 Pt. 2 (paras. 14-19 entitled "Authorisation of manufacturers of autogenous vaccines"), Pt. 3 (paras. 20-24 entitled "Authorisation of blood banks"), Pt. 4 (paras. 25-29 entitled "Authorisation of manufacturers of products for administration under the cascade") and Pt. 5 (paras. 30-35 entitled "Authorisation of equine stem cell centres") (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 92
Inspection of premisesN.I.
34. The Secretary of State must inspect the authorised premises, basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
OffencesE+W+S
35. F195. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E65This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F195Sch. 2 Pt. 2 (paras. 14-25 entitled "Authorisation of autogenous vaccines, blood-banks, stem cell centres and products manufactured under the cascade") substituted for Sch. 2 Pt. 2 (paras. 14-19 entitled "Authorisation of manufacturers of autogenous vaccines"), Pt. 3 (paras. 20-24 entitled "Authorisation of blood banks"), Pt. 4 (paras. 25-29 entitled "Authorisation of manufacturers of products for administration under the cascade") and Pt. 5 (paras. 30-35 entitled "Authorisation of equine stem cell centres") (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 92
OffencesN.I.
35. It is an offence to fail to comply with—
(a)paragraph 4(3);
(b)paragraph 11;
(c)paragraph 14(4);
(d)paragraph 16;
(e)paragraph 17;
(f)paragraph 18;
(g)paragraph 20(6) or (7);
(h)paragraph 21;
(i)paragraph 22;
(j)paragraph 23;
(k)paragraph 25(5);
(l)paragraph 26;
(m)paragraph 27;
(n)paragraph 28;
(o)paragraph 30(4) or (6);
(p)paragraph 31;
(q)paragraph 32; or
(r)paragraph 33.
Extent Information
E219This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Regulation 7
SCHEDULE 3U.K.Classification and supply, wholesale dealers and sheep dip
PART 1U.K.Classification and supply of authorised veterinary medicinal products
Classification of veterinary medicinal productsE+W+S
1.—(1) There shall be the following categories of authorised veterinary medicinal products—
(a)Prescription Only Medicine–Veterinarian (abbreviated to POM-V);
(b)Prescription Only Medicine–Veterinarian, Pharmacist, Suitably Qualified Person (abbreviated to POM-VPS);
(c)Non-Food Animal–Veterinarian, Pharmacist, Suitably Qualified Person (abbreviated to NFA-VPS);
(d)Authorised Veterinary Medicine–General Sales List (abbreviated to AVM-GSL).
(2) The Secretary of State must specify the classification of the veterinary medicinal product when granting the initial marketing authorisation.
(3) The Secretary of State may change the classification after the marketing authorisation has been granted, either at the request of the marketing authorisation holder or in accordance with paragraph 37 of Schedule 1 (compulsory variation).
(4) When granting the marketing authorisation the Secretary of State must classify the following as POM-V—
(a)products containing narcotic or psychotropic substances;
(b)products intended for administration following a diagnosis or clinical assessment by a veterinary surgeon;
[F197(c)products containing an antimicrobial;
(d)products for the purpose of euthanasia;
(e)products with a hormonal or thyrostatic function;
(f)products containing beta-agonists].
(5) When granting the marketing authorisation the Secretary of State must classify the following as POM-V or POM-VPS—
(a)products for food-producing animals;
(b)products in respect of which special precautions must be taken in order to avoid any unnecessary risk to—
(i)the target species;
(ii)the person administering the products to the animal; and
(iii)the environment;
(c)products that may cause effects that impede or interfere with subsequent diagnostic or therapeutic measures; F198...
(d)new veterinary medicinal products containing an active substance that has not been included in an authorised veterinary medicinal product for five years;
[F199(e)immunological veterinary medicinal products.]
(6) The requirement in sub-paragraph (5)(a) relating to veterinary medicinal products for food-producing animals does not apply if all the following criteria are met—
(a)the administration of the veterinary medicinal product is restricted to formulations requiring no particular knowledge or skill in using the product;
(b)the veterinary medicinal product does not present a direct or indirect risk, even if administered incorrectly, to the animal or animals treated, to the person administering the product or to the environment;
(c)the summary of product characteristics of the veterinary medicinal product does not contain any warnings of potential serious side effects deriving from its correct use;
(d)neither the veterinary medicinal product nor any other product containing the same active substance has previously been the subject of frequent serious [F200adverse event] reporting;
(e)the summary of product characteristics does not refer to contra-indications related to other veterinary medicinal products commonly used without prescription;
(f)the veterinary medicinal product is not subject to special storage conditions;
(g)there is no risk for consumer safety as regards residues in food obtained from treated animals even where the veterinary medicinal products are used incorrectly; and
(h)there is no risk to human or animal health as regards the development of resistance to [F201antibiotics] or anthelmintic substances even where the veterinary medicinal products containing those substances are used incorrectly.
Extent Information
E66This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F197Sch. 3 para. 1(4)(c)-(f) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 95(a)
F198Word in Sch. 3 para. 1(5)(c) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 95(b)(i)
F199Sch. 3 para. 1(5)(e) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 95(b)(ii)
F200Words in Sch. 3 para. 1(6)(d) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 95(c)(i)
F201Word in Sch. 3 para. 1(6)(h) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 95(c)(ii)
Classification of veterinary medicinal productsN.I.
1.—(1) There shall be the following categories of authorised veterinary medicinal products—
(a)Prescription Only Medicine–Veterinarian (abbreviated to POM-V);
(b)Prescription Only Medicine–Veterinarian, Pharmacist, Suitably Qualified Person (abbreviated to POM-VPS);
(c)Non-Food Animal–Veterinarian, Pharmacist, Suitably Qualified Person (abbreviated to NFA-VPS);
(d)Authorised Veterinary Medicine–General Sales List (abbreviated to AVM-GSL).
(2) The Secretary of State must specify the classification of the veterinary medicinal product when granting the initial marketing authorisation.
(3) The Secretary of State may change the classification after the marketing authorisation has been granted, either at the request of the marketing authorisation holder or in accordance with paragraph 37 of Schedule 1 (compulsory variation).
(4) When granting the marketing authorisation the Secretary of State must classify the following as POM-V—
(a)products containing narcotic or psychotropic substances;
(b)products intended for administration following a diagnosis or clinical assessment by a veterinary surgeon.
(5) When granting the marketing authorisation the Secretary of State must classify the following as POM-V or POM-VPS—
(a)products for food-producing animals;
(b)products in respect of which special precautions must be taken in order to avoid any unnecessary risk to—
(i)the target species;
(ii)the person administering the products to the animal; and
(iii)the environment;
(c)products that may cause effects that impede or interfere with subsequent diagnostic or therapeutic measures; and
(d)new veterinary medicinal products containing an active substance that has not been included in an authorised veterinary medicinal product for five years.
(6) The requirement in sub-paragraph (5)(a) relating to veterinary medicinal products for food-producing animals does not apply if all the following criteria are met—
(a)the administration of the veterinary medicinal product is restricted to formulations requiring no particular knowledge or skill in using the product;
(b)the veterinary medicinal product does not present a direct or indirect risk, even if administered incorrectly, to the animal or animals treated, to the person administering the product or to the environment;
(c)the summary of product characteristics of the veterinary medicinal product does not contain any warnings of potential serious side effects deriving from its correct use;
(d)neither the veterinary medicinal product nor any other product containing the same active substance has previously been the subject of frequent serious adverse reaction reporting;
(e)the summary of product characteristics does not refer to contra-indications related to other veterinary medicinal products commonly used without prescription;
(f)the veterinary medicinal product is not subject to special storage conditions;
(g)there is no risk for consumer safety as regards residues in food obtained from treated animals even where the veterinary medicinal products are used incorrectly; and
(h)there is no risk to human or animal health as regards the development of resistance to antimicrobials or anthelmintic substances even where the veterinary medicinal products containing those substances are used incorrectly.
Extent Information
E220This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Wholesale supply of veterinary medicinal productsE+W+S
2.—(1) Only a holder F202... of a manufacturing authorisation or the holder of a wholesale dealer’s authorisation granted by the Secretary of State may supply a veterinary medicinal product wholesale, or be in possession of it for that purpose.
(2) A person mentioned in sub-paragraph (1) may only supply a veterinary medicinal product if—
(a)the authorisation in question relates to that product, and
(b)the supply [F203is to the holder of a manufacturing authorisation or] is to another person who is entitled to supply that product under these Regulations, either wholesale or retail.
[F204(3) If the supply is to a veterinary surgeon, a pharmacist or a suitably qualified person, it must be to premises registered (or authorised as the case may be) in accordance with paragraph 8(1), paragraph 10(1) or paragraph 14(4).]
(4) It is immaterial whether or not the supply is for profit.
(5) This paragraph does not apply in relation to a retailer of veterinary medicinal products who supplies another retailer with such products for the purpose of alleviating a temporary supply shortage that could be detrimental to animal welfare.
(6) A wholesale dealer may break open any package (other than the immediate packaging) of a veterinary medicinal product.
Extent Information
E67This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F202Words in Sch. 3 para. 2(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 96(a) (with reg. 202)
F203Words in Sch. 3 para. 2(2)(b) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 96(b)
F204Sch. 3 para. 2(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 96(c)
Wholesale supply of veterinary medicinal productsN.I.
2.—(1) Only a holder of a marketing authorisation, the holder of a manufacturing authorisation or the holder of a wholesale dealer’s authorisation granted by the Secretary of State may supply a veterinary medicinal product wholesale, or be in possession of it for that purpose.
(2) A person mentioned in sub-paragraph (1) may only supply a veterinary medicinal product if—
(a)the authorisation in question relates to that product, and
(b)the supply is to another person who is entitled to supply that product under these Regulations, either wholesale or retail.
(3) If the supply is to a suitably qualified person, it must be to the premises approved in accordance with paragraph 14.
(4) It is immaterial whether or not the supply is for profit.
(5) This paragraph does not apply in relation to a retailer of veterinary medicinal products who supplies another retailer with such products for the purpose of alleviating a temporary supply shortage that could be detrimental to animal welfare.
(6) A wholesale dealer may break open any package (other than the immediate packaging) of a veterinary medicinal product.
Extent Information
E221This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Retail supply of veterinary medicinal productsE+W+S
3.—(1) This paragraph applies in relation to retail supply of veterinary medicinal products.
(2) A veterinary medicinal product classified as POM-V may only be supplied by a veterinary surgeon or a pharmacist and must be supplied in accordance with a prescription from a veterinary surgeon.
(3) A veterinary medicinal product classified as POM-VPS may only be supplied by—
(a)a veterinary surgeon;
(b)a pharmacist; or
(c)a suitably qualified person in accordance with paragraph 14,
and must be in accordance with a prescription from one of those persons.
(4) A veterinary medicinal product classified as NFA-VPS may be supplied without prescription, but may only be supplied by—
(a)a veterinary surgeon;
(b)a pharmacist; or
(c)a suitably qualified person in accordance with paragraph 14.
(5) There are no restrictions on the supply of AVM-GSL products.
(6) In this paragraph—
[F205(a)“retail supply” means a supply whether or not for payment to the owner or keeper of an animal for administration to that animal; and]
(b)a person may supply a product irrespective of who owns it.
Extent Information
E68This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F205Sch. 3 para. 3(6)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 97
Retail supply of veterinary medicinal productsN.I.
3.—(1) This paragraph applies in relation to retail supply of veterinary medicinal products.
(2) A veterinary medicinal product classified as POM-V may only be supplied by a veterinary surgeon or a pharmacist and must be supplied in accordance with a prescription from a veterinary surgeon.
(3) A veterinary medicinal product classified as POM-VPS may only be supplied by—
(a)a veterinary surgeon;
(b)a pharmacist; or
(c)a suitably qualified person in accordance with paragraph 14,
and must be in accordance with a prescription from one of those persons.
(4) A veterinary medicinal product classified as NFA-VPS may be supplied without prescription, but may only be supplied by—
(a)a veterinary surgeon;
(b)a pharmacist; or
(c)a suitably qualified person in accordance with paragraph 14.
(5) There are no restrictions on the supply of AVM-GSL products.
(6) In this paragraph—
(a)“retail supply” means any supply other than to or from the holder of a wholesale dealer’s authorisation, and whether or not for payment; and
(b)a person may supply a product irrespective of who owns it.
Extent Information
E222This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F206Supply of samplesE+W+S
3A.—(1) Subject to sub-paragraph (2) a person mentioned in paragraph 2(1) or 3(2) may not supply a veterinary medicinal product for promotional purposes.
(2) Subject to sub-paragraph (3), the person may supply samples of product labelled in a way that clearly identifies them as such to—
(a)sales representatives who are responsible for promoting the product; or
(b)those entitled to supply the product during sponsored events.
(3) Sub-paragraph (2) does not apply in relation to a product containing an antimicrobial substance.]
Textual Amendments
F206Sch. 3 paras. 3A-3E inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 98
[F206Register of online suppliers of veterinary medicinal productsE+W+S
3B.—(1) No person may supply or offer to supply a veterinary medicinal product classified as POM-V, POM-VPS or NFA-VPS by means of the internet to persons in Great Britain unless the person—
(a)is established within Great Britain;
(b)has an address within Great Britain; and
(c)appears on the register maintained under sub-paragraph (2).
(2) The Secretary of State must establish, maintain and publish on a website a register of persons who supply veterinary medicinal products by means of the internet.]
Textual Amendments
F206Sch. 3 paras. 3A-3E inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 98
[F206Application for registrationE+W+S
3C.—(1) An applicant for registration under paragraph 3B must, at least two months before commencing the activity mentioned in paragraph 3B(1) (or in the case of an existing supplier of veterinary medicinal products by means of the internet within two months of the date on which this provision comes into force), submit to the Secretary of State the name and the address within Great Britain of the proposed registration holder.
(2) Information may be submitted to the Secretary of State pursuant to sub-paragraph (1) prior to the date on which this provision comes into force, and in such a case—
(a)as regards an applicant for registration who is not an existing supplier of veterinary medicinal products by means of the internet, the relevant period of two months is to be treated as having started on the date of submission;
(b)as regards an applicant for registration who is an existing supplier of veterinary medicinal products by means of the internet, the information is to be treated as having been submitted within the relevant period of two months.]
Textual Amendments
F206Sch. 3 paras. 3A-3E inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 98
[F206Duties in relation to online supplyE+W+S
3D. Where a person offers to supply a veterinary medicinal product by means of the internet, that person must make available on each part of the website where the product is offered—
(a)the statement “registered internet retailer of veterinary medicines”;
(b)the contact details of the Secretary of State; and
(c)a link to the published register.]
Textual Amendments
F206Sch. 3 paras. 3A-3E inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 98
[F206Retail storage of veterinary medicinal productsE+W+S
3E. A retailer of veterinary medicinal products must store (including during transport) a veterinary medicinal product in accordance with the terms of any specific instructions on the label of the product and in accordance with the relevant summary of product characteristics.]
Textual Amendments
F206Sch. 3 paras. 3A-3E inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 98
Prescriptions by a veterinary surgeonU.K.
4.—(1) A veterinary surgeon who prescribes a veterinary medicinal product classified as POM-V [F207or a veterinary medicinal product under the cascade] must first carry out a clinical assessment of the animal, and the animal must be under that veterinary surgeon’s care.
(2) This does not apply in relation to the administration of such a product to a wild animal where the administration is authorised by the Secretary of State.
Textual Amendments
F207Words in Sch. 3 para. 4(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 99
PrescriptionsE+W+S
5.—(1) A prescription may be [F208verbal] or written, but a veterinary medicinal product classified as POM-V or POM-VPS [F209or a veterinary medicinal product prescribed under the cascade] may only be supplied—
(a)by the person who prescribed it;
(b)under a written prescription that complies with paragraph 6; or
(c)(in the case of POM-VPS) by a suitably qualified person in accordance with paragraph 14(5).
[F210(1A) Where a veterinary medicinal product is supplied in accordance with a prescription which is not a written prescription, the person who prescribes the product must make a record of the reason for prescribing the product.
(1B) A record made in accordance with sub-paragraph (1A) must be kept by the person mentioned in that sub-paragraph for a period of five years from the date on which the product is prescribed]
(2) A person supplying such a product under a written prescription—
(a)may only supply the product specified in that prescription;
(b)must take all reasonable steps to be satisfied that the prescription has been written and signed by a person entitled to prescribe the product; and
(c)must take all reasonable steps to ensure that it is supplied to the person named in the prescription.
(3) No person may alter a written prescription unless authorised to do so by the person who signed it.
[F211(4) No person may submit a written prescription to a retailer on more than one occasion where the prescription is not repeatable.]
Extent Information
E69This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F208Word in Sch. 3 para. 5(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 100(a)(i) (with reg. 203)
F209Words in Sch. 3 para. 5(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 100(a)(ii) (with reg. 203)
F210Sch. 3 para. 5(1A)(1B) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 100(b) (with reg. 203)
F211Sch. 3 para. 5(4) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 100(c) (with reg. 203)
PrescriptionsN.I.
5.—(1) A prescription may be oral or written, but a veterinary medicinal product classified as POM-V or POM-VPS may only be supplied—
(a)by the person who prescribed it;
(b)under a written prescription that complies with paragraph 6; or
(c)(in the case of POM-VPS) by a suitably qualified person in accordance with paragraph 14(5).
(2) A person supplying such a product under a written prescription—
(a)may only supply the product specified in that prescription;
(b)must take all reasonable steps to be satisfied that the prescription has been written and signed by a person entitled to prescribe the product; and
(c)must take all reasonable steps to ensure that it is supplied to the person named in the prescription.
(3) No person may alter a written prescription unless authorised to do so by the person who signed it.
Extent Information
E223This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Written prescriptionsE+W+S
6.—[F212(1) A written prescription must include—
(a)the full name, address and contact details of the person prescribing the product, including that person’s professional registration number (if available);
(b)the full name, address and contact details of the animal owner or keeper;
(c)the identification (including the species) of the animal or group of animals to be treated;
(d)the premises at which the animals are kept if this is different from the address of the owner or keeper;
(e)the issue date;
(f)the signature or electronic signature of the prescriber;
(g)the name and amount of the product prescribed;
(h)the pharmaceutical form and strength of the product;
(i)as regards veterinary medicinal products that are antibiotics which are prescribed for prophylactic purposes or metaphylactic purposes (as the case may be), a statement to that effect;
(j)the dosage regimen;
(k)any warnings necessary to ensure the proper use, including, where relevant, to ensure prudent use of antimicrobials;
(l)the words “It is an offence under the Veterinary Medicines Regulations 2013 for a person to alter a written prescription unless authorised to do so by the person who signed it”;
(m)for food-producing animal species, the withdrawal period or a statement that the withdrawal period is equal to zero days; and
(n)if the prescription relates to a product prescribed under the cascade, a statement to that effect.
(1A) Subject to the professional obligations of a veterinary surgeon to ensure the health and welfare of animals under their care, a veterinary surgeon may only prescribe a veterinary medicinal product that is an antibiotic where satisfied that the circumstances set out in sub-paragraph (1B) apply.
(1B) For the purposes of sub-paragraph (1A) the circumstances are that the product is not—
(a)used routinely;
(b)used to compensate for poor hygiene, inadequate animal husbandry, or poor farm management practices; or
(c)used to promote growth or increase yield.]
(2) A written prescription for a controlled drug as specified in Schedules 2 to 4 of the Misuse of Drugs Regulations 2001(5) is valid for 28 days.
(3) A written prescription for any other drug is valid for six months or such shorter period as may be specified in the prescription.
(4) If the prescription is repeatable it must specify the number of times the veterinary medicinal product may be supplied.
Extent Information
E70This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F212Sch. 3 para. 6(1)-(1B) substituted for Sch. 3 para. 6(1) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 101 (with reg. 204)
Written prescriptionsN.I.
6.—(1) A written prescription must include—
(a)the name, address and telephone number of the person prescribing the product;
(b)the qualifications enabling the person to prescribe the product;
(c)the name and address of the owner or keeper;
(d)the identification (including the species) of the animal or group of animals to be treated;
(e)the premises at which the animals are kept if this is different from the address of the owner or keeper;
(f)the date of the prescription;
(g)the signature or other authentication of the person prescribing the product;
(h)the name and amount of the product prescribed;
(i)the dosage and administration instructions;
(j)any necessary warnings;
(k)the withdrawal period if relevant; and
(l)if it is prescribed under the cascade, a statement to that effect.
(2) A written prescription for a controlled drug as specified in Schedules 2 to 4 of the Misuse of Drugs Regulations 2001(5) is valid for 28 days.
(3) A written prescription for any other drug is valid for six months or such shorter period as may be specified in the prescription.
(4) If the prescription is repeatable it must specify the number of times the veterinary medicinal product may be supplied.
Extent Information
E224This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Duties when a product is prescribed or suppliedU.K.
7.— [F213(1)] A person who prescribes [F214a veterinary medicinal product under the cascade or] a product classified as POM-V or POM-VPS, or supplies a product classified as NFA-VPS—
(a)before doing so, must be satisfied that the person who will use the product is competent to do so safely, and intends to use it for a purpose for which it is authorised;
(b)when doing so, must advise on its safe administration and on any warnings or contra-indications on the label or package leaflet; and
(c)must not prescribe (or, in the case of a NFA-VPS product, supply) more than the minimum amount required for the treatment; but it is a defence to a charge of failing to comply with this paragraph to show that—
(i)the product prescribed or supplied was in a container specified in the marketing authorisation;
(ii)the manufacturer does not supply that veterinary medicinal product in a smaller container; and
(iii)the person prescribing or supplying is not a person authorised to break open the package before supply.
[F215(2) A person who prescribes antimicrobials must ensure that the product is prescribed for the most limited period that is consistent with the risk to be addressed.]
Textual Amendments
F213Sch. 3 para. 7 renumbered as Sch. 3 para. 7(1) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 102(a)
F214Words in Sch. 3 para. 7(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 102(b)
F215Sch. 3 para. 7(2) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 102(c)
[F216Duties in relation to prescribing of antibiotic veterinary medicinal productsE+W+S
7A.—(1) Subject to sub-paragraphs (2) and (3) a veterinary surgeon may not prescribe a veterinary medicinal product which is an antibiotic for prophylactic purposes.
(2) Without prejudice to paragraph 6(1A), a veterinary surgeon may only prescribe a veterinary medicinal product which is an antibiotic for administration to an animal for prophylactic purposes in exceptional circumstances where the risk of an infection or of an infectious disease is very high and where the consequences of not prescribing the product are likely to be severe.
(3) Subject to sub-paragraph (2), a veterinary surgeon may only prescribe a veterinary medicinal product which is an antibiotic for administration to a group of animals for prophylactic purposes where the circumstances set out in sub-paragraph (4) apply.
(4) For the purposes of sub-paragraph (3) the circumstances are that—
(a)the rationale for prescribing the product to the group of animals is clearly recorded by the veterinary surgeon prescribing it; and
(b)a management review is carried out by a veterinary surgeon at, or as soon as reasonably practicable after, administration of the product in order to identify factors and implement measures for the purpose of eliminating the need for any future such administration.
(5) A veterinary surgeon who prescribes a veterinary medicinal product which is an antibiotic must make a record of the satisfaction of the relevant conditions for the purposes of its use in accordance with this paragraph and keep that documentation for at least five years.]
Textual Amendments
F216Sch. 3 para. 7A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 103
Supply by a veterinary surgeon from registered premisesU.K.
8.—(1) A veterinary surgeon may only supply a veterinary medicinal product from practice premises registered with the Royal College of Veterinary Surgeons as veterinary practice premises at which veterinary medicinal products are stored or supplied.
(2) This paragraph does not apply in relation to a veterinary medicinal product classified as AVM-GSL.
(3) The Royal College of Veterinary Surgeons must, on request, supply the Secretary of State with a copy of the register of veterinary practice premises.
(4) The Secretary of State must, from time to time, inspect premises registered under sub-paragraph (1), basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
(5) Where an inspection under sub-paragraph (4) reveals significant breaches of these Regulations the Secretary of State may require the Royal College of Veterinary Surgeons to remove the premises from the register maintained under sub-paragraph (1).
(6) Where the Secretary of State requires the removal of premises from the register the veterinary surgeon concerned may appeal using the procedure in regulation 30.
(7) Where premises have been removed from the register under sub-paragraph (5) they may not be re-registered without the approval of the Secretary of State.
(8) The Secretary of State may only grant approval under sub-paragraph (7) after a further inspection of the premises.
Supply by a veterinary surgeonU.K.
9.—(1) A veterinary surgeon supplying a veterinary medicinal product (other than one classified as AVM-GSL) must be present when it is handed over unless the veterinary surgeon—
(a)authorises each transaction individually before the product is supplied; and
(b)is satisfied that the person handing it over is competent to do so.
(2) A veterinary surgeon or a person acting under a veterinary surgeon’s responsibility may open any package containing a veterinary medicinal product.
Supply by a pharmacistE+W+S
10.—(1) A pharmacist may only supply a veterinary medicinal product classified as POM-V, POM-VPS or NFA-VPS [F217, or prescribed under the cascade,] from—
(a)premises registered as a pharmacy with the General Pharmaceutical Council or with the Pharmaceutical Society of Northern Ireland;
(b)premises registered with the Royal College of Veterinary Surgeons as being premises from which a veterinary surgeon supplies veterinary medicinal products; or
(c)(in the case of a veterinary medicinal product classified as POM-VPS or NFA-VPS) from premises [F218authorised] under paragraph 14.
(2) A pharmacist supplying a veterinary medicinal product (other than one classified as AVM-GSL) must be present when it is handed over unless the pharmacist—
(a)authorises each transaction individually before the product is supplied; and
(b)is satisfied that the person handing it over is competent to do so.
(3) A pharmacist may supply any veterinary medicinal product prepared in a pharmacy in accordance with the prescriptions of a pharmacopoeia and intended to be supplied directly to the end-user.
(4) A pharmacist may supply a homeopathic remedy prepared extemporaneously by a pharmacist in a registered pharmacy (as well as any other homeopathic remedy permitted to be supplied by a pharmacist under these Regulations) provided that it is prepared in accordance with paragraph 63 of Schedule 1 and intended to be supplied directly to the end user.
(5) A pharmacist may break open any package containing a veterinary medicinal product for the purposes of supply other than the immediate packaging of an injectable product.
Extent Information
E71This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F217Words in Sch. 3 para. 10(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 104(a)
F218Word in Sch. 3 para. 10(1)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 104(b)
Supply by a pharmacistN.I.
10.—(1) A pharmacist may only supply a veterinary medicinal product classified as POM-V, POM-VPS or NFA-VPS from—
(a)premises registered as a pharmacy with the General Pharmaceutical Council or with the Pharmaceutical Society of Northern Ireland;
(b)premises registered with the Royal College of Veterinary Surgeons as being premises from which a veterinary surgeon supplies veterinary medicinal products; or
(c)(in the case of a veterinary medicinal product classified as POM-VPS or NFA-VPS) from premises approved under paragraph 14.
(2) A pharmacist supplying a veterinary medicinal product (other than one classified as AVM-GSL) must be present when it is handed over unless the pharmacist—
(a)authorises each transaction individually before the product is supplied; and
(b)is satisfied that the person handing it over is competent to do so.
(3) A pharmacist may supply any veterinary medicinal product prepared in a pharmacy in accordance with the prescriptions of a pharmacopoeia and intended to be supplied directly to the end-user.
(4) A pharmacist may supply a homeopathic remedy prepared extemporaneously by a pharmacist in a registered pharmacy (as well as any other homeopathic remedy permitted to be supplied by a pharmacist under these Regulations) provided that it is prepared in accordance with paragraph 63 of Schedule 1 and intended to be supplied directly to the end user.
(5) A pharmacist may break open any package containing a veterinary medicinal product for the purposes of supply other than the immediate packaging of an injectable product.
Extent Information
E225This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Supply of a [F219medicinal premix]E+W+S
11.—(1) This paragraph applies in relation to the supply of a [F220medicinal premix].
(2) F221... An authorised manufacturer of the product or an authorised wholesale dealer may only supply such a [F222medicinal premix] to—
(a)a veterinary surgeon, pharmacist or, in the case of a product classified as POM-VPS, a suitably qualified person;
(b)an [F223authorised] [F224intermediate feedingstuffs] manufacturer; or
(c)an [F223authorised] feedingstuffs manufacturer if the approval permits the rate of incorporation specified on the label of that [F222medicinal premix] (if the manufacturer is the end-user the supply must be in accordance with a [F225medicated feedingstuffs prescription]).
(3) A veterinary surgeon, pharmacist or, in the case of a product classified as POM-VPS, a suitably qualified person may only supply such a [F226medicinal premix] to—
(a)an [F223authorised] [F227intermediate feedingstuffs] manufacturer; or
(b)an [F223authorised] feedingstuffs manufacturer if the [F228authorisation] permits the rate of incorporation specified on the label of that [F226medicinal premix] (if the manufacturer is the end user the supply must be in accordance with a prescription [F229for medicated feedingstuffs]).
[F230(4) This paragraph does not apply in relation to a feedingstuffs manufacturer approved to incorporate a medicinal premix who supplies another such feedingstuffs manufacturer with medicinal premix where the purpose of that supply is to alleviate a temporary supply shortage that could be detrimental to animal welfare.]
Extent Information
E72This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F219Words in Sch. 3 para. 11 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(f)
F220Words in Sch. 3 para. 11(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(b)
F221Words in Sch. 3 para. 11(2) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(c)(ii)
F222Words in Sch. 3 para. 11(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(c)(i)
F223Word in Sch. 3 para. 11 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(a)
F224Words in Sch. 3 para. 11(2)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(c)(iii)
F225Words in Sch. 3 para. 11(2)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(c)(iv)
F226Words in Sch. 3 para. 11(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(d)(i)
F227Words in Sch. 3 para. 11(3)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(d)(ii)
F228Word in Sch. 3 Sch. 3 para. 11(3)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(d)(iii)(aa)
F229Words in Sch. 3 para. 11(3)(b) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(d)(iii)(bb)
F230Sch. 3 para. 11(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 105(e)
Supply of a veterinary medicinal product for incorporation into feedingstuffsN.I.
11.—(1) This paragraph applies in relation to the supply of a veterinary medicinal product intended for incorporation into feedingstuffs.
(2) The marketing authorisation holder, an authorised manufacturer of the product or an authorised wholesale dealer may only supply such a veterinary medicinal product to—
(a)a veterinary surgeon, pharmacist or, in the case of a product classified as POM-VPS, a suitably qualified person;
(b)an approved premixture manufacturer; or
(c)an approved feedingstuffs manufacturer if the approval permits the rate of incorporation specified on the label of that veterinary medicinal product (if the manufacturer is the end-user the supply must be in accordance with a prescription).
(3) A veterinary surgeon, pharmacist or, in the case of a product classified as POM-VPS, a suitably qualified person may only supply such a veterinary medicinal product to—
(a)an approved premixture manufacturer; or
(b)an approved feedingstuffs manufacturer if the approval permits the rate of incorporation specified on the label of that veterinary medicinal product (if the manufacturer is the end user the supply must be in accordance with a prescription).
(4) An approved premixture manufacturer or an approved feedingstuffs manufacturer may only supply such a veterinary medicinal product to another approved premixture manufacturer or approved feedingstuff manufacturer if the amount supplied does not exceed five per cent in terms of value of veterinary medicinal product incorporated annually by the person supplying the veterinary medicinal product.
Extent Information
E226This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Labelling at the time of retail supplyU.K.
12.—(1) If a veterinary medicinal product is supplied in a container specified in the marketing authorisation, it must not be supplied if any information on the outer packaging (or, if there is no outer packaging, the immediate packaging) is not clearly visible at the time of supply or has been changed in any way.
(2) Sub-paragraph (1) does not apply to a veterinary surgeon who amends a label, or a pharmacist who amends it in accordance with a prescription from a veterinary surgeon, provided that the unamended information remains clearly visible.
(3) If a veterinary medicinal product is supplied in a container other than that specified in the marketing authorisation, the person supplying the veterinary medicinal product must ensure that the container is suitably labelled and must supply sufficient written information (which may include a copy of the summary of product characteristics or the package leaflet) to enable the product to be used safely.
Supply of veterinary medicinal products for use under the cascadeE+W+S
13.—(1) A veterinary medicinal product supplied for administration under the cascade may only be supplied in accordance with a prescription from a veterinary surgeon.
(2) Unless the veterinary surgeon who prescribed the veterinary medicinal product both supplies the product and administers it to the animal in person, the person supplying it must label it (or ensure that it is labelled) with at least the following information—
(a)the name and address of the pharmacy, [F231veterinary practice premises] or [F232authorised] premises supplying the veterinary medicinal product;
(b)the name of the veterinary surgeon who has prescribed the product;
(c)the name and address of the animal owner;
(d)the identification (including the species) of the animal or group of animals;
(e)the date of supply;
(f)the expiry date of the product, if applicable;
(g)the name or description of the product, which should include at least the name and quantity of active ingredients;
(h)dosage and administration instructions;
(i)any special storage precautions;
(j)any necessary warnings for the user, target species, administration or disposal of the product;
(k)the withdrawal period, if relevant; and
(l)the words “Keep out of reach of children” and “For animal treatment only”.
Extent Information
E73This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F231Words in Sch. 3 para. 13(2)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 106(a)
F232Word in Sch. 3 para. 13(2)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 106(b)
Supply of veterinary medicinal products for use under the cascadeN.I.
13.—(1) A veterinary medicinal product supplied for administration under the cascade may only be supplied in accordance with a prescription from a veterinary surgeon.
(2) Unless the veterinary surgeon who prescribed the veterinary medicinal product both supplies the product and administers it to the animal in person, the person supplying it must label it (or ensure that it is labelled) with at least the following information—
(a)the name and address of the pharmacy, veterinary surgery or approved premises supplying the veterinary medicinal product;
(b)the name of the veterinary surgeon who has prescribed the product;
(c)the name and address of the animal owner;
(d)the identification (including the species) of the animal or group of animals;
(e)the date of supply;
(f)the expiry date of the product, if applicable;
(g)the name or description of the product, which should include at least the name and quantity of active ingredients;
(h)dosage and administration instructions;
(i)any special storage precautions;
(j)any necessary warnings for the user, target species, administration or disposal of the product;
(k)the withdrawal period, if relevant; and
(l)the words “Keep out of reach of children” and “For animal treatment only”.
Extent Information
E227This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Supply by a suitably qualified personE+W+S
14.—(1) The Secretary of State may recognise bodies that are suitable to maintain a register for suitably qualified persons to prescribe and supply veterinary medicinal products classified as POM-VPS and NFA-VPS.
(2) In order to recognise such a body, the Secretary of State must be satisfied that the body—
(a)has in place a system for ensuring that persons applying for registration have adequate training to act as a suitably qualified person under these Regulations;
(b)has adequate standards in deciding whether or not to register someone as a suitably qualified person;
(c)maintains a programme of continuing professional development for persons registered with it;
(d)operates an adequate appeal system if it intends to refuse to register anyone with appropriate qualifications or to remove anyone from the register.
(3) For the purposes of these Regulations, a suitably qualified person is a person who has passed examinations specified by such a body, and is registered with such a body as a suitably qualified person.
(4) A suitably qualified person may only supply a veterinary medicinal product classified as POM-VPS, NFA-VPS or AVM-GSL, and may only supply it from—
(a)premises [F233authorised] by the Secretary of State as being suitable for the storage and supply of veterinary medicinal products by a suitably qualified person;
(b)premises registered as a pharmacy with the General Pharmaceutical Council or with the Pharmaceutical Society of Northern Ireland; or
(c)practice premises registered under these Regulations as being premises from which a veterinary surgeon supplies veterinary medicinal products.
[F234(5) A suitably qualified person who supplies a product classified as POM-VPS or NFA-VPS must be present when it is handed over unless the suitably qualified person—
(a)authorises each transaction individually before the product is supplied; and
(b)is satisfied that the person handing it over is competent to do so.]
(6) A suitably qualified person supplying products from premises [F233authorised] under this regulation by the Secretary of State who considers that the premises no longer comply with the [F235authorisation] must notify the Secretary of State without unreasonable delay.
(7) The Secretary of State may issue a Code of Practice for suitably qualified persons [F236and bodies recognised under this paragraph], and a body recognised under this paragraph must take appropriate action in accordance with any disciplinary code that applies to that body if a suitably qualified person registered with it does not comply with the Code of Practice.
(8) The Secretary of State must publish a list of—
(a)suitably qualified persons; and
(b)the trading names and the addresses of premises [F233authorised] under this paragraph(6).
(9) A suitably qualified person may break open any package (other than the immediate packaging) of a veterinary medicinal product.
(10) The Secretary of State may suspend or revoke the [F235authorisation] of [F233authorised] premises on being satisfied that they are no longer suitable for the storage and supply of veterinary medicinal products.
[F237(11) The Secretary of State must, from time to time, inspect premises authorised under sub-paragraph (4)(a) basing the frequency of the inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
(12) The Secretary of State may suspend or revoke recognition of a body mentioned in sub-paragraph (1) where the body fails to comply with a provision of any Code of Practice issued under this paragraph.]
Extent Information
E74This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F233Word in Sch. 3 para. 14 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 107(a)
F234Sch. 3 para. 14(5) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 107(c)
F235Word in Sch. 3 para. 14 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 107(b)
F236Words in Sch. 3 para. 14(7) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 107(d)
F237Sch. 3 para. 14(11)(12) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 107(e)
Supply by a suitably qualified personN.I.
14.—(1) The Secretary of State may recognise bodies that are suitable to maintain a register for suitably qualified persons to prescribe and supply veterinary medicinal products classified as POM-VPS and NFA-VPS.
(2) In order to recognise such a body, the Secretary of State must be satisfied that the body—
(a)has in place a system for ensuring that persons applying for registration have adequate training to act as a suitably qualified person under these Regulations;
(b)has adequate standards in deciding whether or not to register someone as a suitably qualified person;
(c)maintains a programme of continuing professional development for persons registered with it;
(d)operates an adequate appeal system if it intends to refuse to register anyone with appropriate qualifications or to remove anyone from the register.
(3) For the purposes of these Regulations, a suitably qualified person is a person who has passed examinations specified by such a body, and is registered with such a body as a suitably qualified person.
(4) A suitably qualified person may only supply a veterinary medicinal product classified as POM-VPS, NFA-VPS or AVM-GSL, and may only supply it from—
(a)premises approved by the Secretary of State as being suitable for the storage and supply of veterinary medicinal products by a suitably qualified person;
(b)premises registered as a pharmacy with the General Pharmaceutical Council or with the Pharmaceutical Society of Northern Ireland; or
(c)practice premises registered under these Regulations as being premises from which a veterinary surgeon supplies veterinary medicinal products.
(5) A suitably qualified person who supplies a product classified as POM-VPS or NFA-VPS must either—
(a)hand over or despatch the product personally;
(b)ensure that, when the product is handed over or despatched, the suitably qualified person is in a position to intervene if necessary; or
(c)check the product after it has been allocated for supply to a customer, and be satisfied that the person handing over or dispatching it is competent to do so.
(6) A suitably qualified person supplying products from premises approved under this regulation by the Secretary of State who considers that the premises no longer comply with the approval must notify the Secretary of State without unreasonable delay.
(7) The Secretary of State may issue a Code of Practice for suitably qualified persons, and a body recognised under this paragraph must take appropriate action in accordance with any disciplinary code that applies to that body if a suitably qualified person registered with it does not comply with the Code of Practice.
(8) The Secretary of State must publish a list of—
(a)suitably qualified persons; and
(b)the trading names and the addresses of premises approved under this paragraph(6).
(9) A suitably qualified person may break open any package (other than the immediate packaging) of a veterinary medicinal product.
(10) The Secretary of State may suspend or revoke the approval of approved premises on being satisfied that they are no longer suitable for the storage and supply of veterinary medicinal products.
Extent Information
E228This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F238AuditE+W+S
15.—(1) At least once a year, a retailer of prescription only veterinary medicinal products must carry out a detailed audit of stock and compare the incoming and outgoing veterinary medicinal products recorded with products currently held and make a record of this audit.
(2) Where, as a result of the audit mentioned in sub-paragraph (1), the retailer identifies a discrepancy the retailer must make a record of the fact.
(3) The retailer must keep the records mentioned in sub-paragraphs (1) and (2) for a period of five years from the date of the audit and the Secretary of State may require the retailer to provide a copy of them at any time within that period.]
Extent Information
E75This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F238Sch. 3 para. 15 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 108
Annual auditN.I.
15. At least once a year every person entitled to supply a veterinary medicinal product on prescription must carry out a detailed audit, and incoming and outgoing veterinary medicinal products must be reconciled with products currently held in stock, any discrepancies being recorded.
Extent Information
E229This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
PART 2U.K.Requirements for a wholesale dealer’s authorisation
[F239Wholesale dealer’s authorisationE+W+S
16. No person may carry out any wholesale dealing in veterinary medicinal products otherwise than in accordance with an authorisation granted under paragraph 18(2) (a “wholesale dealer’s authorisation”).]
Extent Information
E76This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F239Sch. 3 para. 16 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 109
ApplicationN.I.
16. An application for a wholesale dealer’s authorisation must be made to the Secretary of State.
Extent Information
E230This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F240Application for authorisationE+W+S
17.—(1) An application for a wholesale dealer’s authorisation (which must be submitted to the Secretary of State electronically) must include the matters mentioned in sub-paragraph (2).
(2) For the purposes of sub-paragraph (1) the matters are—
(a)the name of the person who will hold the wholesale dealer’s authorisation and that person’s address or registered place of business;
(b)the names and addresses of the sites from which wholesale dealing of veterinary medicinal products is to take place;
(c)evidence that the sites mentioned in paragraph (b) are—
(i)weatherproof;
(ii)secure and lockable;
(iii)clean;
(iv)free from contaminants;
(v)designed with designated areas for the receipt of veterinary medicinal products; and
(vi)where the veterinary medicinal products for which the authorisation is sought are subject to specific storage requirements, capable of fulfilling those requirements;
(d)the name of the person nominated to act in accordance with good distribution practice (the “wholesale qualified person”);
(e)the qualifications and a description of the relevant experience of the wholesale qualified person;
(f)a description of the veterinary medicinal products proposed to be dealt in under the authorisation;
(g)evidence that the proposed holder of the authorisation has available to it the services of technically competent staff;
(h)evidence that the proposed holder of the authorisation has in place—
(i)an effective emergency recall plan; and
(ii)a quality system;
(i)a declaration that the applicant complies with good distribution practice and any relevant legislation;
(j)a declaration that any site mentioned in paragraph (b) is ready for inspection.]
Extent Information
E77This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F240Sch. 3 para. 17 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 110
Time limitsN.I.
17. The Secretary of State must process an application for a wholesale dealer’s authorisation within 90 days of receiving it.
Extent Information
E231This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F241Procedure and time limits for authorisationsE+W+S
18.—(1) The Secretary of State must inspect the sites mentioned in paragraph 17(2)(b) within 90 days of validating the application.
(2) Where the Secretary of State is satisfied, following the inspection mentioned in sub-paragraph (1) that—
(a)the sites are suitable for the intended purposes; and
(b)the applicant has—
(i)suitable and sufficient staff and facilities for the storage of veterinary medicinal products; and
(ii)a documented quality system in place,
the Secretary of State must grant the wholesale dealer’s authorisation.
(3) Where the Secretary of State is not satisfied in relation to one or more of the matters mentioned in sub-paragraph (2), the Secretary of State may—
(a)reject the application; or
(b)grant a conditional wholesale dealer’s authorisation for a period specified by the Secretary of State until the deficiency has been addressed.
(4) The Secretary of State may extend the period for which a conditional wholesale dealer’s authorisation is granted under sub-paragraph (3)(b).
(5) Where a conditional wholesale dealer’s authorisation is granted under sub-paragraph (3)(b) and the deficiency is addressed within the specified period to the satisfaction of the Secretary of State, the authorisation continues to have effect without those conditions.]
Extent Information
E78This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F241Sch. 3 para. 18 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 111
Granting the authorisationN.I.
18.—(1) The Secretary of State must grant a wholesale dealer’s authorisation on being satisfied that this paragraph is complied with.
(2) The authorised site must be—
(a)weatherproof;
(b)secure and lockable;
(c)clean; and
(d)free from contaminants.
(3) If the veterinary medicinal products covered by the authorisation are subject to specific storage conditions, the site must be capable of fulfilling those requirements.
(4) The authorisation holder must—
(a)have the services of technically competent staff; and
(b)have an effective emergency recall plan.
Extent Information
E232This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F242Periodic inspections and suspension etc. for lack of useE+W+S
19.—(1) The Secretary of State must, from time to time, inspect the sites from which wholesale dealing of veterinary medicinal products takes place pursuant to a wholesale dealer’s authorisation basing the frequency of the inspection on the risks associated with each site’s history and the nature of the products handled at the site.
(2) The Secretary of State may suspend, vary or revoke a wholesale dealer’s authorisation if, in respect of any one of the sites covered by that authorisation, the holder does not deal in veterinary medicinal products from that site for five years.]
Extent Information
E79This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F242Sch. 3 para. 19 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 112
The authorisationN.I.
19.—(1) The wholesale dealer’s authorisation must specify—
(a)the types of veterinary medicinal products and pharmaceutical forms that may be dealt in;
(b)the place where they are to be stored;
(c)the name and address of the person holding the authorisation;
(d)the address of the premises to which it relates;
(e)the name of the qualified person nominated to act under the Guidelines on Good Distribution Practice for Human Use(7).
(2) It may cover more than one site.
(3) It lapses if the holder does not deal in veterinary medicinal products for five years.
(4) The holder of a wholesale dealer’s authorisation must notify the Secretary of State, and if necessary apply for a variation of the authorisation, before making a material alteration to the premises or facilities used under the authorisation, or in the operations for which they are used.
Extent Information
E233This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F243Application for variation to the authorisationE+W+S
19A.—(1) The holder of a wholesale dealer’s authorisation must notify the Secretary of State, and apply for a variation of the authorisation, before making a material alteration to the premises or facilities used under the authorisation or the operations for which the premises or facilities are used or where there is a change in the personnel carrying out the role of wholesale qualified person.
(2) The Secretary of State must process an application under sub-paragraph (1) within 30 days of receiving it unless the Secretary of State notifies the applicant in writing that the time has been extended to 90 days.
(3) The Secretary of State must grant the application under sub-paragraph (1) if satisfied in respect of the matters in paragraph 18(2) as regards the proposed variation.
(4) The Secretary of State may inspect any site to which the wholesale dealer’s authorisation or proposed variation relates in connection with the application.
(5) Where the Secretary of State is not satisfied for the purposes of sub-paragraph (3), the Secretary of State may—
(a)reject the application; or
(b)grant a conditional variation to the wholesale dealer’s authorisation for a period specified by the Secretary of State until the deficiency has been addressed.
(6) The Secretary of State may extend the period for which a conditional variation to the wholesale dealer’s authorisation is granted under sub-paragraph (5)(b).
(7) Where a conditional variation to the wholesale dealer’s authorisation is granted under sub-paragraph (5)(b) and the deficiency is addressed within the specified period to the satisfaction of the Secretary of State, the authorisation continues to have effect as so varied without those conditions.]
Textual Amendments
F243Sch. 3 para. 19A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 113
Suspension, variation or revocation of the authorisationE+W+S
20. The Secretary of State may suspend, vary or revoke a wholesale dealer’s authorisation if the holder—
(a)has not complied with these Regulations; or
[F244(b)no longer has suitable premises, equipment or technically competent staff]
Extent Information
E80This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F244Sch. 3 para. 20(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 114
Suspension, variation or revocation of the authorisationN.I.
20. The Secretary of State may suspend, vary or revoke a wholesale dealer’s authorisation if the holder—
(a)has not complied with these Regulations; or
(b)no longer has suitable premises or equipment.
Extent Information
E234This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Duties on the holder of a wholesale dealer’s authorisationE+W+S
21. The holder of a wholesale dealer’s authorisation must—
(a)store veterinary medicinal products in accordance with the terms of the marketing authorisation for each product;
[F245(b)comply with good distribution practice;]
F246(c). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(d)supply information and samples to the Secretary of State on demand [F247; and
(e)notify the Secretary of State (and in relation to paragraph (ii), the holder of the relevant marketing authorisation) where it has reason to suspect—
(i)a threat to the continued supply of a veterinary medicinal product;
(ii)that it has been offered veterinary medicinal products which are counterfeit].
Extent Information
E81This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F245Sch. 3 para. 21(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 115(a)
F246Sch. 3 para. 21(c) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 115(b)
F247Sch. 3 para. 21(e) and word inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 115(c)
Duties on the holder of a wholesale dealer’s authorisationN.I.
21. The holder of a wholesale dealer’s authorisation must—
(a)store veterinary medicinal products in accordance with the terms of the marketing authorisation for each product;
(b)comply with the Guidelines on Good Distribution Practice of Medicinal Products for Human Use as if the veterinary medicinal products were authorised human medicinal products;
(c)carry out a detailed stock audit at least once a year; and
(d)supply information and samples to the Secretary of State on demand.
Extent Information
E235This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F248Register of authorised wholesale dealersE+W+S
21A. The Secretary of State must establish, maintain and publish on a website a register of authorised wholesale dealers and their sites.]
Textual Amendments
F248Sch. 3 paras. 21A-21F inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 116
[F248Documentation accompanying veterinary medicinal products supplied wholesaleE+W+S
21B.—(1) This paragraph applies in relation to wholesale supply of veterinary medicinal products.
(2) The holder of a wholesale dealer’s authorisation must ensure that a document accompanies each consignment of veterinary medicinal products specifying—
(a)the name of the veterinary medicinal product;
(b)the strength and pharmaceutical form;
(c)the date on which the veterinary medicinal product was supplied;
(d)the quantity of product supplied;
(e)the batch number;
(f)the expiry date;
(g)the name and address of the wholesale dealer supplying the product;
(h)the means by which the product was transported and the required conditions of storage;
(i)the name of the person to whom the product was supplied and the address to which it is to be delivered.
(3) The holder of a wholesale dealer’s authorisation must make a record of the information mentioned in sub-paragraph (2) and must keep it for at least five years.]
Textual Amendments
F248Sch. 3 paras. 21A-21F inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 116
[F248Recalled, counterfeit or returned productsE+W+S
21C.—(1) The holder of a wholesale dealer’s authorisation must comply with any requirement by the Secretary of State to recall a veterinary medicinal product and must record the details of the recall operation.
(2) The holder of a wholesale dealer’s authorisation must record any veterinary medicinal product which is—
(a)recalled (whether or not the holder physically receives the recalled product);
(b)discovered to be counterfeit; or
(c)returned.
(3) Where any veterinary medicinal product is recalled or returned and physically received, the wholesale qualified person must assess the product received in order to determine whether the product has been stored (including during transport) in accordance with the summary of product characteristics.
(4) Where a recalled or returned veterinary medicinal product has not been stored (including during transport) in accordance with the summary of product characteristics or where it is not possible for the wholesale qualified person to determine whether the product has been stored in accordance with the summary of product characteristics, the product may not be re-sold.
(5) Any veterinary medicinal products which may not be re-sold must be identified, held separately and destroyed and the holder of a wholesale dealer’s authorisation must develop a suitable procedure to set out the steps to be taken in accordance with this sub-paragraph.
(6) The holder of a wholesale dealer’s authorisation must keep any information recorded under this paragraph for five years.]
Textual Amendments
F248Sch. 3 paras. 21A-21F inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 116
[F248AuditE+W+S
21D.—(1) At least once a year, the holder of a wholesale dealer’s authorisation must carry out a detailed audit of stock and compare the incoming and outgoing veterinary medicinal products recorded with products currently held and record the results of the audit in written form.
(2) Where, as a result of the audit mentioned in sub-paragraph (1), the holder identifies a discrepancy the holder must—
(a)make a record of that fact,
(b)conduct an investigation for the purpose of discovering the cause of the discrepancy, and
(c)maintain records of that investigation.
(3) The holder must keep the records mentioned in sub-paragraphs (1) and (2) for a period of five years from the date of the audit and the Secretary of State may require the holder to provide a copy of them at any time within that period.]
Textual Amendments
F248Sch. 3 paras. 21A-21F inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 116
[F248Contractual arrangements between holders of wholesale dealer’s authorisationsE+W+S
21E. Where the holder of a wholesale dealer’s authorisation contracts out any wholesale dealing activities to another such holder, the arrangement must record in writing the responsibilities of each party in relation to their respective roles in the supply process and, in particular, in connection with the recall of a veterinary medicinal product under paragraph 21C.]
Textual Amendments
F248Sch. 3 paras. 21A-21F inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 116
[F248Self-inspection programmeE+W+S
21F.—(1) The holder of a wholesale dealer’s authorisation must have in place a self-inspection programme which ensures that every aspect of its business is inspected at least once a year in order to ensure that it is complying with good distribution practice.
(2) Where, as a result of the self-inspection mentioned in sub-paragraph (1), the holder identifies any non-compliance the holder must—
(a)make a record of that fact,
(b)conduct an investigation for the purpose of discovering the cause of the non-compliance, and
(c)maintain records of that investigation.
(3) The holder must keep the records mentioned in sub-paragraph (2) for a period of five years from the date of the audit and the Secretary of State may require the holder to provide a copy of them at any time within that period.]
Textual Amendments
F248Sch. 3 paras. 21A-21F inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 116
PART 3U.K.Sheep dip
Supply of sheep dipU.K.
22.—(1) A person who supplies by retail sheep dip which contains a veterinary medicinal product must supply it in accordance with this paragraph.
(2) The supply must be to a person (or a person acting on that person’s behalf) who is qualified to use it in accordance with paragraph 23.
(3) The supplier must make a record of that person’s certificate or award number as soon as is reasonably practicable, and keep it for at least three years.
(4) If the active ingredient of the veterinary medicinal product is an organophosphorus compound, the supplier must give to the buyer—
(a)a double-sided laminated notice meeting the specifications in the following sub-paragraph (unless the notice has been provided to the buyer within the previous twelve months and the supplier knows or has reasonable cause to believe that the buyer still has it available for use); and
(b)two pairs of gloves either as described in the notice or providing demonstrably superior protection to the proposed user against exposure to the dip than would be provided by gloves as so described.
(5) The notice must be at least A4 size with a laminated transparent cover and must tell the user of the sheep dip—
(a)to read and act in accordance with the label, including instructions on measuring and diluting concentrate;
(b)that sheep dip is absorbed through the skin;
(c)always to wear the recommended protective clothing, including gloves, and have spare protective clothing available;
(d)always to wash protective clothing before taking it off; and
(e)to direct any questions to the supplier or manufacturer.
(6) The notice must contain a diagram showing recommended protective clothing.
Use of sheep dipE+W+S
23.—(1) [F249No person may use sheep dip which contains a veterinary medicinal product unless they hold, or they are acting under the supervision and in the presence of a person who holds, either]—
(a)a Certificate of Competence in the Safe Use of Sheep Dips showing that Parts 1 and 2 or units 1 and 2 of the assessment referred to in the Certificate have been satisfactorily completed; or
(b)NPTC Level 2 Award in the Safe Use of Sheep Dip (QCF).
(2) The certificate must be issued—
(a)in England, Wales and Northern Ireland; by—
(i)the National Proficiency Tests Council;
(ii)NPTC Part of the City & Guilds Group; or
(iii)City and Guilds NPTC;
(b)in Scotland, by one of those organisations or the Scottish Skills Testing Service.
Extent Information
E82This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F249Words in Sch. 3 para. 23(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 117
Use of sheep dipN.I.
23.—(1) No person may use sheep dip which contains a veterinary medicinal product unless the person is acting under the supervision and in the presence of, a person who holds either—
(a)a Certificate of Competence in the Safe Use of Sheep Dips showing that Parts 1 and 2 or units 1 and 2 of the assessment referred to in the Certificate have been satisfactorily completed; or
(b)NPTC Level 2 Award in the Safe Use of Sheep Dip (QCF).
(2) The certificate must be issued—
(a)in England, Wales and Northern Ireland; by—
(i)the National Proficiency Tests Council;
(ii)NPTC Part of the City & Guilds Group; or
(iii)City and Guilds NPTC;
(b)in Scotland, by one of those organisations or the Scottish Skills Testing Service.
Extent Information
E236This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
OffencesE+W+S
24. It is an offence to fail to comply with—
(a)paragraph 2;
(b)paragraph 3;
[F250(ba)paragraph 3A;
(bb)paragraph 3B;
(bc)paragraph 3C;
(bd)paragraph 3D;
(be)paragraph 3E;]
(c)paragraph 4(1);
(d)paragraph 5;
[F251(da)paragraph 6;]
(e)paragraph 7;
[F252(ea)paragraph 7A;]
(f)paragraph 8(1);
(g)paragraph 9(1);
(h)paragraph 10;
(i)paragraph 11;
(j)paragraph 12(1) or (3);
(k)paragraph 13;
(l)paragraph 14(4), (5) or (6);
(m)paragraph 15;
[F253(ma)paragraph 16;]
F254(n). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(o)paragraph 21;
[F255(oa)paragraph 21B;
(ob)paragraph 21C;
(oc)paragraph 21D;
(od)paragraph 21E;
(oe)paragraph 21F;]
(p)paragraph 22; or
(q)paragraph 23(1).
Extent Information
E83This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F250Sch. 3 paras. 24(ba)-(be) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 118(a)
F251Sch. 3 para. 24(da) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 118(b)
F252Sch. 3 para. 24(ea) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 118(c)
F253Sch. 3 para. 24(ma) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 118(d)
F254Sch. 3 para. 24(n) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 118(e)
F255Sch. 3 paras. 24(oa)-(oe) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 118(f)
OffencesN.I.
24. It is an offence to fail to comply with—
(a)paragraph 2;
(b)paragraph 3;
(c)paragraph 4(1);
(d)paragraph 5;
(e)paragraph 7;
(f)paragraph 8(1);
(g)paragraph 9(1);
(h)paragraph 10;
(i)paragraph 11;
(j)paragraph 12(1) or (3);
(k)paragraph 13;
(l)paragraph 14(4), (5) or (6);
(m)paragraph 15;
(n)paragraph 19(4);
(o)paragraph 21;
(p)paragraph 22; or
(q)paragraph 23(1).
Extent Information
E237This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Regulation 8
SCHEDULE 4U.K.Administration of a veterinary medicinal product outside the terms of a marketing authorisation
Administration under the cascadeE+W+S
1.—(1) A veterinary surgeon acting under this paragraph who prescribes a veterinary medicinal product may either administer it personally or may direct another person to do so under the responsibility of the veterinary surgeon.
(2) If there is no authorised veterinary medicinal product in the United Kingdom for a condition the veterinary surgeon responsible for the animal may, in particular to avoid unacceptable suffering, treat the animal concerned with the following (“the cascade”), cascaded in the following order—
(a)a veterinary medicinal product authorised in the United Kingdom for use with another animal species, or for another condition in the same species; or
(b)if there is no such product that is suitable, either—
(i)a human medicinal product authorised in the United Kingdom; or
(ii)a veterinary medicinal product not authorised in the United Kingdom but authorised in another [F256country] for use with any animal species (in the case of a food-producing animal, it must be a food-producing species); or
(c)if there is no such product that is suitable, a veterinary medicinal product prepared extemporaneously by a pharmacist, a veterinary surgeon or a person holding a manufacturing authorisation authorising the manufacture of that type of product.
(3) In the case of a veterinary medicinal product imported from another [F257country], if the veterinary surgeon has not obtained a certificate from the Secretary of State under regulation 25(5) permitting importation, the veterinary surgeon must obtain a certificate from the Secretary of State before administration.
(4) [F258All substances] included in a medicinal product administered to a food-producing animal under the cascade must [F259be substances for which a maximum residue limit has been established under Regulation (EC) No 470/2009 of the European Parliament and of the Council] [F260or substances which do not fall within the scope of Regulation (EC) No 470/2009 of the European Parliament and of the Council].
[F261(5) Where a substance mentioned in sub-paragraph (4) is administered, the maximum residue limits established in accordance with Regulation (EC) No 470/2009 of the European Parliament and of the Council must be complied with.]
Extent Information
E84This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F256Word in Sch. 4 para. 1(2)(b)(ii) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(a)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F257Word in Sch. 4 para. 1(3) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(a)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F258Words in Sch. 4 para. 1(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 120(a)(i)
F259Words in Sch. 4 para. 1(4) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(a)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F260Words in Sch. 4 para. 1(4) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 120(a)(ii)
F261Sch. 4 para. 1(5) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 120(b)
Administration under the cascadeN.I.
1.—(1) A veterinary surgeon acting under this paragraph who prescribes a veterinary medicinal product may either administer it personally or may direct another person to do so under the responsibility of the veterinary surgeon.
(2) If there is no authorised veterinary medicinal product in [F600Northern Ireland] for a condition the veterinary surgeon responsible for the animal may, in particular to avoid unacceptable suffering, treat the animal concerned with the following (“the cascade”), cascaded in the following order—
(a)a veterinary medicinal product authorised in [F600Northern Ireland] for use with another animal species, or for another condition in the same species; or
(b)if there is no such product that is suitable, either—
(i)a human medicinal product authorised in the United Kingdom; or
(ii)a veterinary medicinal product not authorised in [F600Northern Ireland] but authorised in another member State for use with any animal species (in the case of a food-producing animal, it must be a food-producing species); or
(c)if there is no such product that is suitable, a veterinary medicinal product prepared extemporaneously by a pharmacist, a veterinary surgeon or a person holding a manufacturing authorisation authorising the manufacture of that type of product.
(3) In the case of a veterinary medicinal product imported from another member State, if the veterinary surgeon has not obtained a certificate from the Secretary of State under regulation 25(5) permitting importation, the veterinary surgeon must obtain a certificate from the Secretary of State before administration.
(4) Any pharmacologically active substances included in a medicinal product administered to a food-producing animal under the cascade must be listed in Table 1 in the Annex to Commission Regulation (EU) No 37/2010.
Extent Information
E238This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F600Words in Sch. 4 para. 1 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(a)
Withdrawal periodsE+W+S
2.—(1) A veterinary surgeon prescribing or administering a veterinary medicinal product to a food-producing animal under the cascade must specify an appropriate withdrawal period.
[F262(2) The withdrawal period must ensure that—
(a)where there is a maximum residue limit established for the active substance for the treated species under Regulation (EC) No 470/2009 of the European Parliament and of the Council, the level of residue of the active substance does not exceed that limit; and
(b)where there is no maximum residue limit for the treated species established under Regulation (EC) No 470/2009 of the European Parliament and of the Council but one is established for the substance itself, the level of residue of the active substance does not exceed the level determined by reference to Commission Implementing Regulation (EU) 2018/470 on detailed rules on the maximum residue limit to be considered for control purposes for foodstuffs derived from animals which have been treated in the EU under Article 11 of Directive 2001/82/EC.]
(3) In any event, unless the Secretary of State has specified in writing a different withdrawal period for a particular veterinary medicinal product, the withdrawal period (irrespective of whether or not a maximum residue limit [F263has been established]) must not be less than—
[F264(a)for eggs—
(i)the longest withdrawal period in the summary of product characteristics for any species multiplied by a factor of 1.5; or
(ii)14 days, if the product is not authorised for animals producing eggs for human consumption;]
[F265(b)for milk—
(i)the longest withdrawal period in the summary of product characteristics for any species multiplied by a factor of 1.5;
(ii)7 days, if the veterinary medicinal product is not authorised for animals producing milk for human consumption; or
(iii)1 day, if the medicinal product has a zero-hour withdrawal period;]
[F266(c)for meat and offal from food-producing mammals, poultry and farmed game-birds—
(i)the longest withdrawal period provided in its summary of product characteristics for meat and offal, multiplied by a factor of 1.5;
(ii)28 days if the veterinary medicinal product is not authorised for food-producing animals; or
(iii)1 day, if the veterinary medicinal product has a zero-day withdrawal period;]
[F267(d)for aquatic species producing meat for human consumption—
(i)the longest withdrawal period for any of the aquatic species in the summary of product characteristics multiplied by a factor of 1.5 and expressed as degree-days;
(ii)if the medicinal product is authorised for food-producing terrestrial animal species, the longest withdrawal period for any of the food-producing animal species in the summary of product characteristics multiplied by a factor of 50 and expressed as degree-days; or
(iii)25 degree-days if the highest withdrawal period for any animal species is zero.]
[F268(4) For the purposes of sub-paragraph (3)—
(a)if the calculation of a withdrawal period results in a fraction of days, the withdrawal period must be rounded to the nearest number of days, with any half of a day being rounded upwards;
(b)in relation to the calculation of the withdrawal period for milk, if the calculation of the period results in a milk withdrawal period not divisible by 12, the withdrawal period must be rounded up to the nearest multiple of 12 hours.]
Extent Information
E85This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F262Sch. 4 para. 2(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 121(a)
F263Words in Sch. 4 para. 2(3) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F264Sch. 4 para. 2(3)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 121(b)(i)
F265Sch. 4 para. 2(3)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 121(b)(ii)
F266Sch. 4 para. 2(3)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 121(b)(iii)
F267Sch. 4 para. 2(3)(d) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 121(b)(iv)
F268Sch. 4 para. 2(4) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 121(c)
Withdrawal periodsN.I.
2.—(1) A veterinary surgeon prescribing or administering a veterinary medicinal product to a food-producing animal under the cascade must specify an appropriate withdrawal period.
(2) The withdrawal period must ensure that, if there is a maximum residue limit specified for the active substance in Table 1 in the Annex to Commission Regulation (EU) No 37/2010, the level of residue of the active substance does not exceed that limit.
(3) In any event, unless the Secretary of State has specified in writing a different withdrawal period for a particular veterinary medicinal product, the withdrawal period (irrespective of whether or not a maximum residue limit is specified in Table 1 in the Annex to Commission Regulation (EU) No 37/2010) must not be less than—
(a)7 days for eggs;
(b)7 days for milk;
(c)28 days for meat from poultry and mammals including fat and offal;
(d)500 degree days(8) for fish meat.
Extent Information
E239This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Administration to food-producing horsesU.K.
3.—(1) If there is no authorised veterinary medicinal product for a food-producing horse (as shown on its horse passport) and treatment under the cascade is unsuitable, substances may be administered in accordance with Commission Regulation (EC) No 122/2013 (establishing, in accordance with Directive 2001/82/EC of the European Parliament and of the Council on the Community code relating to veterinary medicinal products, a list of substances essential for the treatment of equidae (9)).
(2) The person administering the substance must comply with Article 3(2) of Commission Regulation (EC) No 122/2013 (recording the details of the treatment in the animal’s passport).
Immunological products for serious epizootic disease [F269or emerging disease]E+W+S
4. In the event of serious epizootic diseases [F270or emerging diseases], the Secretary of State may permit in writing the administration of immunological veterinary medicinal products without a marketing authorisation, in the absence of a suitable medicinal product F271... and may publicise any permit as the Secretary of State sees fit.
Extent Information
E86This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F269Words in Sch. 4 para. 4 heading inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 122(b)
F270Words in Sch. 4 para. 4 inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 122(a)
F271Words in Sch. 4 para. 4 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(c) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Immunological products for serious epizootic diseaseN.I.
4. In the event of serious epizootic diseases, the Secretary of State may permit in writing the administration of immunological veterinary medicinal products without a marketing authorisation, in the absence of a suitable medicinal product and after informing the Commission of the detailed conditions of use and may publicise any permit as the Secretary of State sees fit.
Extent Information
E240This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Immunological products for an imported or exported animalE+W+S
5. If an animal is imported from, or exported to, [F272another] country, the Secretary of State may permit the administration to that animal of an immunological veterinary medicinal product that is not covered by a marketing authorisation in the United Kingdom but is authorised under the legislation of [F273that other] country.
Extent Information
E87This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F272Word in Sch. 4 para. 5 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(d)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F273Words in Sch. 4 para. 5 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(d)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Immunological products for an imported or exported animalN.I.
5. If an animal is imported from, or exported to, a third country, the Secretary of State may permit the administration to that animal of an immunological veterinary medicinal product that is not covered by a marketing authorisation in [F601Northern Ireland] but is authorised under the legislation of the third country.
Extent Information
E241This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F601Words in Sch. 4 para. 5 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(b)
Administration by veterinary surgeons from other [F274countries] E+W+S
6.—(1) Veterinary surgeons practising in another [F275country with equivalent medicines regulation standards to those of the United Kingdom] may bring into the United Kingdom and administer to animals small quantities of veterinary medicinal products that are not authorised for use in the United Kingdom if—
(a)the quantity does not exceed the requirements for the treatment of specific animals;
(b)the product is authorised in the [F276country] in which the veterinary surgeon is established;
(c)the product is transported by the veterinary surgeon in the original manufacturer’s packaging;
(d)in the case of administration to food-producing animals, there is a veterinary medicinal product authorised in the United Kingdom that has the same qualitative and quantitative composition in terms of active substances;
(e)the veterinary surgeon is acquainted with the Code of Professional Conduct for veterinary surgeons issued by the Royal College of Veterinary Surgeons(10).
(2) The veterinary surgeon must only supply to the owner or keeper enough veterinary medicinal product to complete the treatment of animals concerned.
(3) The veterinary surgeon must—
(a)ensure that the withdrawal period specified on the label of the product is complied with, or the United Kingdom withdrawal period for the equivalent product authorised in the United Kingdom if this is longer than the one on the label; and
(b)keep detailed records of the animals treated, the diagnosis or clinical assessment, the products administered, the dosage administered, the duration of treatment and the withdrawal period applied, and must keep them in the United Kingdom for at least three years.
(4) The overall range and quantity of veterinary medicinal products carried by the veterinary surgeon must not exceed that generally required for the daily needs of good veterinary practice.
(5) This paragraph does not apply in relation to immunological veterinary medicinal products.
Extent Information
E88This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F274Word in Sch. 4 para. 6 heading substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(e)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F275Words in Sch. 4 para. 6(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(e)(ii)(aa) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F276Word in Sch. 4 para. 6(1)(b) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(e)(ii)(bb) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Administration by veterinary surgeons from F602... member StatesN.I.
6.—(1) Veterinary surgeons practising in [F603a] member State may bring into [F604Northern Ireland] and administer to animals small quantities of veterinary medicinal products that are not authorised for use in [F604Northern Ireland] if—
(a)the quantity does not exceed the requirements for the treatment of specific animals;
(b)the product is authorised in the member State in which the veterinary surgeon is established;
(c)the product is transported by the veterinary surgeon in the original manufacturer’s packaging;
(d)in the case of administration to food-producing animals, there is a veterinary medicinal product authorised in [F605Northern Ireland] that has the same qualitative and quantitative composition in terms of active substances;
(e)the veterinary surgeon is acquainted with the Code of Professional Conduct for veterinary surgeons issued by the Royal College of Veterinary Surgeons(10).
(2) The veterinary surgeon must only supply to the owner or keeper enough veterinary medicinal product to complete the treatment of animals concerned.
(3) The veterinary surgeon must—
[F606(a)ensure that the withdrawal period specified on the label of the product is complied with, or the Northern Ireland withdrawal period for the equivalent product authorised in Northern Ireland if this is longer than the one on the label; and]
(b)keep detailed records of the animals treated, the diagnosis or clinical assessment, the products administered, the dosage administered, the duration of treatment and the withdrawal period applied, and must keep them in [F607Northern Ireland] for at least three years.
(4) The overall range and quantity of veterinary medicinal products carried by the veterinary surgeon must not exceed that generally required for the daily needs of good veterinary practice.
(5) This paragraph does not apply in relation to immunological veterinary medicinal products.
Extent Information
E242This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F602Word in Sch. 4 para. 6 heading omitted (N.I.) (31.12.2020) by virtue of The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(i)
F603Word in Sch. 4 para. 6(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(ii)
F604Words in Sch. 4 para. 6(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(iii)
F605Words in Sch. 4 para. 6(1)(d) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(iii)
F606Sch. 4 para. 6(3)(a) substituted (N.I) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(iv)
F607Words in Sch. 4 para. 6(3)(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(14)(c)(iii)
[F277Administration of autogenous vaccinesE+W+S
6A.—(1) An autogenous vaccine may only be administered to animals in exceptional circumstances where no suitable immunological veterinary medicinal product has been authorised in relation to the target species and indication.
(2) Where a vaccine is used in accordance with sub-paragraph (1) it must be administered in accordance with a prescription under the cascade.]
Textual Amendments
F277Sch. 4 para. 6A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 123
Treatment in exceptional circumstancesE+W+S
7.—(1) Where the health situation so requires, and where there is no suitable veterinary medicinal product available either as an authorised product or under the cascade, a veterinary surgeon may treat an animal with a medicinal product authorised in [F278another] country; but a veterinary surgeon who has not obtained a certificate from the Secretary of State under regulation 25(5) permitting importation must obtain a certificate from the Secretary of State before treating the animal.
(2) The certificate may be granted subject to any condition the Secretary of State thinks fit.
Extent Information
E89This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F278Word in Sch. 4 para. 7(1) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(34)(f) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Treatment in exceptional circumstancesN.I.
7.—(1) Where the health situation so requires, and where there is no suitable veterinary medicinal product available either as an authorised product or under the cascade, a veterinary surgeon may treat an animal with a medicinal product authorised in a third country; but a veterinary surgeon who has not obtained a certificate from the Secretary of State under regulation 25(5) permitting importation must obtain a certificate from the Secretary of State before treating the animal.
(2) The certificate may be granted subject to any condition the Secretary of State thinks fit.
Extent Information
E243This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Administration of a homeopathic remedyU.K.
8.—(1) A registered homeopathic remedy or a homeopathic remedy prepared and supplied by a pharmacist under paragraph 10 of Schedule 3 may be administered to an animal by anyone, subject to any restrictions specified in its registration.
(2) A homeopathic remedy that was on the market before 1st January 1994 may be administered by anyone.
(3) A veterinary surgeon may administer, either personally or under the veterinary surgeon’s responsibility—
(a)a homeopathic remedy authorised for human use, or
(b)a homeopathic remedy prepared extemporaneously by a veterinary surgeon, a pharmacist or a person holding a manufacturing authorisation authorising the manufacture of that type of product.
Administration under an animal test certificateE+W+S
9.—(1) A medicinal product may be administered in accordance with an animal test certificate granted for [F279clinical trials] by the Secretary of State.
(2) An application for an animal test certificate may be refused if this is necessary for the protection of animal or public health or the environment, and the animal test certificate may be varied, suspended or revoked in the same way as a marketing authorisation.
(3) The holder of an animal test certificate may not supply a product for administration that is not within the terms of the animal test certificate.
(4) The holder of an animal test certificate test who becomes aware of any [F280adverse event] following the administration of a product under an animal test certificate must report the reaction to the Secretary of State within [F28130 days] of learning of it.
Extent Information
E90This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F279Words in Sch. 4 para. 9(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 124(a)
F280Words in Sch. 4 para. 9(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 124(b)(i)
F281Words in Sch. 4 para. 9(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 124(b)(ii)
Administration under an animal test certificateN.I.
9.—(1) A medicinal product may be administered in accordance with an animal test certificate granted for research purposes by the Secretary of State.
(2) An application for an animal test certificate may be refused if this is necessary for the protection of animal or public health or the environment, and the animal test certificate may be varied, suspended or revoked in the same way as a marketing authorisation.
(3) The holder of an animal test certificate may not supply a product for administration that is not within the terms of the animal test certificate.
(4) The holder of an animal test certificate test who becomes aware of any serious adverse reaction following the administration of a product under an animal test certificate must report the reaction to the Secretary of State within 15 days of learning of it.
Extent Information
E244This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F282Misuse of the cascadeE+W+S
9A. A person must not promote or facilitate any purported use of the cascade which is not in accordance with this Schedule.]
Textual Amendments
F282Sch. 4 para. 9A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 125
OffencesE+W+S
10. It is an offence to fail to comply with—
[F283(za)paragraph 2;]
(a)paragraph 3(2);
(b)paragraph 6; F284...
[F285(ba)paragraph 6A;]
(c)paragraph 9(3) or [F286(4); or]
[F287(d)paragraph 9A.]
Extent Information
E91This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F283Sch. 4 para. 10(za) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 126(a)
F284Word in Sch. 4 para. 10(b) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 126(b)
F285Sch. 4 para. 10(ba) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 126(c)
F286Words in Sch. 4 para. 10(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 126(d)
F287Sch. 4 para. 10(d) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 126(e)
OffencesN.I.
10. It is an offence to fail to comply with—
(a)paragraph 3(2);
(b)paragraph 6; or
(c)paragraph 9(3) or (4).
Extent Information
E245This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Regulation 14
SCHEDULE 5U.K.Medicated feedingstuffs and specified feed additives
Scope and interpretationU.K.
1.—(1) This Schedule applies in relation to the following (referred to in this Schedule as “specified feed additives”) when used as feed additives—
(a)coccidiostats;
(b)histomonostats; and
(c)all other zootechnical additives except—
(i)digestibility enhancers;
(ii)gut flora stabilisers; and
(iii)substances incorporated with the intention of favourably affecting the environment.
(2) It also applies in relation to the manufacture and placing on the market of feedingstuffs containing a veterinary medicinal product.
(3) In this Schedule—
[F288“animal keeper” means any natural or legal person responsible for animals, whether on a permanent or a temporary basis;
“batch” means an identifiable quantity of feed determined to have common characteristics whether in relation to origin, variety, type of packaging, packer, consignor or labelling and, in the case of a production process, a unit of production from a single plant using uniform production parameters or a number of such units when produced in continuous order and stored together;
“cross-contamination” means contamination of a non-target feed with an active substance originating from the previous use of the relevant facilities or equipment;
“distributor” means a feed business operator distributing specified feed additives, intermediate feedingstuff or complete feed containing specified feed additives, or intermediate feedingstuff or complete feed containing medicinal premixes;
“feed business” means any undertaking whether for profit or not and whether public or private, carrying out any operation of production, manufacture, processing, storage, transport or distribution of feed including any producer producing, processing or storing feed for feeding to animals on their own holding;
“feed business operator” means any person responsible for ensuring that the requirements of this Schedule are met within the feed business under that person’s control;
“non-target feed” means feed, whether medicated or not which is not intended to contain a specific active substance;
“premises” means any unit of a feed business;]
[F289“premixture” means a mixture of a veterinary medicinal product or a specified feed additive with feedingstuffs materials, intended for further mixing with feedingstuffs before being fed to animals;]
“zootechnical additive” means any additive used to maintain animals in good health or favourably affect their performance.
Textual Amendments
F288Words in Sch. 5 para. 1(3) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 128(b)
F289Words in Sch. 5 para. 1(3) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 128(a)
Enforcement of Regulation (EC) No 178/2002U.K.
2.—(1) For the purposes of [F290Regulation (EC) No 178/2002] the competent authority is the Secretary of State.
(2) No person may fail to comply with any of the following provisions of that Regulation—
(a)Article 11 (requirements relating to imports);
(b)Article 12 (requirements relating to exports);
(c)Article 15(1) (prohibition on the placing on the market or feeding unsafe feedingstuffs);
(d)Article 16 so far as it prohibits misleading labelling, advertising or presentation of feedingstuffs;
(e)Article 18(2) and (3) (requirements of traceability) in so far as it relates to feed business operators; and
(f)Article 20 (responsibilities of feed business operators).
Textual Amendments
F290Words in Sch. 5 para. 2(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(a)
Enforcement of Regulation (EC) No 1831/2003E+W+S
3.—(1) For the purposes of [F291Regulation (EC) No 1831/2003] the competent authority is the Secretary of State.
(2) An authorisation under Article 3(2) of that Regulation must be in writing.
(3) No person may possess a specified feed additive, or [F292an intermediate feedingstuff] or feedingstuffs containing a specified feed additive, unless the specified feed additive has been authorised under Regulation (EC) No 1831/2003 or is for export to [F293another] country.
(4) No person may fail to comply with any of the following provisions of that Regulation—
(a)Article 3(1) or Article 3(3) (the authorisation, conditions of use and labelling of specified feed additives);
(b)Article 12(1) or (2) (conditions relating to specified feed additives);
(c)Article 16(1) (labelling);
(d)Article 16(3) (additional labelling requirement);
(e)Article 16(4) (premixtures containing specified feed additives);
(f)Article 16(5) (packaging).
Extent Information
E92This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F291Words in Sch. 5 para. 3(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(b)
F292Words in Sch. 5 para. 3(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 129
F293Word in Sch. 5 para. 3(3) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Enforcement of Regulation (EC) No 1831/2003N.I.
3.—(1) For the purposes of [F608Regulation (EC) No 1831/2003] the competent authority is the Secretary of State.
(2) An authorisation under Article 3(2) of that Regulation must be in writing.
(3) No person may possess a specified feed additive, or a premixture or feedingstuffs containing a specified feed additive, unless the specified feed additive has been authorised under Regulation (EC) No 1831/2003 or is for export to a third country.
(4) No person may fail to comply with any of the following provisions of that Regulation—
(a)Article 3(1) or Article 3(3) (the authorisation, conditions of use and labelling of specified feed additives);
(b)Article 12(1) or (2) (conditions relating to specified feed additives);
(c)Article 16(1) (labelling);
(d)Article 16(3) (additional labelling requirement);
(e)Article 16(4) (premixtures containing specified feed additives);
(f)Article 16(5) (packaging).
Extent Information
E246This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F608Words in Sch. 5 para. 3(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(b)
Enforcement of [F294Regulation (EU) 2017/625] E+W+S
4. For the purposes of [F295 Regulation (EU) 2017/625] the competent authority is the Secretary of State.
Extent Information
E93This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F294Words in Sch. 5 para. 4 heading substituted (E.) (14.12.2019) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) Regulations 2019 (S.I. 2019/1488), regs. 1(1), 27(d)(ii); and said words substituted (W.) (31.1.2020) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) (Wales) Regulations 2020 (S.I. 2020/44), regs. 1(2), 24(1)(d)(ii); and said words substituted (S.N.I.) (31.12.2020) by The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(4)(b)
F295Words in Sch. 5 para. 4 substituted (E.) (14.12.2019) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) Regulations 2019 (S.I. 2019/1488), regs. 1(1), 27(d)(ii); and said words substituted (W.) (31.1.2020) by The Official Controls (Animals, Feed and Food, Plant Health Fees etc.) (Wales) Regulations 2020 (S.I. 2020/44), regs. 1(2), 24(1)(d)(i); and said words substituted (S.N.I.) (31.12.2020) by The Official Controls (Animals, Feed and Food, Plant Health etc.) (Amendment) (EU Exit) (No. 2) Regulations 2020 (S.I. 2020/1631), regs. 1(2), 3(4)(b)
[F609Enforcement of Regulation (EU) 2017/625N.I.
4. For the purposes of Regulation (EU) 2017/625 the competent authority is the Secretary of State.]
Extent Information
E247This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
Enforcement of Regulation (EC) No 183/2005U.K.
5.—(1) For the purposes of [F296Regulation (EC) No 183/2005] the competent authority is the Secretary of State.
(2) No person may fail to comply with any of the following provisions of that Regulation—
(a)Article 5(2), (5) or (6) (specific obligations);
(b)Article 6(1) as read with (2) and (3) (HACCP system);
(c)Article 7(1) (documents concerning the HACCP system);
(d)Article 9(2) (official controls, notification and registration);
(e)Article 10(1) (approval of feed business establishments);
(f)Article 11 (prohibition on operating without approval or registration);
(g)Article 17(2) (exemption from on-site visits);
(h)Article 18(3) (declaration of compliance);
(i)Article 23(1) (conditions relating to imports from third countries);
(j)Article 25 (feedingstuffs produced for export to third countries).
(3) A manufacturer must ensure that, so far as is reasonably practicable, the active ingredient is evenly incorporated throughout the feedingstuffs.
(4) In the case of the refusal, suspension or revocation of an approval under the Regulation the appeals procedure relating to a manufacturing authorisation in regulation 30 applies.
Textual Amendments
F296Words in Sch. 5 para. 5(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(d)
Enforcement of Regulation (EC) No 767/2009U.K.
6. No person may contravene Article 8 of Regulation (EC) No 767/2009 of the European Parliament and of the Council in relation to feedingstuffs containing specified feed additives.
[F297Authorisation] of manufacturers and distributors of feedingstuffs containing [F298medicinal premixes]E+W+S
7.—(1) For the purposes of Directive 90/167/EEC laying down the conditions governing the preparation, placing on the market and use of medicated feedingstuffs in the Community(11) the competent authority is the Secretary of State.
(2) No person may incorporate a [F299medicinal premix] into [F300an intermediate feedingstuff] or feedingstuff, or act as a distributor of [F301intermediate feedingstuffs] or feedingstuffs containing a [F299medicinal premix], without being [F302authorised] to do so by the Secretary of State.
(3) The conditions which govern [F303authorisation] of feed business [F304premises] under Regulation (EC) No 183/2005 laying down requirements for feed hygiene(12) also govern [F303authorisation] of manufacturers and distributors under sub-paragraph (2).
(4) The Secretary of State shall conduct inspections of manufacturers and distributors [F305authorised] under sub-paragraph (2) basing the frequency of inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
[F306(5) A manufacturer must ensure that, so far as is reasonably practical the medicinal premix is evenly incorporated and homogeneously dispersed throughout the feedingstuffs, taking into account the specific properties of the medicinal premix and the mixing technology employed.]
(6) The provisions of this paragraph do not apply in relation to any person breeding or selling ornamental fish not intended for human consumption provided that the person does not use more than a total of 1kg of [F307medicinal premix] annually for that purpose.
(7) In the case of the refusal, suspension or revocation of an [F308authorisation] under this paragraph the appeals procedure relating to a manufacturing authorisation in regulation 30 applies.
Extent Information
E94This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F297Word in Sch. 5 para. 7 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(g)(i)
F298Words in Sch. 5 para. 7 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(g)(ii)
F299Words in Sch. 5 para. 7(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(a)(i)
F300Words in Sch. 5 para. 7(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(a)(ii)
F301Words in Sch. 5 para. 7(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(a)(iii)
F302Word in Sch. 5 para. 7(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(a)(iv)
F303Word in Sch. 5 para. 7(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(b)(i)
F304Word in Sch. 5 para. 7(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(b)(ii)
F305Word in Sch. 5 para. 7(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(c)
F306Sch. 5 para. 7(5) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(d)
F307Words in Sch. 5 para. 7(6) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(e)
F308Word in Sch. 5 para. 7(7) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 130(f)
Approval of manufacturers and distributors of feedingstuffs containing veterinary medicinal productsN.I.
7.—(1) For the purposes of Directive 90/167/EEC laying down the conditions governing the preparation, placing on the market and use of medicated feedingstuffs in the Community(11) the competent authority is the Secretary of State.
(2) No person may incorporate a veterinary medicinal product into a premixture or feedingstuff, or act as a distributor of premixtures or feedingstuffs containing a veterinary medicinal product, without being approved to do so by the Secretary of State.
(3) The conditions which govern approval of feed business establishments under Regulation (EC) No 183/2005 laying down requirements for feed hygiene(12) also govern approval of manufacturers and distributors under sub-paragraph (2).
(4) The Secretary of State shall conduct inspections of manufacturers and distributors approved under sub-paragraph (2) basing the frequency of inspection on the risks associated with each premises’ history and the nature of the products handled at the premises.
(5) A manufacturer must ensure that, so far as is reasonably practical, the veterinary medicinal product is evenly incorporated throughout the feedingstuffs.
(6) The provisions of this paragraph do not apply in relation to any person breeding or selling ornamental fish not intended for human consumption provided that the person does not use more than a total of 1kg of veterinary medicinal product annually for that purpose.
(7) In the case of the refusal, suspension or revocation of an approval under this paragraph the appeals procedure relating to a manufacturing authorisation in regulation 30 applies.
Extent Information
E248This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Incorporation of a [F309medicinal premix into an intermediate feedingstuff]E+W+S
8. Any person who incorporates a [F310medicinal premix] into [F311an intermediate feedingstuff]—
(a)must do so in accordance with the summary of product characteristics, and must take account of any interactions listed there; and
(b)must ensure that the [F310medicinal premix] does not contain the same active substance as any other additive.
Extent Information
E95This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F309Words in Sch. 5 para. 8 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 131(c)
F310Words in Sch. 5 para. 8 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 131(a)
F311Words in Sch. 5 para. 8 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 131(b)
Incorporation of a veterinary medicinal product into a premixtureN.I.
8. Any person who incorporates a veterinary medicinal product into a premixture—
(a)must do so in accordance with the summary of product characteristics, and must take account of any interactions listed there; and
(b)must ensure that the veterinary medicinal product does not contain the same active substance as any other additive.
Extent Information
E249This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Top dressingU.K.
9. No person may promote or label any veterinary medicinal product, or anything containing a veterinary medicinal product, as being suitable for top dressing (that is, sprinkling it on to feedingstuffs without thoroughly incorporating it) unless the summary of product characteristics specifically permits this use.
Incorporation of a [F312medicinal premix] into feedingstuffsE+W+S
10. Any person who incorporates a [F313medicinal premix] (or [F314an intermediate feedingstuff]) into feedingstuffs—
(a)must do so in accordance with the summary of product characteristics, and must take account of any interactions listed there;
(b)must ensure that the [F313medicinal premix] does not contain the same active substance as any other additive;
(c)must ensure that the [F313medicinal premix] is incorporated in accordance with its marketing authorisation (unless it has been prescribed under the cascade) and the [F315medicated feedingstuff prescription];
(d)must ensure that the daily dose of the [F313medicinal premix] is contained in a quantity of medicated feedingstuffs corresponding to at least half the daily feedingstuffs ration of the animals treated or, in the case of ruminants, corresponding to at least half the daily requirements of non-mineral complementary feedingstuffs.
Extent Information
E96This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F312Words in Sch. 5 para. 10 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 132(d)
F313Words in Sch. 5 para. 10 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 132(a)
F314Words in Sch. 5 para. 10 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 132(b)
F315Words in Sch. 5 para. 10(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 132(c)
Incorporation of a veterinary medicinal product into feedingstuffsN.I.
10. Any person who incorporates a veterinary medicinal product (or a premixture containing a veterinary medicinal product) into feedingstuffs—
(a)must do so in accordance with the summary of product characteristics, and must take account of any interactions listed there;
(b)must ensure that the veterinary medicinal product does not contain the same active substance as any other additive;
(c)must ensure that the veterinary medicinal product is incorporated in accordance with its marketing authorisation (unless it has been prescribed under the cascade) and the prescription;
(d)must ensure that the daily dose of the veterinary medicinal product is contained in a quantity of medicated feedingstuffs corresponding to at least half the daily feedingstuffs ration of the animals treated or, in the case of ruminants, corresponding to at least half the daily requirements of non-mineral complementary feedingstuffs.
Extent Information
E250This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Additional record keeping requirements relating to [F316medicinal premixes] E+W+S
11.—(1) Any person who—
(a)incorporates a [F317medicinal premix] into [F318an intermediate feedingstuff];
(b)incorporates [F319an intermediate feedingstuff] containing a [F320medicinal premix] into feedingstuffs; or
(c)incorporates a [F321medicinal premix] into feedingstuffs,
must make a daily record of—
(d)the types and quantities of all [F322medicinal premixes] (and specified feed additives, if any) and [F323intermediate feedingstuffs] used in the manufacturing process; and
(e)the quantity of feedingstuffs and [F324intermediate feedingstuffs] containing [F325medicinal premix] manufactured that day.
(2) An [F326authorised] distributor must make a daily record of—
(a)the types and quantities of all [F327intermediate feedingstuffs] and feedingstuffs containing [F328medicinal premixes] bought and sold that day; and
(b)the quantity held.
(3) A manufacturer and distributor must also record, as soon as reasonably practicable, for each consignment supplied—
(a)the date of delivery;
(b)the name and address of each consignee (or, in the case of a manufacturer supplying to a distributor, the name and address of the distributor);
(c)the type of feedingstuffs or [F329intermediate feedingstuffs] supplied;
(d)the quantity;
(e)the type of [F330medicinal premix] incorporated into the feedingstuffs; F331...
(f)the expiry date [F332; and
(g)the batch number.]
(4) Records must be kept for five years.
Textual Amendments
F316Words in Sch. 5 para. 11 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(d)
F317Words in Sch. 5 para. 11(1)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(i)(aa)
F318Words in Sch. 5 para. 11(1)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(i)(bb)
F319Words in Sch. 5 para. 11(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(ii)(aa)
F320Words in Sch. 5 para. 11(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(ii)(bb)
F321Words in Sch. 5 para. 11(1)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(iii)
F322Words in Sch. 5 para. 11(1)(d) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(iv)(aa)
F323Words in Sch. 5 para. 11(1)(d) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(iv)(bb)
F324Words in Sch. 5 para. 11(1)(e) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(v)(aa)
F325Words in Sch. 5 para. 11(1)(e) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(a)(v)(bb)
F326Word in Sch. 5 para. 11(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(b)(i)
F327Words in Sch. 5 para. 11(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(b)(ii)
F328Words in Sch. 5 para. 11(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(b)(iii)
F329Words in Sch. 5 para. 11(3)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(c)(i)
F330Words in Sch. 5 para. 11(3)(e) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(c)(ii)
F331Word in Sch. 5 para. 11(3) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(c)(iii)
F332Sch. 5 para. 11(3)(g) and word inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 133(c)(iv)
Additional record keeping requirements relating to veterinary medicinal productsN.I.
11.—(1) Any person who—
(a)incorporates a veterinary medicinal product into a premixture;
(b)incorporates a premixture containing a veterinary medicinal product into feedingstuffs; or
(c)incorporates a veterinary medicinal product into feedingstuffs,
must make a daily record of—
(d)the types and quantities of all veterinary medicinal products (and specified feed additives, if any) and premixture used in the manufacturing process; and
(e)the quantity of feedingstuffs and premixture containing veterinary medicinal product manufactured that day.
(2) An approved distributor must make a daily record of—
(a)the types and quantities of all premixtures and feedingstuffs containing veterinary medicinal products bought and sold that day; and
(b)the quantity held.
(3) A manufacturer and distributor must also record, as soon as reasonably practicable, for each consignment supplied—
(a)the date of delivery;
(b)the name and address of each consignee (or, in the case of a manufacturer supplying to a distributor, the name and address of the distributor);
(c)the type of feedingstuffs or premixture supplied;
(d)the quantity;
(e)the type of veterinary medicinal product incorporated into the feedingstuffs; and
(f)the expiry date.
(4) Records must be kept for five years.
Labelling [F333an intermediate feedingstuff] containing a [F334medicinal premix]E+W+S
12.—(1) [F335An intermediate feedingstuff] containing a [F336medicinal premix] must be clearly and legibly labelled with the following—
(a)the words “[F337INTERMEDIATE FEEDINGSTUFF]” (or, if it is to be labelled as “complementary feedingstuffs” under legislation implementing Council Directive 79/373/EEC on the marketing of compound feedingstuffs(13), “MEDICATED COMPLEMENTARY FEEDINGSTUFFS” ) in upper case letters;
(b)the proprietary name of the [F338medicinal premix] and the authorisation number;
(c)the name and amount of the active substance (mg/kg) in the [F339intermediate feedingstuff];
(d)the range of acceptable inclusion rates of the [F340intermediate feedingstuff] into the final feedingstuffs, the range of acceptable levels of the active ingredients in the final feedingstuffs and the words “refer to the [F341medicated feedingstuffs prescription] for the exact inclusion rate” or equivalent wording;
(e)warnings and contra-indications;
[F342(ea)a statement that the product must be used in accordance with its summary of product characteristics;
(eb)the contact details (including a free helpline number) for the supplier of the product;
(ec)the words “inappropriate disposal of this product poses a serious threat to the environment”;
(ed)in the case of a product containing an antibiotic, the words “inappropriate disposal of this product may contribute to antimicrobial resistance”;]
(f)the withdrawal period, and a statement that, if the [F343medicated feedingstuffs prescription] requires a longer withdrawal period, that is the one that applies;
(g)the expiry date;
(h)any special storage instructions [F344required by the marketing authorisation];
(i)where a [F345medicated feedingstuffs prescription] is required, a statement to this effect.
(2) If there is more than one [F346medicinal premix] used, the longest withdrawal period must be shown on the label.
(3) If the [F347intermediate feedingstuff] also contains a specified feed additive to which this Schedule applies it must also contain the information required under Article 16 of Regulation (EC) No 1831/2003(14).
(4) No person may supply such [F348an intermediate feedingstuff] unless it is labelled in accordance with this paragraph.
Extent Information
E97This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F333Words in Sch. 5 para. 12 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(e)(i) (with reg. 205)
F334Words in Sch. 5 para. 12 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(e)(ii) (with reg. 205)
F335Words in Sch. 5 para. 12(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(i)(aa) (with reg. 205)
F336Words in Sch. 5 para. 12(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(i)(bb) (with reg. 205)
F337Words in Sch. 5 para. 12(1)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(ii) (with reg. 205)
F338Words in Sch. 5 para. 12(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(iii) (with reg. 205)
F339Words in Sch. 5 para. 12(1)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(iv) (with reg. 205)
F340Words in Sch. 5 para. 12(1)(d) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(v)(aa) (with reg. 205)
F341Words in Sch. 5 para. 12(1)(d) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(v)(bb) (with reg. 205)
F342Sch. 5 para. 12(1)(ea)-(ed) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(vi) (with reg. 205)
F343Words in Sch. 5 para. 12(1)(f) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(vii) (with reg. 205)
F344Words in Sch. 5 para. 12(1)(h) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(viii) (with reg. 205)
F345Words in Sch. 5 para. 12(1)(i) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(a)(ix) (with reg. 205)
F346Words in Sch. 5 para. 12(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(b) (with reg. 205)
F347Words in Sch. 5 para. 12(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(c) (with reg. 205)
F348Words in Sch. 5 para. 12(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 134(d) (with reg. 205)
Labelling a premixture containing a veterinary medicinal productN.I.
12.—(1) A premixture containing a veterinary medicinal product must be clearly and legibly labelled with the following—
(a)the words “MEDICATED PREMIXTURE” (or, if it is to be labelled as “complementary feedingstuffs” under legislation implementing Council Directive 79/373/EEC on the marketing of compound feedingstuffs(13), “MEDICATED COMPLEMENTARY FEEDINGSTUFFS” ) in upper case letters;
(b)the proprietary name of the veterinary medicinal product and the authorisation number;
(c)the name and amount of the active substance (mg/kg) in the premixture;
(d)the range of acceptable inclusion rates of the premixture into the final feedingstuffs, the range of acceptable levels of the active ingredients in the final feedingstuffs and the words “refer to the prescription for the exact inclusion rate” or equivalent wording;
(e)warnings and contra-indications;
(f)the withdrawal period, and a statement that, if the prescription requires a longer withdrawal period, that is the one that applies;
(g)the expiry date;
(h)any special storage instructions;
(i)where a prescription is required, a statement to this effect.
(2) If there is more than one veterinary medicinal product used, the longest withdrawal period must be shown on the label.
(3) If the premixture also contains a specified feed additive to which this Schedule applies it must also contain the information required under Article 16 of Regulation (EC) No 1831/2003(14).
(4) No person may supply such a premixture unless it is labelled in accordance with this paragraph.
Extent Information
E251This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Labelling of feedingstuffs containing a specified feed additiveU.K.
13. No person may contravene the labelling requirements of Article 15 and Article 17 of Regulation (EC) No 767/2009 of the European Parliament and of the Council.
Labelling of feedingstuffs containing a [F349medicinal premix]E+W+S
14.—(1) Feedingstuffs containing a [F350medicinal premix] must be clearly and legibly labelled with the following—
(a)the words “MEDICATED COMPLETE FEED” in upper case letters, or where feedingstuffs are to be labelled as a complementary feedingstuff and intended to be fed to animals without further mixing with feed materials, the words “MEDICATED COMPLEMENTARY FEEDINGSTUFF”;
(b)the proprietary name, authorisation number and inclusion rate (kg/tonne or mg/kg) of the [F350medicinal premix] incorporated into the feedingstuffs;
(c)the name and amount of the active substance (mg/kg) in the feedingstuffs;
(d)the species of animal for which the feedingstuffs are intended;
(e)warnings and contra-indications;
[F351(ea)the contact details (including a free helpline number) for the supplier of the product;
(eb)the words “inappropriate disposal of this product poses a serious threat to the environment”;
(ec)in the case of a product containing an antibiotic, the words “inappropriate disposal of this product may contribute to antimicrobial resistance”;]
(f)the withdrawal period, and a statement that, if the [F352medicated feedingstuffs prescription] requires a longer withdrawal period, that is the one that applies;
[F353(fa)the batch number;]
(g)the expiry date;
(h)any special storage instructions required by the marketing authorisation;
(i)a statement to the effect that the feedingstuffs must only be fed in accordance with its [F354medicated feedingstuffs prescription];
(j)the name and [F355authorisation] number of the manufacturer or the distributor.
(2) If there is more than one [F356medicinal premix] used, the longest withdrawal period must be shown on the label.
(3) If the feedingstuff also contains a specified feed additive to which this Schedule applies it must also contain the information required by Articles 15 and 17 of Regulation (EC) No 767/2009 of the European Parliament and of the Council.
(4) No person may supply feedingstuffs [F357containing a medicinal premix] unless they are labelled in accordance with this paragraph.
Extent Information
E98This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F349Words in Sch. 5 para. 14 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(d) (with reg. 205)
F350Words in Sch. 5 para. 14(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(a)(i) (with reg. 205)
F351Sch. 5 para. 14(1)(ea)-(ec) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(a)(ii) (with reg. 205)
F352Words in Sch. 5 para. 14(1)(f) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(a)(iii) (with reg. 205)
F353Sch. 5 para. 14(1)(fa) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(a)(iv) (with reg. 205)
F354Words in Sch. 5 para. 14(1)(i) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(a)(v) (with reg. 205)
F355Word in Sch. 5 para. 14(1)(j) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(a)(vi) (with reg. 205)
F356Words in Sch. 5 para. 14(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(b) (with reg. 205)
F357Words in Sch. 5 para. 14(4) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 135(c) (with reg. 205)
Labelling of feedingstuffs containing a veterinary medicinal productN.I.
14.—(1) Feedingstuffs containing a veterinary medicinal product must be clearly and legibly labelled with the following—
(a)the words “MEDICATED COMPLETE FEED” in upper case letters, or where feedingstuffs are to be labelled as a complementary feedingstuff and intended to be fed to animals without further mixing with feed materials, the words “MEDICATED COMPLEMENTARY FEEDINGSTUFF”;
(b)the proprietary name, authorisation number and inclusion rate (kg/tonne or mg/kg) of the veterinary medicinal product incorporated into the feedingstuffs;
(c)the name and amount of the active substance (mg/kg) in the feedingstuffs;
(d)the species of animal for which the feedingstuffs are intended;
(e)warnings and contra-indications;
(f)the withdrawal period, and a statement that, if the prescription requires a longer withdrawal period, that is the one that applies;
(g)the expiry date;
(h)any special storage instructions required by the marketing authorisation;
(i)a statement to the effect that the feedingstuffs must only be fed in accordance with its prescription;
(j)the name and approval number of the manufacturer or the distributor.
(2) If there is more than one veterinary medicinal product used, the longest withdrawal period must be shown on the label.
(3) If the feedingstuff also contains a specified feed additive to which this Schedule applies it must also contain the information required by Articles 15 and 17 of Regulation (EC) No 767/2009 of the European Parliament and of the Council.
(4) No person may supply feedingstuffs unless they are labelled in accordance with this paragraph.
Extent Information
E252This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Supply of specified feed additivesE+W+S
15.—(1) No person other than the person who manufactured a specified feed additive or an [F358authorised] distributor may supply a specified feed additive.
(2) The person who manufactured the specified feed additive may only supply it to—
(a)an [F358authorised] distributor;
(b)an [F358authorised] [F359intermediate feedingstuff] manufacturer or an [F358authorised] complementary feedingstuffs manufacturer; or
(c)a feedingstuff manufacturer [F358authorised] to mix a specified feed additive directly into feedingstuff.
(3) An [F358authorised] distributor may only supply it to—
(a)another [F358authorised] distributor;
(b)an [F358authorised] [F360intermediate feedingstuff] manufacturer or an [F358authorised] complementary feedingstuffs manufacturer; or
(c)a feedingstuff manufacturer [F358authorised] to mix a specified feed additive directly into feedingstuff.
Extent Information
E99This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F358Word in Sch. 5 para. 15 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 136(a)
F359Words in Sch. 5 para. 15(2)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 136(b)
F360Words in Sch. 5 para. 15(3)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 136(c)
Supply of specified feed additivesN.I.
15.—(1) No person other than the person who manufactured a specified feed additive or an approved distributor may supply a specified feed additive.
(2) The person who manufactured the specified feed additive may only supply it to—
(a)an approved distributor;
(b)an approved premixture manufacturer or an approved complementary feedingstuffs manufacturer; or
(c)a feedingstuff manufacturer approved to mix a specified feed additive directly into feedingstuff.
(3) An approved distributor may only supply it to—
(a)another approved distributor;
(b)an approved premixture manufacturer or an approved complementary feedingstuffs manufacturer; or
(c)a feedingstuff manufacturer approved to mix a specified feed additive directly into feedingstuff.
Extent Information
E253This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Supply of [F361intermediate feedingstuff or specified feed additive]E+W+S
16.—(1) No person other than the person who manufactured [F362an intermediate feedingstuff or specified feed additive] or an [F363authorised] distributor may supply [F362an intermediate feedingstuff or specified feed additive].
(2) The person who manufactured the [F364intermediate feedingstuff or specified feed additive] may only supply it to—
(a)an [F363authorised] distributor; or
(b)a feedingstuff manufacturer [F363authorised] to incorporate that [F364intermediate feedingstuff or specified feed additive].
(3) An [F363authorised] distributor may only supply it to—
(a)another [F363authorised] distributor; or
(b)a feedingstuff manufacturer [F363authorised] to incorporate that [F365intermediate feedingstuff or specified feed additive].
Extent Information
E100This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F361Words in Sch. 5 para. 16 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 137(e)
F362Words in Sch. 5 para. 16(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 137(b)
F363Word in Sch. 5 para. 16 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 137(a)
F364Words in Sch. 5 para. 16(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 137(c)
F365Words in Sch. 5 para. 16(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 137(d)
Supply of premixtureN.I.
16.—(1) No person other than the person who manufactured a premixture or an approved distributor may supply a premixture.
(2) The person who manufactured the premixture may only supply it to—
(a)an approved distributor; or
(b)a feedingstuff manufacturer approved to incorporate that premixture.
(3) An approved distributor may only supply it to—
(a)another approved distributor; or
(b)a feedingstuff manufacturer approved to incorporate that premixture.
Extent Information
E254This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Supply of a complementary feedingstuffE+W+S
17.—(1) No person other than—
(a)the person who manufactured a complementary feedingstuff containing a specified feed additive; or
(b)an [F366authorised] distributor
may supply a complementary feedingstuff containing a specified feed additive.
(2) The person who manufactured such complementary feedingstuff may only supply it to—
(a)an [F366authorised] distributor; or
(b)a feedingstuff manufacturer registered to incorporate that complementary feedingstuff or [F366authorised] to incorporate [F367an intermediate feedingstuff].
(3) An [F366authorised] distributor may only supply it to—
(a)another [F366authorised] distributor, or
(b)a feedingstuff manufacturer registered to incorporate that complementary feedingstuff or [F366authorised] to incorporate [F368an intermediate feedingstuff].
F369(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E101This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F366Word in Sch. 5 para. 17 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 138(a)
F367Words in Sch. 5 para. 17(2)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 138(b)
F368Words in Sch. 5 para. 17(3)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 138(c)
F369Sch. 5 para. 17(4) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 138(d)
Supply of a complementary feedingstuffN.I.
17.—(1) No person other than—
(a)the person who manufactured a complementary feedingstuff containing a specified feed additive; or
(b)an approved distributor
may supply a complementary feedingstuff containing a specified feed additive.
(2) The person who manufactured such complementary feedingstuff may only supply it to—
(a)an approved distributor; or
(b)a feedingstuff manufacturer registered to incorporate that complementary feedingstuff or approved to incorporate a premixture.
(3) An approved distributor may only supply it to—
(a)another approved distributor, or
(b)a feedingstuff manufacturer registered to incorporate that complementary feedingstuff or approved to incorporate a premixture.
(4) In this paragraph “complementary feedingstuff” has the meaning given in Article 3 of Regulation EC No 767/2009.
Extent Information
E255This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Supply of feedingstuffs containing a [F370medicinal premix]E+W+S
18.—(1) No person other than the person who manufactured the feedingstuffs or an [F371authorised] distributor may supply feedingstuffs containing a [F372medicinal premix].
(2) The person who manufactured the feedingstuff may only supply it to—
(a)an [F371authorised] distributor; or
(b)[F373an animal keeper] for feeding to those animals.
(3) A distributor may only supply it to—
(a)another [F371authorised] distributor; or
(b)[F374an animal keeper] for feeding to those animals.
(4) Supply to [F375an animal keeper] must be in accordance with a written [F376medicated feedingstuff prescription] as specified in the following paragraph.
(5) If a [F377medicated feedingstuff prescription] is for a period of longer than one month, the supplier may not provide more than one month’s supply at any one time.
(6) No manufacturer or distributor may supply a feedingstuff to anyone not specified in this paragraph, or otherwise than in accordance with this paragraph.
(7) The person supplying the feedingstuff must keep the [F378medicated feedingstuff prescription] for five years.
[F379(8) Nothing in this paragraph prevents a commercial feed manufacturer from incorporating a medicinal premix with a feedingstuff in advance of receiving a written prescription for that feedingstuff.]
Extent Information
E102This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F370Words in Sch. 5 para. 18 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(i)
F371Word in Sch. 5 para. 18 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(a)
F372Words in Sch. 5 para. 18(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(b)
F373Words in Sch. 5 para. 18(2)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(c)
F374Words in Sch. 5 para. 18(3)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(d)
F375Words in Sch. 5 para. 18(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(e)(i)
F376Words in Sch. 5 para. 18(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(e)(ii)
F377Words in Sch. 5 para. 18(5) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(f)
F378Words in Sch. 5 para. 18(7) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(g)
F379Sch. 5 para. 18(8) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 139(h)
Supply of feedingstuffs containing a veterinary medicinal productN.I.
18.—(1) No person other than the person who manufactured the feedingstuffs or an approved distributor may supply feedingstuffs containing a veterinary medicinal product.
(2) The person who manufactured the feedingstuff may only supply it to—
(a)an approved distributor; or
(b)a person who keeps animals for feeding to those animals.
(3) A distributor may only supply it to—
(a)another approved distributor; or
(b)a person who keeps animals for feeding to those animals.
(4) Supply to a person who keeps animals must be in accordance with a written prescription as specified in the following paragraph.
(5) If a prescription is for a period of longer than one month, the supplier may not provide more than one month’s supply at any one time.
(6) No manufacturer or distributor may supply a feedingstuff to anyone not specified in this paragraph, or otherwise than in accordance with this paragraph.
(7) The person supplying the feedingstuff must keep the prescription for five years.
Extent Information
E256This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F380Medicated feedingstuff prescriptions] for feedingstuffs containing a [F381medicinal premix]E+W+S
19.—(1) A [F382medicated feedingstuff prescription] for feedingstuffs containing a [F383medicinal premix] must contain the following—
(a)the name and address of the person prescribing the product;
(b)the qualifications enabling the person to prescribe the product;
(c)the name and address of the keeper of the animals to be treated;
(d)the species of animal, identification and number of the animals;
(e)the premises at which the animals are kept if this is different from the address of the keeper;
[F384(ea)the diagnosed disease to be treated or prevented (in the case of immunological veterinary medicinal products or antiparasitics without antimicrobial effects);]
(f)the date of the prescription;
(g)the signature or other authentication of the person prescribing the product;
[F385(h)the name, active substance, amount of the product prescribed and inclusion rate of the medicinal premix and resulting inclusion rate of the active substance;]
(i)the dosage and administration instructions;
(j)any necessary warnings;
[F386(ja)a statement that the prescription may not be re-used;]
(k)the withdrawal period;
(l)the manufacturer or the distributor of the feedingstuffs (who must be [F387authorised] for the purpose) [F388, whichever is the supplier to the end user];
(m)if the validity exceeds one month, a statement that not more than 31 days’ supply may be provided at any time;
(n)the name, type and quantity of feedingstuffs to be used;
[F389(na)the overall amount of feedingstuff to be supplied under the prescription;]
F390(o). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(p)any special instructions;
(q)the percentage of the prescribed feedingstuffs to be added to the daily ration; and
(r)if it is prescribed under the cascade, a statement to that effect.
(2) It is valid for three months or such shorter period as may be specified in the [F391medicated feedingstuff prescription].
[F392(2A) In the case of a prescription to which sub-paragraph (1) applies which relates to an antibiotic, the time between a prescription being issued and the course of treatment starting must be no more than five working days.
(2B) Subject to paragraph 7A in Schedule 3, a prescription for a medicated feedingstuff containing a medicinal premix which includes an antibiotic may not be issued for prophylactic purposes.]
[F393(3) In relation to food-producing animals a medicated feedingstuffs prescription may not confer authority for more than one course of treatment.]
Extent Information
E103This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F380Words in Sch. 5 para. 19 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(e)(i) (with reg. 206)
F381Words in Sch. 5 para. 19 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(e)(ii) (with reg. 206)
F382Words in Sch. 5 para. 19(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(i) (with reg. 206)
F383Words in Sch. 5 para. 19(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(ii) (with reg. 206)
F384Sch. 5 para. 19(1)(ea) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(iii) (with reg. 206)
F385Sch. 5 para. 19(1)(h) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(iv) (with reg. 206)
F386Sch. 5 para. 19(1)(ja) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(v) (with reg. 206)
F387Word in Sch. 5 para. 19(1)(l) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(vi)(aa) (with reg. 206)
F388Words in Sch. 5 para. 19(1)(l) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(vi)(bb) (with reg. 206)
F389Sch. 5 para. 19(1)(na) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(vii) (with reg. 206)
F390Sch. 5 para. 19(1)(o) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(a)(viii) (with reg. 206)
F391Words in Sch. 5 para. 19(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(b) (with reg. 206)
F392Sch. 5 para. 19(2A)(2B) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(c) (with reg. 206)
F393Sch. 5 para. 19(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 140(d) (with reg. 206)
Prescriptions for feedingstuffs containing a veterinary medicinal productN.I.
19.—(1) A prescription for feedingstuffs containing a veterinary medicinal product must contain the following—
(a)the name and address of the person prescribing the product;
(b)the qualifications enabling the person to prescribe the product;
(c)the name and address of the keeper of the animals to be treated;
(d)the species of animal, identification and number of the animals;
(e)the premises at which the animals are kept if this is different from the address of the keeper;
(f)the date of the prescription;
(g)the signature or other authentication of the person prescribing the product;
(h)the name and amount of the product prescribed;
(i)the dosage and administration instructions;
(j)any necessary warnings;
(k)the withdrawal period;
(l)the manufacturer or the distributor of the feedingstuffs (who must be approved for the purpose);
(m)if the validity exceeds one month, a statement that not more than 31 days’ supply may be provided at any time;
(n)the name, type and quantity of feedingstuffs to be used;
(o)the inclusion rate of the veterinary medicinal product and the resulting inclusion rate of the active substance;
(p)any special instructions;
(q)the percentage of the prescribed feedingstuffs to be added to the daily ration; and
(r)if it is prescribed under the cascade, a statement to that effect.
(2) It is valid for three months or such shorter period as may be specified in the prescription.
(3) It must be sufficient for only one course of treatment.
Extent Information
E257This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Writing the [F394medicated feedingstuff prescription]E+W+S
20.—(1) The person who writes the [F395medicated feedingstuff prescription] must—
(a)give a copy to the person incorporating the [F396medicinal premix] into the feedingstuffs or to the distributor of the feedingstuffs [F397, whichever is the supplier to the end user];
(b)give one copy to the keeper of the animals to be treated;
(c)keep a copy.
(2) The person must be satisfied that—
(a)there is no undesirable interaction between the [F398medicinal premix] and any feed additive used in the feedingstuffs; and
(b)the active substance of the [F398medicinal premix] is not the same as an active substance in any feed additive used in the feedingstuffs.
[F399(3) The person must prescribe a [F400medicinal premix] authorised for incorporation in feedingstuffs but may, if there is no [F400medicinal premix] authorised for a condition in a particular species—
(a)prescribe a [F400medicinal premix] authorised for another species or for another condition in the same species, and
(b)prescribe more than one [F400medicinal premix],
provided all [F401medicinal premixes] prescribed are authorised for incorporation in feedingstuffs.]
Extent Information
E104This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F394Words in Sch. 5 para. 20 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 141(d)
F395Words in Sch. 5 para. 20(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 141(a)(i)
F396Words in Sch. 5 para. 20(1)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 141(a)(ii)(aa)
F397Words in Sch. 5 para. 20(1)(a) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 141(a)(ii)(bb)
F398Words in Sch. 5 para. 20(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 141(b)
F399Sch. 5 para. 20(3) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(2)
F400Words in Sch. 5 para. 20(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 141(c)(i)
F401Words in Sch. 5 para. 20(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 141(c)(ii)
Writing the prescriptionN.I.
20.—(1) The person who writes the prescription must—
(a)give a copy to the person incorporating the veterinary medicinal product into the feedingstuffs or to the distributor of the feedingstuffs;
(b)give one copy to the keeper of the animals to be treated;
(c)keep a copy.
(2) The person must be satisfied that—
(a)there is no undesirable interaction between the veterinary medicinal product and any feed additive used in the feedingstuffs; and
(b)the active substance of the veterinary medicinal product is not the same as an active substance in any feed additive used in the feedingstuffs.
[F610(3) The person must prescribe a veterinary medicinal product authorised for incorporation in feedingstuffs but may, if there is no veterinary medicinal product authorised for a condition in a particular species—
(a)prescribe a veterinary medicinal product authorised for another species or for another condition in the same species, and
(b)prescribe more than one veterinary medicinal product,
provided all veterinary medicinal products prescribed are authorised for incorporation in feedingstuffs.]
Extent Information
E258This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F610Sch. 5 para. 20(3) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(2)
PossessionE+W+S
21.—(1) No person other than a person holding the appropriate [F402authorisation] under this Schedule may be in possession of any—
(a)specified feed additive or [F403medicinal premix] to which this Schedule applies;
(b)[F404intermediate feedingstuffs] containing such an additive or a [F405medicinal premix]; or
(c)feedingstuffs or complementary feedingstuffs containing [F406a medicinal premix] unless supplied under these Regulations.
(2) No person other than a manufacturer or distributor may be in possession of feedingstuffs incorporating a [F407medicinal premix] unless it has been supplied under a [F408medicated feedingstuffs prescription].
Extent Information
E105This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F402Word in Sch. 5 para. 21(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 142(a)(i)
F403Words in Sch. 5 para. 21(1)(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 142(a)(ii)
F404Words in Sch. 5 para. 21(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 142(a)(iii)(aa)
F405Words in Sch. 5 para. 21(1)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 142(a)(iii)(bb)
F406Words in Sch. 5 para. 21(1)(c) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 142(a)(iv)
F407Words in Sch. 5 para. 21(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 142(b)(i)
F408Words in Sch. 5 para. 21(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 142(b)(ii)
PossessionN.I.
21.—(1) No person other than a person holding the appropriate approval under this Schedule may be in possession of any—
(a)specified feed additive or veterinary medicinal product to which this Schedule applies;
(b)premixtures containing such an additive or a veterinary medicinal product; or
(c)feedingstuffs or complementary feedingstuffs containing such an additive or a veterinary medicinal product unless supplied under these Regulations.
(2) No person other than a manufacturer or distributor may be in possession of feedingstuffs incorporating a veterinary medicinal product unless it has been supplied under a prescription.
Extent Information
E259This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Sampling and analysisE+W+S
22.—(1) If any enforcement action is taken under this Schedule based on a sample, that sample must have been taken and analysed in accordance with [F409Regulation (EC) No 152/2009 laying down methods of sampling and analysis for the official control of feedingstuffs.]
(2) Unless otherwise specified in the marketing authorisation, it is a defence if the active ingredient in the medicated feedingstuff sample is within the following tolerances—
[F410Tolerance table for medicated feedingstuff
| Level of active ingredient specified on the label | Tolerance |
|---|---|
| ≤500mg/kg | ±30% |
| ˃500mg/kg ≤5g/kg | ±20% |
| ˃5g/kg | ±10%] |
(3) Unless otherwise specified in the Commission Regulation authorising the specified feed additive in question, it is a defence if the active ingredient of a specified feed additive in a feedingstuff sample is within the tolerances set out in Articles 11(5) and paragraph 2(e) of Annex IV to Regulation (EC) No 767/2009 of the European Parliament and of the Council.
Extent Information
E106This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F409Words in Sch. 5 para. 22(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(e)
F410Sch. 5 para. 22(2) Table substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 143 (with reg. 207)
Sampling and analysisN.I.
22.—(1) If any enforcement action is taken under this Schedule based on a sample, that sample must have been taken and analysed in accordance with [F611Regulation (EC) No 152/2009 laying down methods of sampling and analysis for the official control of feedingstuffs.]
(2) Unless otherwise specified in the marketing authorisation, it is a defence if the active ingredient in the medicated feedingstuff sample is within the following tolerances—
Tolerance table for medicated feedingstuff
| Level of active ingredient specified on the label | Tolerance |
|---|---|
| ≤50 mg/kg | ± 50% |
| >50 mg/kg ≤ 500 mg/kg | ± 40% |
| >500 mg/kg ≤ 5g/kg | ± 30% |
| >5g/kg ≤50g/kg | ± 20% |
| >50g/kg | ± 10% |
(3) Unless otherwise specified in the Commission Regulation authorising the specified feed additive in question, it is a defence if the active ingredient of a specified feed additive in a feedingstuff sample is within the tolerances set out in Articles 11(5) and paragraph 2(e) of Annex IV to Regulation (EC) No 767/2009 of the European Parliament and of the Council.
Extent Information
E260This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F611Words in Sch. 5 para. 22(1) substituted (26.3.2019) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(a), 2(4)(e)
[F411Sampling for cross-contaminationE+W+S
22A.—(1) A feed business operator must ensure that cross-contamination of non-target feeds is as low as is reasonably achievable.
(2) A feed business operator must analyse samples of non-target feeds in order to determine whether cross-contamination into non-target feed has occurred.
(3) Where as a result of the process mentioned in sub-paragraph (2) it is determined that a cross-contamination rate has occurred which is 1% or more but less than 3% compared to the authorised maximum content, the feed business operator must make a record of this cross-contamination.
(4) Where as a result of the process mentioned in sub-paragraph (2) it is determined that a cross-contamination rate has occurred of 3% or more compared to the authorised maximum content, the feed business operator must conduct an investigation in order to discover the cause of the occurrence and make a record of the fact and any conclusions.
(5) The feed business operator must keep the records under sub-paragraphs (3) and (4) for at least five years.
(6) Upon request of the Secretary of State, the feed business operator must provide any information in the feed business operator’s possession relating to the matters mentioned in this paragraph.]
Textual Amendments
F411Sch. 5 para. 22A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 144 (with reg. 208)
StorageE+W+S
23. No person may store a [F412medicinal premix] intended for incorporation into feedingstuffs, or [F413an intermediate feedingstuff] or feedingstuffs containing a [F412medicinal premix], except in—
(a)a suitable storage area that is locked when not in use; or
(b)a hermetic container designed to store those products.
Extent Information
E107This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F412Words in Sch. 5 para. 23 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 145(a)
F413Words in Sch. 5 para. 23 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 145(b)
StorageN.I.
23. No person may store a veterinary medicinal product intended for incorporation into feedingstuffs, or a premixture or feedingstuffs containing a veterinary medicinal product, except in—
(a)a suitable storage area that is locked when not in use; or
(b)a hermetic container designed to store those products.
Extent Information
E261This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Packages and other containersE+W+S
24. No person may place on the market feedingstuffs containing a [F414medicinal premix] except in packages or containers that are sealed in such a way that, when the package or container is opened, the seal is damaged.
Extent Information
E108This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F414Words in Sch. 5 para. 24 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 146
Packages and other containersN.I.
24. No person may place on the market feedingstuffs containing a veterinary medicinal product except in packages or containers that are sealed in such a way that, when the package or container is opened, the seal is damaged.
Extent Information
E262This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
TransportE+W+S
25.—(1) No person may transport feedingstuffs by road tankers or in bulk unless the labelling requirements are set out in a document accompanying the feedingstuffs, and the transporter must hand over details when delivering the feedingstuffs unless these have already been provided to the purchaser.
(2) Any person transporting feedingstuffs containing [F415medicinal premixes] or specified feed additives in road tankers or similar containers must ensure that the vehicle or container is cleaned before any re-use if this is necessary to prevent undesirable interaction or contamination.
(3) In the case of feedingstuffs containing a [F416medicinal premix or specified feed additive] the transporter must ensure that the vehicle is accompanied by documentation stating this.
(4) Any person operating an undertaking transporting feedingstuffs containing [F417medicinal premixes] or specified feed additives must give written instructions to drivers on how to load and unload vehicles so as to avoid cross-contamination, and take reasonable steps to ensure that the driver complies with those instructions.
Extent Information
E109This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F415Words in Sch. 5 para. 25(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 147(a)
F416Words in Sch. 5 para. 25(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 147(b)
F417Words in Sch. 5 para. 25(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 147(c)
TransportN.I.
25.—(1) No person may transport feedingstuffs by road tankers or in bulk unless the labelling requirements are set out in a document accompanying the feedingstuffs, and the transporter must hand over details when delivering the feedingstuffs unless these have already been provided to the purchaser.
(2) Any person transporting feedingstuffs containing veterinary medicinal products or specified feed additives in road tankers or similar containers must ensure that the vehicle or container is cleaned before any re-use if this is necessary to prevent undesirable interaction or contamination.
(3) In the case of feedingstuffs containing a veterinary medicinal product the transporter must ensure that the vehicle is accompanied by documentation stating this.
(4) Any person operating an undertaking transporting feedingstuffs containing veterinary medicinal products or specified feed additives must give written instructions to drivers on how to load and unload vehicles so as to avoid cross-contamination, and take reasonable steps to ensure that the driver complies with those instructions.
Extent Information
E263This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Possession, placing on the market and use of feedingstuffsE+W+S
26.—(1) No person may possess, place on the market or feed to animals any feedingstuffs incorporating [F418medicinal premixes] or specified feed additives unless they have been incorporated in accordance with this Schedule.
(2) No person may feed to any animal, or buy, possess or supply for the purpose of feeding to any animal, any feedingstuff containing a [F419medicinal premix] or specified feed additive unless—
(a)that [F419medicinal premix] or specified feed additive is authorised for that species of animal and for the purpose for which it is used; or
(b)in the case of a [F419medicinal premix], it was prescribed for that animal.
[F420(2A) An animal keeper must ensure that any product to which this Schedule applies is appropriately stored in accordance with its authorisation.
(2B) An animal keeper must ensure in respect of any such product that—
(a)no cross-contamination occurs between products held by the keeper;
(b)no product contaminates any feedingstuff or feed material;
(c)no product escapes into the environment; and
(d)a product is administered only to correctly identified animals mentioned on the medicated feedingstuffs prescription.
(2C) An animal keeper must comply with the withdrawal period in relation to any such product.]
(3) This paragraph does not apply in relation to feedingstuffs if the [F421medicinal premix] has been incorporated in accordance with an animal test certificate or the feedingstuff has been imported in accordance with this Schedule.
Extent Information
E110This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F418Words in Sch. 5 para. 26(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 148(a)
F419Words in Sch. 5 para. 26(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 148(b)
F420Sch. 5 para. 26(2A)-(2C) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 148(c)
F421Words in Sch. 5 para. 26(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 148(d)
Possession, placing on the market and use of feedingstuffsN.I.
26.—(1) No person may possess, place on the market or feed to animals any feedingstuffs incorporating veterinary medicinal products or specified feed additives unless they have been incorporated in accordance with this Schedule.
(2) No person may feed to any animal, or buy, possess or supply for the purpose of feeding to any animal, any feedingstuff containing a veterinary medicinal product or specified feed additive unless—
(a)that veterinary medicinal product or specified feed additive is authorised for that species of animal and for the purpose for which it is used; or
(b)in the case of a veterinary medicinal product, it was prescribed for that animal.
(3) This paragraph does not apply in relation to feedingstuffs if the veterinary medicinal product has been incorporated in accordance with an animal test certificate or the feedingstuff has been imported in accordance with this Schedule.
Extent Information
E264This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F422Unused and expired medicated feedingstuffsE+W+S
26A. No person may feed medicated feedingstuffs which have passed their expiry date to an animal.]
Textual Amendments
F422Sch. 5 para. 26A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 149
Imports from third countriesE+W+S
F42327. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E111This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F423Sch. 5 para. 27 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(b) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
Imports from third countriesN.I.
27. No person may import a feedingstuff containing a veterinary medicinal product from a third country.
Extent Information
E265This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Trade between [F424countries]E+W+S
28. No person may bring in from another [F425country] a feedingstuff containing a veterinary medicinal product unless—
F426(a). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(b)it only contains a veterinary medicinal product that has the same quantitative and qualitative composition as a [F427medicinal premix] authorised in [F428Great Britain].
Extent Information
E112This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F424Word in Sch. 5 para. 28 heading substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(c)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F425Word in Sch. 5 para. 28 substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(c)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F426Sch. 5 para. 28(a) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(c)(iii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F427Words in Sch. 5 para. 28(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 150
F428Words in Sch. 5 para. 28(b) substituted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(9)
Trade [F612with] member StatesN.I.
28. No person may bring in from [F613a] member State a feedingstuff containing a veterinary medicinal product unless—
(a)the feedingstuff has been manufactured in accordance with the provisions of Council Directive 90/167/EEC (laying down the conditions governing the preparation, placing on the market and use of medicated feedingstuffs in the Community(15)) and Regulation (EC) No 183/2005 of the European Parliament and of the Council laying down requirements for food hygiene; and
(b)it only contains a veterinary medicinal product that has the same quantitative and qualitative composition as a veterinary medicinal product authorised in [F614Northern Ireland].
Extent Information
E266This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F612Word in Sch. 5 para. 28 heading substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(c)(i)
F613Word in Sch. 5 para. 28 substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(c)(ii)
F614Words in Sch. 5 para. 28(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(c)(iii)
Import for incorporation into [F429intermediate feedingstuffs] or feedingstuffs for exportE+W+S
29.—[F430(1) A manufacturer of [F431intermediate feedingstuffs] or feedingstuffs who imports a veterinary medicinal product authorised in another F432... country for the purposes of incorporating it into [F431intermediate feedingstuffs] or feedingstuffs for export does not commit an offence under regulation 43(q) (importation of an unauthorised veterinary medicinal product) or regulation 43(r) (possession of an unauthorised veterinary medicinal product)]
(2) No person may place that [F433intermediate feedingstuff] or feedingstuff on the market in the United Kingdom once the veterinary medicinal product has been incorporated into it.
Extent Information
E113This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F429Words in Sch. 5 para. 29 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 151(c)
F430Sch. 5 para. 29(1) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(3)
F431Words in Sch. 5 para. 29(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 151(a)
F432Words in Sch. 5 para. 29(1) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(d) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F433Words in Sch. 5 para. 29(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 151(b)
Import for incorporation into premixture or feedingstuffs for exportN.I.
29.—[F615(1) A manufacturer of premixture or feedingstuffs who imports a veterinary medicinal product authorised in [F616a] Member State or third country for the purposes of incorporating it into premixture or feedingstuffs for export does not commit an offence under regulation 43(q) (importation of an unauthorised veterinary medicinal product) or regulation 43(r) (possession of an unauthorised veterinary medicinal product)]
(2) No person may place that premixture or feedingstuff on the market in [F617Northern Ireland] once the veterinary medicinal product has been incorporated into it.
Extent Information
E267This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F615Sch. 5 para. 29(1) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(3)
F616Word in Sch. 5 para. 29(1) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(d)(i)
F617Words in Sch. 5 para. 29(2) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(15)(d)(ii)
Animals on domestic premisesE+W+S
30.—(1) The requirements of paragraph 7 ([F434authorisation] of manufacturers and distributors of feedingstuffs containing [F435medicinal premix]) do not apply in relation to a person who incorporates a [F435medicinal premix] into feedingstuffs in domestic premises for feeding, on those premises—
(a)non-food-producing animals; or
(b)food-producing animals provided that the animals or products from those animals are not sold or supplied commercially.
(2) Notwithstanding paragraphs 16 and 18 of this Schedule, a veterinary surgeon, a pharmacist or a suitably qualified person who is registered in accordance with paragraph 14 of Schedule 3 may be supplied with and may supply [F436an intermediate feedingstuff] containing a [F437medicinal premix], or feedingstuffs containing a [F437medicinal premix], to such a producer.
(3) The requirement for a written prescription does not apply in relation to such supply, but the provisions of Schedule 3 relating to supply of a veterinary medicinal product apply in relation to the supply of [F438intermediate feedingstuffs] and feedingstuffs in the same way as they apply to a veterinary medicinal product.
Extent Information
E114This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F434Word in Sch. 5 para. 30(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 152(a)(i)
F435Words in Sch. 5 para. 30(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 152(a)(ii)
F436Words in Sch. 5 para. 30(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 152(b)(i)
F437Words in Sch. 5 para. 30(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 152(b)(ii)
F438Words in Sch. 5 para. 30(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 152(c)
Animals on domestic premisesN.I.
30.—(1) The requirements of paragraph 7 (approval of manufacturers and distributors of feedingstuffs containing veterinary medicinal product) do not apply in relation to a person who incorporates a veterinary medicinal product into feedingstuffs in domestic premises for feeding, on those premises—
(a)non-food-producing animals; or
(b)food-producing animals provided that the animals or products from those animals are not sold or supplied commercially.
(2) Notwithstanding paragraphs 16 and 18 of this Schedule, a veterinary surgeon, a pharmacist or a suitably qualified person who is registered in accordance with paragraph 14 of Schedule 3 may be supplied with and may supply a premixture containing a veterinary medicinal product, or feedingstuffs containing a veterinary medicinal product, to such a producer.
(3) The requirement for a written prescription does not apply in relation to such supply, but the provisions of Schedule 3 relating to supply of a veterinary medicinal product apply in relation to the supply of premixture and feedingstuffs in the same way as they apply to a veterinary medicinal product.
Extent Information
E268This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
OffencesE+W+S
31. It is an offence to fail to comply with—
(a)paragraph 2(2);
(b)paragraph 3(3) or (4);
(c)paragraph 5(2) or (3);
(d)paragraph 6;
(e)paragraph 7(2) or (5);
(f)paragraph 8;
(g)paragraph 9;
(h)paragraph 10;
(i)paragraph 11;
(j)paragraph 12(4);
(k)paragraph 13;
(l)paragraph 14(4);
(m)paragraph 15;
(n)paragraph 16;
(o)paragraph 17;
(p)paragraph 18;
[F439(pa)paragraph 19;]
(q)paragraph [F44020];
(r)paragraph 21;
[F441(ra)paragraph 22A;]
(s)paragraph 23;
(t)paragraph 24;
(u)paragraph 25;
(v)paragraph 26(1) [F442, (2), (2A), (2B) or (2C)];
[F443(va)paragraph 26A;]
F444(w). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(x)paragraph 28; or
(y)paragraph 29(2).
Extent Information
E115This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F439Sch. 5 para. 31(pa) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 153(a)
F440Word in Sch. 5 para. 31(q) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(4)
F441Sch. 5 para. 31(ra) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 153(b)
F442Words in Sch. 5 para. 31(v) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 153(c)
F443Sch. 5 para. 31(va) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 153(d)
F444Sch. 5 para. 31(w) omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(35)(e) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
OffencesN.I.
31. It is an offence to fail to comply with—
(a)paragraph 2(2);
(b)paragraph 3(3) or (4);
(c)paragraph 5(2) or (3);
(d)paragraph 6;
(e)paragraph 7(2) or (5);
(f)paragraph 8;
(g)paragraph 9;
(h)paragraph 10;
(i)paragraph 11;
(j)paragraph 12(4);
(k)paragraph 13;
(l)paragraph 14(4);
(m)paragraph 15;
(n)paragraph 16;
(o)paragraph 17;
(p)paragraph 18;
(q)paragraph [F61820] ;
(r)paragraph 21;
(s)paragraph 23;
(t)paragraph 24;
(u)paragraph 25;
(v)paragraph 26(1) or (2);
(w)paragraph 27;
(x)paragraph 28; or
(y)paragraph 29(2).
Extent Information
E269This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F618Word in Sch. 5 para. 31(q) substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 3(4)
Regulation 15(4)
SCHEDULE 6U.K.Exemptions for small pet animals
Animals to which this Schedule appliesU.K.
1. This Schedule applies in relation to veterinary medicinal products intended solely for the following animals kept exclusively as a pet—
(a)aquarium animals;
(b)cage birds;
(c)ferrets;
(d)homing pigeons;
(e)rabbits;
(f)small rodents; and
(g)terrarium animals.
Placing on the market, importing and administering the productU.K.
2. A veterinary medicinal product intended solely for an animal to which this Schedule applies may be placed on the market, imported or administered without a marketing authorisation if it complies with this Schedule [F445and the manufacturer appears on the register maintained under paragraph 3A].
Textual Amendments
F445Words in Sch. 6 para. 2 inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 155
ManufactureE+W+S
3.—[F446(1)] The product must have been manufactured by—
(a)the holder of a manufacturing authorisation if manufactured in [F447Great Britain];
F448(b). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F449(c). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(d)in the case of any other country, a manufacturer whose premises have been inspected and approved by an officer of the Secretary of State.
[F450(2) Sub-paragraph (1)(d) does not apply where the United Kingdom has a formal agreement with the exporting country that includes mutual recognition of good manufacturing practice or where the Secretary of State is satisfied that the exporting country requires manufacturers of veterinary medicinal products to apply standards of good manufacturing practice which are at least equivalent to those in Great Britain.]
Extent Information
E116This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F446Sch. 6 para. 3 renumbered as Sch. 6 para. 3(1) (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(a) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)); 2020 c. 1, Sch. 5 para. 1(1)
F447Words in Sch. 6 para. 3(1)(a) substittued (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(b)(i) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(e)(i)); 2020 c. 1, Sch. 5 para. 1(1)
F448Sch. 6 para. 3(1)(b) omitted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(e)(i)); 2020 c. 1, Sch. 5 para. 1(1)
F449Sch. 6 para. 3(1)(c) omitted (E.W.S.) (31.12.2020) by The Veterinary Medicines and Animals and Animal Products (Examination of Residues and Maximum Residue Limits) (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/676), regs. 1(2)(b), 3(36)(b)(ii) (as amended by S.I. 2020/1461, regs. 1(2)(a), 3(2)(b)(3)(e)(i)); 2020 c. 1, Sch. 5 para. 1(1)
F450Sch. 6 para. 3(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 156
ManufactureN.I.
3. The product must have been manufactured by—
(a)the holder of a manufacturing authorisation if manufactured in the United Kingdom;
(b)the holder of a manufacturing authorisation issued under Directive 2001/82/EC if manufactured in [F619a] member State;
(c)in the case of Australia, Canada, New Zealand, or Switzerland, the holder of an authorisation from the competent authority permitting the manufacture of medicinal products;
(d)in the case of any other country, a manufacturer whose premises have been inspected and approved by an officer of the Secretary of State.
Extent Information
E270This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F619Word in Sch. 6 para. 3(b) substituted (N.I.) (31.12.2020) by The Animals (Health, Identification, Trade and Veterinary Medicines) (Amendment) (EU Exit) Regulations (Northern Ireland) 2020 (S.R. 2020/353), regs. 1(3), 10(16)
[F451Register of persons placing veterinary medicinal products on the market (small pet animals)E+W+S
3A.—(1) A person placing the product on the market must be registered in accordance with this paragraph.
(2) An application for registration in respect of that person must be submitted under sub-paragraph (4)—
(a)at least two months before that person places the product on the market, or
(b)where that person has already placed the product on the market, within six months of the date on which this provision comes into force.
(3) Information may be submitted to the Secretary of State pursuant to sub-paragraph (2) prior to the date on which this provision comes into force, and in such a case—
(a)as regards an applicant for registration who has not already placed the product on the market, the period of two months is to be treated as having started on the date of submission;
(b)as regards an applicant for registration who has already placed the product on the market, the information is to be treated as having been submitted within the period of six months.
(4) An application for registration must be made to the Secretary of State electronically and must include—
(a)the name and address of the person placing the product on the market;
(b)the individual making the application in respect of that person;
(c)the telephone number and email address of the individual mentioned in sub-paragraph (b);
(d)the name and address of the manufacturer of the product;
(e)the brand name of the product;
(f)the names and quantities of the active substances;
(g)the method and (where applicable) route of administration;
(h)the dosage instructions;
(i)the category of animal mentioned in paragraph 1 for which the product is intended.
(5) For the purposes of sub-paragraph (1) the Secretary of State must establish and maintain a register of persons placing on the market products to which this Schedule applies.
(6) The particulars entered on the register must include the name and the address of the person mentioned in sub-paragraph (1).]
Textual Amendments
F451Sch. 6 paras. 3A, 3B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 157
[F451Persons registered in accordance with paragraph 3A: annual returnE+W+S
3B. At least once each calendar year a person registered under paragraph 3A must notify the Secretary of State in writing of the following in respect of each product placed on the market—
(a)the name and registered address of the person (if different from that listed on the register);
(b)the individual designated for the purpose of making the annual return under this paragraph;
(c)the telephone number and email address of the individual mentioned in sub-paragraph (b);
(d)the name and address of the manufacturer of the product;
(e)the brand name of the product;
(f)the names and quantities of the active substances;
(g)the method and (where applicable) route of administration;
(h)the dosage instructions;
(i)the category of animal mentioned in paragraph 1 for which the product is intended.]
Textual Amendments
F451Sch. 6 paras. 3A, 3B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 157
Approval of the active substanceE+W+S
4.—(1) The Secretary of State may approve an active substance for use in a veterinary medicinal product manufactured under this Schedule.
(2) The Secretary of State may not grant an approval if the active substance requires veterinary control.
(3) The approval must specify the species of animals for which it is approved, and may specify how the active substance or a product containing it is to be administered.
(4) The Secretary of State may suspend or revoke the approval (or limit it to a smaller number of species) if—
(a)it is demonstrated that the substance requires veterinary control;
(b)[F452adverse events] are reported making suspension or revocation necessary; or
(c)it is demonstrated that the substance—
(i)is carcinogenic;
(ii)is genotoxic; or
(iii)shows developmental toxicity (including teratogenicity).
(5) The procedure for the refusal, suspension or revocation of an approval under this paragraph is the same as the procedure for a marketing authorisation.
Extent Information
E117This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F452Words in Sch. 6 para. 4(4)(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 158
Approval of the active substanceN.I.
4.—(1) The Secretary of State may approve an active substance for use in a veterinary medicinal product manufactured under this Schedule.
(2) The Secretary of State may not grant an approval if the active substance requires veterinary control.
(3) The approval must specify the species of animals for which it is approved, and may specify how the active substance or a product containing it is to be administered.
(4) The Secretary of State may suspend or revoke the approval (or limit it to a smaller number of species) if—
(a)it is demonstrated that the substance requires veterinary control;
(b)serious adverse reactions are reported making suspension or revocation necessary; or
(c)it is demonstrated that the substance—
(i)is carcinogenic;
(ii)is genotoxic; or
(iii)shows developmental toxicity (including teratogenicity).
(5) The procedure for the refusal, suspension or revocation of an approval under this paragraph is the same as the procedure for a marketing authorisation.
Extent Information
E271This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
The productU.K.
5.—(1) The active substance in the veterinary medicinal product must be approved under paragraph 4.
(2) The veterinary medicinal product must not be an antibiotic.
(3) It must not contain any narcotic or psychotropic substance.
(4) It must not be intended for treatments or pathological processes that require a precise prior diagnosis or the use of which may cause effects that impede or interfere with subsequent diagnostic or therapeutic measures.
LabellingU.K.
6.—(1) The product must be clearly labelled as being exempt from the requirements of these Regulations in relation to a marketing authorisation.
(2) The labelling must show the following—
(a)the name of the veterinary product, including, if it is part of the name, its strength and pharmaceutical form;
(b)the authorisation number of the manufacturer;
(c)the name and strength of each active substance;
(d)the route of administration;
(e)the batch number;
(f)the expiry date;
(g)the words “For animal treatment only”;
(h)the contents by weight, volume or number of dose units;
(i)the name and address of the manufacturer or distributor;
(j)the target species;
(k)the words “Keep out of reach of children”;
(l)storage instructions;
(m)the shelf-life after the immediate packaging has been opened for the first time;
(n)disposal advice;
(o)full indications, including—
(i)therapeutic indications;
(ii)contra-indications;
(iii)interaction with other medicines and other forms of interaction; and
(p)dosage instructions.
(3) If there is insufficient room on the label, the information may instead be in a package leaflet, but the leaflet must contain all the information in the preceding sub-paragraph other than the batch number and the expiry date, but the label on the product must contain at least the following—
(a)the name of the veterinary medicinal product;
(b)its active substance and its strength;
(c)the route of administration;
(d)the batch number;
(e)the expiry date; and
(f)the words “For animal treatment only”.
AdministrationU.K.
7. The method of administration must be oral or topical or (in the case of a product for fish) by addition to the water.
Pack sizeU.K.
8. The pack size must only be sufficient for a single course of treatment or, in the case of a veterinary medicinal product for aquarium fish, sufficient for a single course of treatment of no more than 7 administrations to an aquarium of 25,000 litres.
[F453Adverse events]E+W+S
9.—(1) The [F454manufacturer or importer] of a veterinary medicinal product must—
(a)notify the Secretary of State within 15 days of learning of any serious [F455adverse events]; and
(b)make a record of each [F456adverse event] and serious [F456adverse event] on becoming aware of it and keep it for three years.
F457(2) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E118This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F453Words in Sch. 6 para. 9 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 159(e)
F454Words in Sch. 6 para. 9 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 159(a)
F455Words in Sch. 6 para. 9(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 159(b)
F456Words in Sch. 6 para. 9(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 159(c)
F457Sch. 6 para. 9(2) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 159(d)
Adverse reactionsN.I.
9.—(1) The manufacturer, importer or retailer of a veterinary medicinal product must—
(a)notify the Secretary of State within 15 days of learning of any serious adverse reactions (as defined in paragraph 57 of Schedule 1); and
(b)make a record of each adverse reaction and serious adverse reaction on becoming aware of it and keep it for three years.
(2) It is an offence to fail to comply with this paragraph.
Extent Information
E272This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F458OffencesE+W+S
10. It is an offence to fail to comply with—
(a)paragraph 3A(1);
(b)paragraph 3B; or
(c)paragraph 9(1).]
Textual Amendments
F458Sch. 6 para. 10 inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 160
Regulation 16
SCHEDULE 7U.K.Fees
PART 1U.K.Introduction
InterpretationE+W+S
1.— [F459(1)] In this Schedule—
F460...
“pharmaceutical product” means any veterinary medicinal product other than an immunological product [F461or a biological veterinary medicinal product that is not immunological];
“simultaneous application” is an application in which, at the time an authorisation for a product is applied for, one or more additional applications are submitted for products that are identical to the first product except that—
(a)
in the case of an immunological product, they have a lesser number of antigens than the first product, but only contain antigens contained in the first product; and
(b)
in the case of a pharmaceutical product, they have different strengths of the active substance,
F462...
[F463(2) For the purposes of this Schedule “manufacturing authorisation” means the following activities—
(a)manufacture or import of an authorised veterinary medicinal product;
(b)manufacture of a product to which paragraph 2 of Schedule 6 relates;
(c)manufacture of a product for administration under the cascade;
(d)manufacture of—
(i)an autogenous vaccine;
(ii)a stem cell product; or
(iii)a blood product for administration to non-food animals.]
Extent Information
E119This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F459Sch. 7 para. 1 renumbered as Sch. 7 para. 1(1) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 162(a)
F460Words in Sch. 7 para. 1 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(a)(i); 2020 c. 1, Sch. 5 para. 1(1)
F461Words in Sch. 7 para. 1(1) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 162(b)
F462Words in Sch. 7 para. 1 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(a)(ii); 2020 c. 1, Sch. 5 para. 1(1)
F463Sch. 7 para. 1(2) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 162(c)
InterpretationN.I.
1. In this Schedule—
“national application” means an application for a marketing authorisation that does not involve another member State;
“pharmaceutical product” means any veterinary medicinal product other than an immunological product;
“simultaneous application” is an application in which, at the time an authorisation for a product is applied for, one or more additional applications are submitted for products that are identical to the first product except that—
(a)
in the case of an immunological product, they have a lesser number of antigens than the first product, but only contain antigens contained in the first product; and
(b)
in the case of a pharmaceutical product, they have different strengths of the active substance,
and, in the case of an application involving more than one member State, the additional applications do not include a member State that was not included in the first application.
Extent Information
E273This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Payment of feesU.K.
2. All fees under this Schedule are payable to the Secretary of State.
Time of paymentU.K.
3. All fees are payable on invoice unless otherwise specified.
Multiple inspectionsU.K.
4. If a site, premises or establishment is inspected for more than one type of authorisation [F464, approval] or registration at the same time, [F465and in relation to the same legal entity,] the fee is the sum of—
(a)the highest fee payable; and
(b)50% of each of the other fees.
Textual Amendments
F464Word in Sch. 7 para. 4 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 163(a)
F465Words in Sch. 7 para. 4 inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 163(b)
Expenses for inspections outside the United KingdomU.K.
5. Whenever premises outside the United Kingdom are inspected, the travel and subsistence costs of the inspectors and interpreters’ fees are payable in addition to the inspection fee specified.
TranslationU.K.
6. All translation costs are charged additionally.
PART 2U.K.Fees relating to marketing authorisations
[F466Application for a marketing authorisation for a pharmaceutical [F467, immunological or biological that is not immunological] veterinary medicinal product]E+W+S
7. The following table sets out the fees relating to a pharmaceutical [F468, immunological or biological that is not immunological] veterinary medicinal product for—
(a)[F469an application] for a marketing authorisation that is—
(i)a full application under Part 1 of Schedule 1; [F470or]
(ii)a bibliographic application [F471for a pharmaceutical veterinary medicinal product]; F472...
F473(iii). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F474(b). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F475(c). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
| [F476Application | Fee (£) per authorisation |
|---|---|
| Base fee | 27,995 |
| Fee for 1st additional strength | 4,590 |
| Fee for each subsequent additional strength | 1,465] |
Extent Information
E120This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F466Sch. 7 para. 7 heading substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(i); 2020 c. 1, Sch. 5 para. 1(1)
F467Words in Sch. 7 para. 7 heading inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 164(d)
F468Words in Sch. 7 para. 7 inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 164(a)
F469Words in Sch. 7 para. 7(a) substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(ii); 2020 c. 1, Sch. 5 para. 1(1)
F470Word in Sch. 7 para. 7(a)(i) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 164(b)(i)
F471Words in Sch. 7 para. 7(a)(ii) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 164(b)(ii)(aa)
F472Word in Sch. 7 para. 7(a)(ii) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 164(b)(ii)(bb)
F473Sch. 7 para. 7(a)(iii) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 164(b)(iii)
F474Sch. 7 para. 7(b) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(iii); 2020 c. 1, Sch. 5 para. 1(1)
F475Sch. 7 para. 7(c) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(b)(iii); 2020 c. 1, Sch. 5 para. 1(1)
F476Sch. 7 para. 7(c) Table substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 164(c)
Specified pharmaceutical applicationsN.I.
7. The following table sets out the fees relating to a pharmaceutical veterinary medicinal product for—
(a)a national application for a marketing authorisation that is—
(i)a full application under Part 1 of Schedule 1;
(ii)a bibliographic application; or
(iii)an application based on pharmacological equivalence;
(b)an application for a marketing authorisation using the decentralised procedure where the United Kingdom is a concerned member State;
(c)an application for the mutual recognition of a product authorised in another member State.
| Application | Full national application under Part 1 of Schedule 1 (£) | Bibliographic national application (£) | Pharmacologically equivalent national application | Decentralised application where the UK is a concerned member State or recognition of a product authorised in another member State (£) | ||
|---|---|---|---|---|---|---|
| Reference product authorised in UK (£) | Reference product not authorised in UK (£) | |||||
| Base Fee: | 13,530 | 12,115 | 7,195 | 9,220 | 6,515 | |
| Additional fee if any of the target species is a food-producing animal: | 3,905 | 3,585 | 2,155 | 2,760 | 1,415 | |
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom— | ||||||
| food-producing animal: | 7,465 | 6,595 | 5,885 | 7,495 | 2,630 | |
| non-food-producing animal: | 6,525 | 5,855 | 5,590 | 7,155 | 2,295 | |
| Additional fee for each additional pack type: | 740 | 740 | 605 | 775 | 330 | |
| Additional fee for each additional active ingredient (food-producing animal): | 6,465 | 6,125 | 4,040 | 5,165 | 2,085 | |
| Additional fee for each additional active ingredient (non-food-producing animal): | 4,310 | 4,105 | 3,235 | 4,135 | 1,475 | |
| Additional fee if there is more than one target species, for each additional species (food-producing animal): | 3,970 | 3,565 | 2,425 | 3,100 | 1,280 Applies for a maximum of 2 additional species | |
| Additional fee if there is more than one target species, for each additional species (non- food-producing animal): | 2,495 | 2,090 | 1,550 | 1,980 | 805 Applies for a maximum of 2 additional species | |
| Additional fee for each additional recommended route of administration (food-producing animal): | 2,695 | 2,490 | 1,620 | 2,070 | 940 | |
| Additional fee for each additional recommended route of administration (non- food-producing animal): | 1,215 | 1,010 | 740 | 945 | 405 | |
| Simultaneous applications: fee for each additional product in the application: | 2,895 | 2,895 | 2,895 | 3,705 | 1,685 | |
Extent Information
E274This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F477Application for a marketing authorisation for specific applicationsE+W+S
7A. The fee for an application for a marketing authorisation which involves one or more of the following is £45,000—
(a)any biotechnical process involving recombinant DNA or the controlled expression of genes;
(b)a veterinary medicinal product containing a new active substance;
(c)a biopharmaceutical product.]
Textual Amendments
F477Sch. 7 para. 7A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 165
Decentralised pharmaceutical application where the United Kingdom is the reference member StateE+W+S
F4788. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E121This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F478Sch. 7 para. 8 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(c); 2020 c. 1, Sch. 5 para. 1(1)
Decentralised pharmaceutical application where the United Kingdom is the reference member StateN.I.
8. The fee for a decentralised application for a pharmaceutical product where the United Kingdom is the reference member State is the same as for a national application as set out in the table in paragraph 7, with the addition of the fees in the following table.
Fees for decentralised pharmaceutical application where the United Kingdom is the reference member State
| Application | Additional fee for a pharmacologically equivalent product (£) | Additional fee otherwise (£) | |
|---|---|---|---|
| Food-producing animal: one member State: | 5,230 | 3,705 | |
| Non-food-producing animal: one member State: | 3,985 | 3,220 | |
| Each additional member State: | 530 | 530 | |
| Simultaneous application: fee for each additional product in the application: | |||
| one member State: | 6,670 | 6,670 | |
| each additional member State: | 120 | 120 | |
Extent Information
E275This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for a marketing authorisation for an immunological or biosimilar productE+W+S
F4799. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E122This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F479Sch. 7 para. 9 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 166
Application for a marketing authorisation for an immunological or biosimilar productN.I.
9.—(1) The fee for a national application for a marketing authorisation relating to an immunological or biosimilar product, a decentralised application where the United Kingdom is the concerned member State or the mutual recognition of a product authorised in another member State is in accordance with the following table.
(2) In this paragraph a biosimilar application means an application made in accordance with Article 13(4) of Directive 2001/82/EC and a biosimilar product means a product which is the subject of such an application.
Fees for specified immunological and biosimilar applications
| Application | National application for a marketing authorisation(£) | Decentralised application where the UK is a concerned member State or recognition of a product authorised in another member State (£) | |
|---|---|---|---|
| 1. Immunological or biosimilar product other than in paragraph 2 below: Base fee: | 11,775 | 5,785 | |
| The following fees are in addition to the base fee— | |||
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom, and for each new combination of active ingredients: | 7,405 | 2,490 | |
| Additional fee for each adjuvant or preservative not previously included in a veterinary medicinal product authorised in the United Kingdom and for each new combination of adjuvants or preservatives: | 1,345 | 675 | |
| More than one antigenic component – fee for each additional component: | 1,350 | 405 | |
| More than one species – fee for each additional species: | 5,380 | 1,615 Applies for a maximum of 2 additional species | |
| More than one route of administration – fee for each additional route of administration: | 5,380 | 1,615 | |
| Simultaneous application - fee for each additional product in the application: | 2,895 | 1,685 | |
| 2. Immunological or product that is identical to a product already authorised in the United Kingdom but with a lesser number of antigens and that only contains antigens contained in that product: | 10,430 | 5,380 | |
Extent Information
E276This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Decentralised immunological application where the United Kingdom is the reference member StateE+W+S
F48010. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E123This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F480Sch. 7 para. 10 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(e); 2020 c. 1, Sch. 5 para. 1(1)
Decentralised immunological application where the United Kingdom is the reference member StateN.I.
10. The fee for a decentralised application for a marketing authorisation for an immunological product where the United Kingdom is the reference member State is the same as for a national application set out in the previous table, with the addition of the fees in the following table—
Fees for decentralised immunological application where the United Kingdom is the reference member State
| Application | Additional fee (£) | |
|---|---|---|
| One member State: | 3,470 | |
| Each additional member State: | 530 | |
| Simultaneous applications: fee for each additional product in the application: | ||
| one member State: | 6,670 | |
| each additional member State: | 120 | |
Extent Information
E277This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F481Application for a marketing authorisation based on informed consentE+W+S
11. The fee for applications for marketing authorisations using identical data submitted simultaneously or on the basis of information provided under paragraph 9 of Schedule 1 is as follows—
| Application | Fee (£) per authorisation |
|---|---|
| Application | 1,465] |
Extent Information
E124This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F481Sch. 7 para. 11 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 167
Applications for a marketing authorisation using data already assessedN.I.
11. The fees for applications for marketing authorisations using identical data submitted simultaneously or on the basis of information provided under Article 13(c) of Directive 2001/82/EC are in accordance with the following table.
Fees for a marketing authorisation using data already assessed
| Application | Fee (£)per authorisation | |
|---|---|---|
| Decentralised application where the United Kingdom is the reference member State— | ||
| one member State: | 4,165 | |
| each additional member State: | 530 | |
| Any other application: | 945 | |
Extent Information
E278This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for an exceptional marketing authorisation (pharmaceutical)U.K.
12. The fee for an application for an exceptional marketing authorisation for a pharmaceutical product is in accordance with the following table.
Fees for an exceptional marketing authorisation for a pharmaceutical product
| Application | Provisional (£) | Limited (£) | |
|---|---|---|---|
| Base Fee: | 12,015 | 6,765 | |
| The following fees are in addition to the base fee— | |||
| Additional fee if any of the target species is a food-producing animal: | 3,905 | 1,952 | |
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom— | |||
| food-producing animal: | 5,850 | 3,732 | |
| non-food-producing animal: | 4,910 | 3,262 | |
| Additional fee for each additional pack type: | 710 | 370 | |
| Additional fee for each additional active ingredient (food-producing animal): | 5,955 | 3,232 | |
| Additional fee for each additional active ingredient (non-food-producing animal): | 3,800 | 2,155 | |
| Additional fee if there is more than one target species, for each additional species (food-producing animal): | 2,965 | 1,985 | |
| Additional fee if there is more than one target species, for each additional species (non-food-producing animal): | 1,485 | 1,247 | |
| Additional fee for each additional recommended route of administration (food-producing animal): | 2,185 | 1,347 | |
| Additional fee for each additional recommended route of administration (non-food-producing animal): | 710 | 608 | |
| Simultaneous applications— fee for each additional product in the application: | 2,895 | 1,447 | |
Fees for an application for an exceptional marketing authorisation [F482(immunological or biological non-immunological)]E+W+S
13. The fee for an application for an exceptional marketing authorisation for an immunological product [F483or a biological veterinary medicinal product that is not immunological] is in accordance with the following table.
Fees for an exceptional marketing authorisation for an immunological product [F483or a biological veterinary medicinal product that is not immunological]
| Application | Provisional (£) | Limited (£) |
|---|---|---|
| Base fee: | 10,810 | 5,887 |
| The following fees are in addition to the base fee— | ||
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom, and for each new combination of active ingredients: | 5,650 | 3,702 |
| Additional fee for each adjuvant or preservative not previously included in a veterinary medicinal product authorised in the United Kingdom and for each new combination of adjuvants or preservatives: | 1,350 | 672 |
| More than one antigenic component – fee for each additional component: | 1,190 | 675 |
| More than one species – fee for each additional species: | 4,060 | 2,690 |
| More than one route of administration – fee for each additional route of administration: | 4,060 | 2,690 |
| Simultaneous application - fee for each additional product in the application: | 2,895 | 1,447 |
Extent Information
E125This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F482Words in Sch. 7 para. 13 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 168(b)
F483Words in Sch. 7 para. 13 inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 168(a)
Fees for an application for an exceptional marketing authorisation (immunological)N.I.
13. The fee for an application for an exceptional marketing authorisation for an immunological product is in accordance with the following table.
Fees for an exceptional marketing authorisation for an immunological product
| Application | Provisional (£) | Limited (£) |
|---|---|---|
| Base fee: | 10,810 | 5,887 |
| The following fees are in addition to the base fee— | ||
| Additional fee for each active ingredient not previously included in a veterinary medicinal product authorised in the United Kingdom, and for each new combination of active ingredients: | 5,650 | 3,702 |
| Additional fee for each adjuvant or preservative not previously included in a veterinary medicinal product authorised in the United Kingdom and for each new combination of adjuvants or preservatives: | 1,350 | 672 |
| More than one antigenic component – fee for each additional component: | 1,190 | 675 |
| More than one species – fee for each additional species: | 4,060 | 2,690 |
| More than one route of administration – fee for each additional route of administration: | 4,060 | 2,690 |
| Simultaneous application - fee for each additional product in the application: | 2,895 | 1,447 |
Extent Information
E279This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Fee for the conversion from an exceptional to a full marketing authorisationU.K.
14. The fee for the conversion of an exceptional marketing authorisation to a full marketing authorisation is £3,000.
Application for a marketing authorisation relating to a parallel importE+W+S
F48415. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E126This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F484Sch. 7 para. 15 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 169
Application for a marketing authorisation relating to a parallel importN.I.
15. The fee for a marketing authorisation for a parallel import is in accordance with the following table.
Parallel imports
| Application | Fee (£) | |
|---|---|---|
| Application where the imported product has been authorised in accordance with the mutual recognition procedure or decentralised procedure, and the United Kingdom is included in these procedures— | ||
| import from one or more member States: | 1,755 | |
| Application to add an additional member State after the marketing authorisation has been granted – fee for each member State: | 455 | |
| Application where the imported product has not been authorised in accordance with the mutual recognition procedure or the decentralised procedure but where the imported product originates from the same manufacturing site as the product authorised in the United Kingdom to which the imported product is considered to be essentially similar: | 2,130 | |
| Any other application – fee for each member State from which the product is imported: | 4,710 | |
Extent Information
E280This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F485Fee for a generic marketing authorisationE+W+S
15A.—(1) The fee for a marketing authorisation in respect of a generic veterinary medicinal product is to be calculated in accordance with the following table.
| Application | Fee (£) per authorisation | |
|---|---|---|
| Hybrid | Standard | |
| Base Fee | 13,950 | 12,390 |
| Fee for 1st additional strength | 4,590 | |
| Fee for each subsequent additional strength | 1,465. | |
(2) In this paragraph “hybrid” means an application to which paragraph 10A of Schedule 1 applies.]
Textual Amendments
F485Sch. 7 para. 15A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 170
Application to change the distribution category of a product authorised through the centralised procedureE+W+S
F48616. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E127This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F486Sch. 7 para. 16 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(h); 2020 c. 1, Sch. 5 para. 1(1)
Application to change the distribution category of a product authorised through the centralised procedureN.I.
16. The fee to change the distribution category of a product authorised through the centralised procedure is £3,135.
Extent Information
E281This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for a variation to a marketing authorisation F487....E+W+S
17.—(1) This paragraph applies in relation to an application for a variation to one or more marketing authorisations except where paragraph F488... 21 applies.
(2) The fees for the variations to which this paragraph applies are set out in the following table.
(3) Where applications are made at the same time seeking an identical change to the terms of more than one marketing authorisation, and those applications are based on identical data, fees are payable as for a grouped variation.
(4) References in this paragraph to a grouped variation being “led” by a particular type of variation indicate that the principal variation in that group is a variation of that type.
| [F489Type of variations | Fee (£) | ||
|---|---|---|---|
| Single variations; one change for each product | |||
| Variation – standard | 2,895 | ||
| Unless the variation is— | |||
| (a) a change of route of administration, or the addition of a new one, of— | |||
| (i) | an immunological product, or a pharmaceutical product for a non-food-producing animal | 5,390 | |
| (ii) | a pharmaceutical product for a food-producing animal | 7,135 | |
| (b) a change of bioavailability | 8,415 | ||
| (c) a change of active substance, where the change is to— | |||
| (i) | use a different biologically active substance with a slightly different molecular structure | 8,415 | |
| (ii) | modify the vector used to produce the antigen or the source material, including a new master cell bank from a different source | 8,415 | |
| (d) a change of pharmacokinetics | 8,415 | ||
| Simultaneous application falling within (a) to (d): fee for each additional product in the application | 1,465 | ||
| Variation – reduced | 885 | ||
| Variation - no assessment | 455 | ||
| Grouped variations | |||
| Variation – standard led | |||
| For the first nine changes | 6,280 | ||
| For each subsequent group of five or fewer changes | 2,250 | ||
| Variation – reduced led: | |||
| For the first nine changes | 1,770 | ||
| For each subsequent group of five or fewer changes | 2,250] | ||
Extent Information
E128This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F487Words in Sch. 7 para. 17 heading omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(i); 2020 c. 1, Sch. 5 para. 1(1)
F488Words in Sch. 7 para. 17(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 171(a)
F489Sch. 7 para. 17 Table substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 171(b)
Application for a variation to a marketing authorisation dealt with under national or mutual recognition variation procedures.N.I.
17.—(1) This paragraph applies in relation to an application for a variation to one or more marketing authorisations except where paragraph 18, 19 or 21 applies.
(2) The fees for the variations to which this paragraph applies are set out in the following table.
(3) Where applications are made at the same time seeking an identical change to the terms of more than one marketing authorisation, and those applications are based on identical data, fees are payable as for a grouped variation.
(4) References in this paragraph to a grouped variation being “led” by a particular type of variation indicate that the principal variation in that group is a variation of that type.
| Type of variation | National | UK is the reference member State | UK is a concerned member State | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Single variations; one change for each product | |||||||||
| Extension: | |||||||||
| Change of strength or potency or the addition of a new strength or potency: | 6,670 | - | 1,998 | ||||||
| Change of pharmaceutical form or the addition of a new pharmaceutical form: | 8,415 | - | 2,301 | ||||||
| Change of route of administration, or the addition of a new one, of— | - | ||||||||
| (i) an immunological product, or a pharmaceutical product for a [F620non-food-producing] animal: | 5,390 | - | 1,737 | ||||||
| (ii) a pharmaceutical product for a food-producing animal: | 7,135 | - | 2,058 | ||||||
| Change or addition of a food producing target species: | 9,620 | - | 2,547 | ||||||
| Change of active substance, including: | 8,415 | - | 2,301 | ||||||
| use of a different salt, ester, complex or derivative of the same therapeutic moiety: | |||||||||
| use of a different biologically active substance with a slightly different molecular structure: | |||||||||
| modification of the vector used to produce the antigen or the source material, including a new master cell bank from a different source: | |||||||||
| use of a new ligand or coupling mechanism for a radiopharmaceutical: | |||||||||
| change of the extraction solvent or change of the ratio of herbal drug to herbal drug preparation: | |||||||||
| Change of bioavailability: | 8,415 | - | 2,301 | ||||||
| Change of pharmacokinetics: | 8,415 | - | 2,301 | ||||||
| Simultaneous application: fee for each additional product in the application: | 2,895 | - | 1,011 | ||||||
| Type II: | 2,895 | 6,030 | 1,872 | ||||||
| Type IB: | 885 | 1,325 | 531 | ||||||
| Type IA: | 455 | 685 | 273 | ||||||
| Grouped variations | |||||||||
| Extension-led: | |||||||||
| The fee for an application for an extension-led grouped variation is the fee for that extension as specified above plus — | |||||||||
| (a) if there is one variation in addition to the extension, the fee for that variation as specified above; or | |||||||||
| (b) if there is more than one variation in addition to the extension, the fee that would be payable for a grouped variation of that type as specified below. | |||||||||
| Type II led: | |||||||||
| For the first nine changes: | 6,280 | 12,060 | 3,768 | ||||||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 2,700 | ||||||
| Type IB led: | |||||||||
| For the first nine changes: | 1,770 | 2,650 | 1,062 | ||||||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 2,700 | ||||||
| Type IA led: | |||||||||
| For the first nine changes: | 885 | 1,325 | 531 | ||||||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 2,700 | ||||||
Extent Information
E282This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Textual Amendments
F620Words in Sch. 7 para. 17 substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 4(2)
Application for a variation to a marketing authorisation dealt with under worksharing proceduresE+W+S
F49018. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Textual Amendments
F490Sch. 7 para. 18 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 172
Application for a variation to a marketing authorisation dealt with under worksharing proceduresN.I.
18.—(1) This paragraph applies in relation to an application for a variation to a marketing authorisation dealt with in accordance with worksharing procedures as set out in Article 20 of Commission Regulation (EC) No 1234/.
(2) The fee for a worksharing application, involving marketing authorisations obtained by a national procedure in the United Kingdom only, is the fee specified in the following table in the column headed “UK Only”.
(3) The fee for a worksharing application, involving marketing authorisations obtained through a national procedure in the United Kingdom and any other member State, is specified in the following table by reference to the United Kingdom’s role in the procedure, as “UK Reference Authority”, “UK Co-Reference Authority” or “Other”.
(4) The fee for a worksharing application, involving at least one marketing authorisation obtained through the mutual recognition or decentralised procedure, is specified in the following table by reference to the United Kingdom’s role in the procedure, as “UK Reference Authority”, “UK Co-Reference Authority” or “UK Concerned member State”.
(5) The fee for any kind of variation where the Agency co-ordinates worksharing is £455 for each marketing authorisation.
| Type of application | UK Only | Where the application involves nationally authorised products in more than one member State | Application involves mutually recognised products | ||||||
|---|---|---|---|---|---|---|---|---|---|
UK Only | UK Reference Authority | UK Co-Reference Authority | Other | UK Reference Authority | UK Co-Reference Authority | UK Concerned member State | |||
Worksharing applications The following fees apply for each change to each product: | |||||||||
| Type II | |||||||||
| For the first nine changes: | 6,240 | 12,060 | 7,485 | 12,060 | 13,265 | 6,745 | 3,372 | ||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 4,500 | 4,500 | 4,500 | 4,500 | 2,700 | ||
| Type IB | |||||||||
| For the first nine changes: | 1,770 | 2,650 | 2,120 | 2,650 | 2,915 | 1,905 | 954 | ||
| For each subsequent group of up to ten changes: | 4,500 | 4,500 | 4,500 | 4,500 | 4,500 | 4,500 | 2,700 | ||
Extent Information
E283This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for an extension dealt with under the decentralised procedure where the United Kingdom is the reference member StateE+W+S
F49119. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E129This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F491Sch. 7 para. 19 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(k); 2020 c. 1, Sch. 5 para. 1(1)
Application for an extension dealt with under the decentralised procedure where the United Kingdom is the reference member StateN.I.
19. The fee for a decentralised application for an extension where the United Kingdom is the reference member State is the same as for a national application as set out in the table in paragraph 17, with the addition of the supplementary fees in the following table (save that, where the application is for the addition of more than one species, only one supplementary fee applies).
Decentralised application for an extension where the United Kingdom is the reference member State
| Application | Supplementary fee (£) | |
|---|---|---|
| Pharmaceutical product for a food-producing animal – one member State: | 3,705 | |
| Pharmaceutical product for a non-food-producing animal – one member State: | 3,220 | |
| Immunological product – one member State: | 3,460 | |
| Each additional member State: | 530 | |
| Simultaneous application: fee for each additional product in the application: | ||
| one member State: | 6,670 | |
| each additional member State: | 120 | |
Extent Information
E284This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Provision of information relating to the recognition of a United Kingdom marketing authorisation or an extensionE+W+S
F49220. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E130This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F492Sch. 7 para. 20 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(k); 2020 c. 1, Sch. 5 para. 1(1)
Provision of information relating to the recognition of a United Kingdom marketing authorisation or an extensionN.I.
20.—(1) Where an application is made for the Secretary of State to provide information to other member States to enable them to recognise a marketing authorisation already granted by the United Kingdom the following fees are payable.
(2) Those fees also apply where a marketing authorisation has been granted in more than one member State, the holder applies for an extension for that marketing authorisation and the United Kingdom acts as reference member State.
(3) Where a valid application to provide information to another member State is received within six months of the original grant of the marketing authorisation, or where the Secretary of State has already provided the information to a member State, and a further valid application is made to provide the information to an additional member State within six months of the date the last information was provided, the fees are—
| Type of application | Fee for a pharmacologically equivalent product(a) | Fee (other products) (£) | ||
|---|---|---|---|---|
(a) This fee is payable if the application for the marketing authorisation was on the basis that the product was pharmacologically equivalent to another veterinary medicinal product. | ||||
| Pharmaceutical product for a food-producing animal – one member State: | 3,940 | 2,440 | ||
| Pharmaceutical product for a non-food-producing animal ‑ one member State: | 2,645 | 1,895 | ||
| Immunological product – one member State: | 2,130 | 2,130 | ||
| Each additional member State: | 535 | 535 | ||
(4) Where the information to be provided relates to a product granted a marketing authorisation using identical data submitted simultaneously or on the basis of information provided under Article 13(c) of Directive 2001/82/EC the fees are—
| Application | Fee (£) | |
|---|---|---|
| Provision of information to— | ||
| one member State: | 4,165 | |
| each additional member State: | 530 | |
(5) In any other case the fees are—
| Type of application | Fee for a pharmacologically equivalent product (£)(a) | Fee (other products) (£) | |
|---|---|---|---|
(a) This fee is payable if the application for the marketing authorisation was on the basis that the product was pharmacologically equivalent to another veterinary medicinal product. | |||
| Pharmaceutical product for a food-producing animal – one member State: | 12,015 | 10,515 | |
| Pharmaceutical product for a non-food-producing animal – one member State: | 8,115 | 7,365 | |
| Immunological product – one member State: | 8,940 | 8,940 | |
| Each additional member State: | 535 | 535 | |
(6) In the case of simultaneous applications, the above fees are payable for each additional product in the application for one member State, with a fee of £115 for each additional product for each additional member State.
Extent Information
E285This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Exception for a variation relating to animal testingE+W+S
21. If the only purpose of a variation is to remove animal testing or to reduce the numbers of animals used in testing, no fee is payable for the variation F493....
Extent Information
E131This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F493Words in Sch. 7 para. 21 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(l); 2020 c. 1, Sch. 5 para. 1(1)
Exception for a variation relating to animal testingN.I.
21. If the only purpose of a variation is to remove animal testing or to reduce the numbers of animals used in testing, no fee is payable for the variation in the case of a national authorisation, and the United Kingdom element of the fee for the variation is not payable for an authorisation obtained through the mutual recognition procedure or the decentralised procedure.
Extent Information
E286This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F494Application for a reassessment of an exceptional marketing authorisation]E+W+S
22.—F495(1) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(2) The fee for the first reassessment of an exceptional marketing authorisation is £305, and the fee for each subsequent reassessment is £1,360.
Extent Information
E132This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F494Sch. 7 para. 22 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 173(b)
F495Sch. 7 para. 22(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 173(a)
Application for the renewal of a national marketing authorisationN.I.
22.—(1) The fee for an application for the renewal of a marketing authorisation is £1,360.
(2) The fee for the first reassessment of an exceptional marketing authorisation is £305, and the fee for each subsequent reassessment is £1,360.
Extent Information
E287This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Application for the renewal of a marketing authorisation obtained through mutual recognition or the decentralised procedureE+W+S
F49623. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E133This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F496Sch. 7 para. 23 omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(n); 2020 c. 1, Sch. 5 para. 1(1)
Application for the renewal of a marketing authorisation obtained through mutual recognition or the decentralised procedureN.I.
23. The fee for an application for the renewal of a marketing authorisation obtained through mutual recognition or the decentralised procedure is —
(a)£1,835 if the United Kingdom is the reference member State; and
(b)£1,225 if the United Kingdom is a concerned member State.
Extent Information
E288This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Registration of a homeopathic remedyE+W+S
24. The fee for an application for the registration of a homeopathic remedy is in accordance with the following table.
Fee for the registration of a homeopathic remedy
| Type of application | Fees(£) | ||
|---|---|---|---|
| If all stocks and the formulation have already been assessed by the Secretary of State— | |||
| not more than five stocks: | 160 | ||
| more than five stocks: | 375 | ||
| If either all the stocks have already been assessed by the Secretary of State but there is a new formulation, or if the formulation has already been assessed by the Secretary of State but one or more of the stocks have not been already assessed— | |||
| not more than five stocks: | 455 | ||
| more than five stocks: | 665 | ||
| If the formulation and at least one of the stocks has not already been assessed by the Secretary of State— | |||
| not more than five stocks: | 760 | ||
| more than five stocks: | 985 | ||
| If the product is already authorised for human use in the United Kingdom, or for human or veterinary use in the United Kingdom F497...— | |||
| not more than five stocks: | 160 | ||
| more than five stocks: | 375 | ||
Extent Information
E134This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F497Words in Sch. 7 para. 24 Table omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(o); 2020 c. 1, Sch. 5 para. 1(1)
Registration of a homeopathic remedyN.I.
24. The fee for an application for the registration of a homeopathic remedy is in accordance with the following table.
Fee for the registration of a homeopathic remedy
| Type of application | Fees(£) | ||
|---|---|---|---|
| If all stocks and the formulation have already been assessed by the Secretary of State— | |||
| not more than five stocks: | 160 | ||
| more than five stocks: | 375 | ||
| If either all the stocks have already been assessed by the Secretary of State but there is a new formulation, or if the formulation has already been assessed by the Secretary of State but one or more of the stocks have not been already assessed— | |||
| not more than five stocks: | 455 | ||
| more than five stocks: | 665 | ||
| If the formulation and at least one of the stocks has not already been assessed by the Secretary of State— | |||
| not more than five stocks: | 760 | ||
| more than five stocks: | 985 | ||
| If the product is already authorised for human use in the United Kingdom, or for human or veterinary use in the United Kingdom or in another member State— | |||
| not more than five stocks: | 160 | ||
| more than five stocks: | 375 | ||
Extent Information
E289This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Renewal of a homeopathic remedyU.K.
[F49825. The fee for the renewal of a homeopathic remedy is £320.]
Textual Amendments
F498Sch. 7 para. 25 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 174
Annual fees for marketing authorisationsU.K.
26.—(1) Within 30 days of receiving a written demand from the Secretary of State, a holder of a marketing authorisation must provide the Secretary of State with a statement of turnover for the previous calendar year.
(2) The annual fee, rounded to the next £1, is—
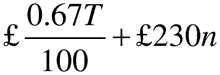
where—
(a)
T is the annual turnover in the previous calendar year;
(b)
and n is the number of active marketing authorisations held at any time during the previous calendar year.
(3) In the case of an authorisation holder with a turnover relating to all marketing authorisations held of less than £230,000, the annual fee, rounded to the next £1 is—
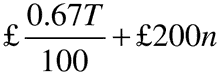
where—
(a)
T is the annual turnover in the previous calendar year;
(b)
and n is the number of active marketing authorisations held at any time during the previous calendar year.
(4) In this paragraph—
“turnover” means the sales value at manufacturers’ prices of all authorised veterinary medicinal products sold or supplied in the United Kingdom;
“manufacturers’ prices” means the prices charged (excluding value added tax) for authorised products by manufacturers to wholesalers, except to the extent that—
(a)
the products are supplied by manufacturers direct to retailers, in which case it means the prices charged for the products by the manufacturers to the retailers reduced by such sum as, in the opinion of the Secretary of State, represents the difference between the prices paid by the retailers and those which could be expected to be charged by the manufacturers to wholesalers according to the practice prevailing during the period in question with regard to such products;
(b)
a marketing authorisation holder sells or supplies products that the marketing authorisation holder has neither manufactured nor obtained from the manufacturer, in which case it means the prices paid by the marketing authorisation holder for those products.
Auditor’s certificateU.K.
27.—(1) The Secretary of State may at any time require an audit certificate in support of a statement of turnover.
(2) If the holder of the marketing authorisation does not provide an audit certificate before the date stipulated in the demand, an additional fee is payable for that year of £11,300 plus an additional £2,245 in respect of each marketing authorisation held.
(3) If the Secretary of State is not satisfied that the audit certificate provides sufficient assurance that the figures fairly present the financial records of the company, the Secretary of State may require the marketing authorisation holder to produce a further certificate and specify what further assurances are needed; and if these are not provided by the required date, the additional fee specified in sub-paragraph (2) is payable.
(4) Nothing in this paragraph limits the powers of an inspector to examine financial records.
PART 3U.K.Fees payable by manufacturers
Application for a manufacturing authorisationE+W+S
28.—[F499(1)] The fee for an application for a manufacturing authorisation for a veterinary medicinal product [F500is £762]
[F501(2) Fees relating to an application for a manufacturing authorisation are payable with the application.]
Textual Amendments
F499Sch. 7 para. 28 renumbered as Sch. 7 para. 28(1) (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 175(a)
F500Words in Sch. 7 para. 28(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 175(b)
F501Sch. 7 para. 28(2) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 175(c)
Application for a manufacturing authorisationN.I.
28. The fee for an application for a manufacturing authorisation for a veterinary medicinal product is—
(a)£3,040; or
(b)£530 if the authorisation only covers veterinary medicinal products manufactured under Schedule 6 (exemptions for small pet animals).
Application for a variation of a manufacturing authorisationE+W+S
29. The fee for an application for the variation of a manufacturing authorisation is—
(a)[F502£684] if the variation requires scientific or pharmaceutical assessment;
[F503(b)£105 if the variation only involves an administrative variation such as a change of ownership.”;]
F504(c). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F505(d). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E135This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F502Sum in Sch. 7 para. 29(a) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 176(a)
F503Sch. 7 para. 29(b) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 176(b)
F504Sch. 7 para. 29(c) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 176(c)
F505Sch. 7 para. 29(d) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 176(c)
Application for a variation of a manufacturing authorisationN.I.
29. The fee for an application for the variation of a manufacturing authorisation is—
(a)£636 if the variation requires scientific or pharmaceutical assessment;
(b)£443 if the variation only involves a change of ownership;
(c)£210 if the authorisation only covers veterinary medicinal products manufactured under Schedule 6 (exemptions for small pet animals); and
(d)otherwise £350.
Extent Information
E290This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F506Inspection of sites authorised to manufacture a product for administration under the cascade]E+W+S
30.—F507(1) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
[F508(2) The fees for the inspection of sites in connection with an authorisation (or an application for authorisation) for the manufacture of unauthorised veterinary medicinal products for administration under the cascade are set out in the following table—
Inspection fees
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 21,416 | 22,710 |
| Major site | 12,850 | 14,144 |
| Standard site | 6,425 | 7,719 |
| Minor site | 4,283 | 5,577] |
F509(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
F510(4) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E136This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F506Sch. 7 para. 30 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 177(d)
F507Sch. 7 para. 30(1) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 177(a)
F508Sch. 7 para. 30(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 177(b)
F509Sch. 7 para. 30(3) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 177(c)
F510Sch. 7 para. 30(4) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 177(c)
Application for an authorisation to manufacture an autogenous vaccine or a product for administration under the cascadeN.I.
30.—(1) The fee for an application for a standard authorisation to manufacture an autogenous vaccine or a veterinary medicinal product for administration under the cascade is—
(a)£3,435 for a site in the United Kingdom;
(b)£3,270 for a site outside the United Kingdom.
(2) The fee for each inspection after a standard authorisation has been granted is (in each case) the same as the fee specified in paragraph (1).
(3) In the case of an application for an individual authorisation to manufacture a single batch of autogenous vaccine, or a single batch of veterinary medicinal product for administration under the cascade the fee is £1,635.
(4) The fee to vary an authorisation is £305 if no further inspection is required, and otherwise is the full application fee.
Extent Information
E291This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F511Autogenous vaccinesE+W+S
30A.—(1) The fee for the scientific assessment of an authorisation (or an application for authorisation) to manufacture an autogenous vaccine is £6,962.
(2) The fees for the inspection of sites in connection with an authorisation (or an application for authorisation) to manufacture autogenous vaccines are set out in the following table—]
Inspection fees
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 21,416 | 22,710 |
| Major site | 12,850 | 14,144 |
| Standard site | 6,425 | 7,719 |
| Minor site | 4,283 | 5,577 |
Textual Amendments
F511Sch. 7 paras. 30A, 30B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 178
[F511Assessment of a variation of an authorisation to manufacture an autogenous vaccineE+W+S
30B. The fee for the scientific assessment of an application for the variation of an authorisation to manufacture an autogenous vaccine is—
(a)£2,895 if the variation requires complex scientific or pharmaceutical assessment;
(b)£885 if the variation requires simple scientific or pharmaceutical assessment;
(c)£455 in relation to an administrative variation.]
Textual Amendments
F511Sch. 7 paras. 30A, 30B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 178
[F512Annual fee (manufacturing authorisations)E+W+S
31. An annual fee of £575 is payable in respect of each manufacturing authorisation held.]
Extent Information
E137This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F512Sch. 7 para. 31 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 179
Annual feesN.I.
31.—(1) An annual fee of £550 is payable in respect of each manufacturing authorisation held (other than as specified in this paragraph).
(2) The annual fee for a manufacturing authorisation for an autogenous vaccine or a veterinary medicinal product for administration under the cascade is 0.67% of the turnover in the previous calendar year rounded to the next £1, with a minimum fee of £10.
(3) There is no annual fee for a manufacturing authorisation for a veterinary medicinal product manufactured in accordance with Schedule 6 for small pet animals.
(4) In this paragraph “turnover” means the sales value at manufacturers’ prices net of value added tax of all authorised veterinary medicinal products sold or supplied in the United Kingdom.
Extent Information
E292This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Site inspections – type of siteU.K.
32. For the purposes of deciding the fee for a site inspection—
“super site” is a site at which 250 or more relevant persons are employed;
“major site” is a site at which 60 or more, but fewer than 250, relevant persons are employed;
“standard site” is a site at which 10 or more, but fewer than 60 relevant persons are employed;
“minor site” is a site at which fewer than 10 relevant persons are employed;
“relevant person” means a person employed on the premises and systems inspected.
Inspection of a site where immunological veterinary medicinal products are manufacturedE+W+S
33. The fees for the inspection of a site where immunological veterinary medicinal products are manufactured are in accordance with the following table.
[F513Sites where immunological veterinary medicinal products are manufactured
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 32,124 | 33,418 |
| Major site | 21,416 | 22,710 |
| Standard site | 10,708 | 12,002 |
| Minor site | 6,425 | 7,719] |
Extent Information
E138This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F513Sch. 7 para. 33 Table substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 180
Inspection of a site where immunological veterinary medicinal products are manufacturedN.I.
33. The fees for the inspection of a site where immunological veterinary medicinal products are manufactured are in accordance with the following table.
Sites where immunological veterinary medicinal products are manufactured
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside United Kingdom | |
| Super site | 24,071 | 22,867 |
| Major site | 16,785 | 15,946 |
| Standard site | 6,661 | 6,327 |
| Minor site | 4,757 | 4,519 |
Extent Information
E293This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Inspection of a site where sterile veterinary medicinal products are manufacturedE+W+S
34. The following fees are payable for the inspection of a site where no immunological veterinary medicinal products are manufactured, but where sterile products are manufactured.
[F514Sites where sterile veterinary medicinal products are manufactured
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 27,841 | 29,135 |
| Major site | 19,274 | 20,569 |
| Standard site | 10,708 | 12,002 |
| Minor site | 6,425 | 7,719] |
Extent Information
E139This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F514Sch. 7 para. 34 Table substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 181
Inspection of a site where sterile veterinary medicinal products are manufacturedN.I.
34. The following fees are payable for the inspection of a site where no immunological veterinary medicinal products are manufactured, but where sterile products are manufactured.
Sites where sterile veterinary medicinal products are manufactured
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 23,324 | 22,157 |
| Major site | 13,010 | 12,359 |
| Standard site | 8,244 | 7,832 |
| Minor site | 5,022 | 4,770 |
Extent Information
E294This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Inspection of a site where no immunological or sterile veterinary medicinal products are manufacturedE+W+S
35. The following fees are payable for the inspection of a site where only non-immunological and non-sterile veterinary medicinal products are manufactured—
[F515Sites where no immunological or sterile veterinary medicinal products are manufactured
| Type of site | Fee (£) | ||
|---|---|---|---|
| United Kingdom site | Site outside the United Kingdom | ||
| Super site | 21,416 | 22,710 | |
| Major site | 12,850 | 14,144 | |
| Standard site | 8,566 | 9,861 | |
| Minor site | 4,283 | 5,577 | |
| If the site is only involved in the manufacture of veterinary medicinal products authorised under Schedule 6 (exemptions for small pet animals)— | |||
| Standard site | 3,212 | 4,507 | |
| Minor site | 2,142 | 3,436] | |
Extent Information
E140This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F515Sch. 7 para. 35 Table substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 182
Inspection of a site where no immunological or sterile veterinary medicinal products are manufacturedN.I.
35. The following fees are payable for the inspection of a site where only non-immunological and non-sterile veterinary medicinal products are manufactured—
Site where no immunological or sterile veterinary medicinal products are manufactured
| Type of site | Fee (£) | ||
|---|---|---|---|
| United Kingdom site | Site outside the United Kingdom | ||
| Super site | 14,180 | 13,471 | |
| Major site | 8,325 | 7,909 | |
| Standard site | 6,854 | 6,511 | |
| Minor site | 3,789 | 3,600 | |
| If the site is only involved in the manufacture of veterinary medicinal products authorised under Schedule 6 (exemptions for small pet animals— | |||
| Standard site | 5,055 | 4,802 | |
| Minor site | 2,728 | 2,592 | |
Extent Information
E295This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Inspection of a site where veterinary medicinal products are assembledE+W+S
36. The following fees are payable for the inspection of a site where the only manufacturing process in relation to veterinary medicinal products is their assembly after the product has been put into its immediate container.
[F516Sites where medicinal products are assembled
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 17,133 | 18,427 |
| Major site | 10,708 | 12,002 |
| Standard site | 6,425 | 7,719 |
| Minor site | 4,283 | 5,577] |
Extent Information
E141This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F516Sch. 7 para. 36 Table substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 183
Inspection of a site where veterinary medicinal products are assembledN.I.
36. The following fees are payable for the inspection of a site where the only manufacturing process in relation to veterinary medicinal products is their assembly after the product has been put into its immediate container.
Site where medicinal products are assembled
| Type of site | Fee (£) | |
|---|---|---|
| United Kingdom site | Site outside the United Kingdom | |
| Super site | 11,025 | 10,474 |
| Major site | 5,949 | 5,652 |
| Standard site | 4,917 | 4,671 |
| Minor site | 2,035 | 1,933 |
Extent Information
E296This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Test sitesE+W+S
37. The fee for the inspection of a test site is [F517£3,212], or [F518£4,507] for a site outside the United Kingdom.
Extent Information
E142This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F517Sum in Sch. 7 para. 37 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 184(a)
F518Sum in Sch. 7 para. 37 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 184(b)
Test sitesN.I.
37. The fee for the inspection of a test site is £3,344, or £3,177 for a site outside the United Kingdom.
Extent Information
E297This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F519Animal blood bank or non-food animal stem cell centre authorisationsE+W+S
38.—(1) The fee for the inspection of a blood bank is—
(a)£3,212 for a site in the United Kingdom; and
(b)£4,507 for a site outside the United Kingdom.
(2) The fee for the inspection of a non-food animal stem cell centre is—
(a)£2,142 for a site in the United Kingdom; and
(b)£3,436 for a site outside the United Kingdom]
Extent Information
E143This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F519Sch. 7 para. 38 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 185
Animal blood bank or equine stem cell centre authorisationsN.I.
38.—(1) The fee for an authorisation to operate a blood bank is—
(a)on a first inspection £3,113; and
(b)on each subsequent inspection—
(i)£3,113 for a site in the United Kingdom; and
(ii)£2,966 for a site outside the United Kingdom.
(2) The fee for an authorisation to operate an equine stem cell centre is £3,427, and £3,092 for each subsequent inspection.
(3) The fee for a variation to an authorisation to operate a blood-bank or equine stem cell centre is £320.
Extent Information
E298This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
PART 4U.K.Fees relating to a wholesale dealer’s authorisation
[F520Application for a wholesale dealer’s authorisationE+W+S
39.—(1) The fee for an application for a wholesale dealer’s authorisation is £344.
(2) Fees relating to an application for a wholesale dealer’s authorisation are payable with the application.]
Extent Information
E144This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F520Sch. 7 para. 39 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 186
Application for a wholesale dealer’s authorisationN.I.
39.—(1) The fee for an application for a wholesale dealer’s authorisation is—
(a)£1,745;
(b)£785 if the application is accompanied by an estimate that the first year’s turnover will be less than £35,000; or
(c)£785 if the authorisation only relates to products classified as AVM-GSL, homeopathic remedies, or products authorised under Schedule 6 (exemptions for small pet animals).
(2) An applicant who has paid a fee of £785 on the grounds of turnover must send a declaration of turnover for the first year of trading on the anniversary of the grant of the authorisation, and if the figure is more than £35,000 must pay the balance of £960 within 30 days.
(3) If the applicant paid £1,745 but the turnover for the first year of trading was lower than £35,000, if the applicant sends a declaration certifying the turnover, the Secretary of State must refund the excess.
(4) Nothing in this paragraph limits the powers of an inspector to examine financial records.
(5) In this paragraph “turnover” means the sales value net of value added tax of all veterinary medicinal products (whether or not authorised for use in the United Kingdom) sold by way of wholesale dealing by the holder in the United Kingdom.
Extent Information
E299This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F521Variation of a wholesale dealer’s authorisationE+W+S
40. The fee for an application to vary a wholesale dealer’s authorisation is—
(a)£265 if the variation requires scientific or pharmaceutical assessment;
(b)£105 for a change of ownership or other administrative variation.]
Extent Information
E145This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F521Sch. 7 para. 40 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 187
Variation of a wholesale dealer’s authorisationN.I.
40. The fee for an application to vary a wholesale dealer’s authorisation is—
(a)£515 if the variation requires scientific or pharmaceutical assessment;
(b)£430 if the variation only involves a change of ownership; and
(c)otherwise £300.
Extent Information
E300This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F522Annual fee for a wholesale dealer’s authorisationE+W+S
41. The annual fee for a wholesale dealer’s authorisation is £427.]
Extent Information
E146This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F522Sch. 7 para. 41 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 188
Annual fee for a wholesale dealer’s authorisationN.I.
41.—(1) The annual fee for a wholesale dealer’s authorisation is—
(a)£483; or
(b)£315, if—
(i)the holder certifies when making the payment that the turnover during the previous year was less than £35,000; or
(ii)the authorisation only relates to products classified as AVM-GSL or homeopathic remedies;
(c)£215 if the authorisation only relates to products authorised under Schedule 6 (exemptions for small pet animals).
(2) In this paragraph “turnover” means the sales value net of value added tax of all veterinary medicinal products (whether or not authorised for use in the United Kingdom) sold by way of wholesale dealing by the holder in the United Kingdom.
Extent Information
E301This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F523Inspection of a wholesale dealer’s sitesE+W+S
42. The fee for inspection of a wholesale dealer’s site is—
(a)£1,177; or
(b)£877 if—
(i)the authorisation only relates to products classified as AVM-GSL or homeopathic remedies; or
(ii)the authorisation only relates to products marketed under Schedule 6 (exemptions for small pet animals).]
Extent Information
E147This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F523Sch. 7 para. 42 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 189
Inspection of a wholesale dealer’s premisesN.I.
42. The fee for the inspection of a wholesale dealer’s premises is—
(a)£3,058; or
(b)£1,442 if—
(i)the authorisation only relates to products classified as AVM-GSL or homeopathic remedies; or
(ii)the turnover relating to all veterinary medicinal products in the calendar year preceding the inspection was less than £35,000;
(c)£830 if the authorisation only relates to products authorised under Schedule 6 (exemptions for small pet animals).
Extent Information
E302This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
PART 5U.K.Fees relating to feedingstuffs
Fees for [F524applications for authorisation] and annual fees relating to feedingstuffs in Great BritainU.K.
43.—(1) Subject to sub-paragraph (3) the fee for the application for [F525authorisation] of [F526premises] manufacturing feedingstuffs and [F525authorisation] of distributors of feedingstuffs in Great Britain is [F527£105].
(2) An annual fee of [F528£122] is payable in respect of any such [F529authorisation].
(3) No fee is payable under sub-paragraph (1) in respect of [F530premises] where specified feed additives are manufactured if a [F531medicinal premix] is manufactured at [F532those premises] in accordance with a manufacturing authorisation.
(4) Fees relating to feedingstuffs are payable with the application F533....
(5) Where more than one manufacturing activity is carried out at one [F534premises by the same legal entity] only one fee (the highest) is payable.
Textual Amendments
F524Words in Sch. 7 para. 43 heading substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(f)
F525Word in Sch. 7 para. 43(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(a)(i)
F526Word in Sch. 7 para. 43(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(a)(ii)
F527Sum in Sch. 7 para. 43(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(a)(iii)
F528Sum in Sch. 7 para. 43(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(b)(i)
F529Word in Sch. 7 para. 43(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(b)(ii)
F530Word in Sch. 7 para. 43(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(c)(i)
F531Words in Sch. 7 para. 43(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(c)(ii)
F532Words in Sch. 7 para. 43(3) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(c)(iii)
F533Words in Sch. 7 para. 43(4) omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(d)
F534Words in Sch. 7 para. 43(5) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 190(e)
Inspection fees relating to feedingstuffs in Great BritainU.K.
44. Fees for the inspection of [F535premises] manufacturing or distributing feedingstuffs in Great Britain are in accordance with the following table.
[F536Inspection Fees
| Type of premises inspected | Fee payable (£) |
|---|---|
| Manufacturer of a specified feed additive (SFA) | 1,610 |
| Manufacturer of an intermediate feedingstuff (including balancers) containing a medicinal premix or an SFA | 976 |
Manufacturer of a feedingstuff for sale containing— a medicinal premix and/or an SFA, and/or an intermediate feedingstuff containing a medicinal premix or an SFA | 841 |
Manufacturer of a feedingstuff for feeding to their own animals only, containing— a medicinal premix and/or an SFA incorporated at a rate of at least 2kg/t, and/or an intermediate feedingstuff containing a medicinal premix and/or an SFA incorporated at a rate of at least 2kg/t | 476 |
Distributor or trader of Schedule 5 products (A distributor of specified feed additives, or intermediate feedingstuffs containing specified feed additives or medicinal premixes; or feedingstuffs containing a medicinal premix) | 350] |
Textual Amendments
F535Word in Sch. 7 para. 44 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 191(a)
F536Sch. 7 para. 44 Table substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 191(b)
Fees payable in relation to feedingstuffs in Northern IrelandE+W+S
F53745. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Extent Information
E148This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F537Sch. 7 para. 45 omitted (E.W.S.) (31.12.2020) by virtue of The Veterinary Medicines and Residues (Amendment) (EU Exit) Regulations 2020 (S.I. 2020/1461), regs. 1(2)(b), 4(10)
Fees payable in relation to feedingstuffs in Northern IrelandN.I.
45.—(1) The annual fees payable for the approval of establishments manufacturing and distributing feedingstuffs in Northern Ireland are in accordance with the following table.
(2) Fees are payable with the application or, for the subsequent annual fee, on invoice.
(3) Where more than one manufacturing activity is carried out at one establishment only the highest fee is payable.
Approval fees
| Type of establishment | Fee payable (£) | |
|---|---|---|
(a) No fee is payable for establishments that already have a manufacturing authority relating to veterinary medicinal products for incorporating into feedingstuffs. | ||
| 1 | Establishment manufacturing a specified feed additive(a): | 545 |
| 2 | Establishment manufacturing a premixture: | 435 |
| 3 | Establishment manufacturing feedingstuffs using specified feed additives and veterinary medicinal products directly at any concentration, or using premixtures or specified feed additive complementary feedingstuffs: | 435 |
| 4 | Establishment manufacturing feedingstuffs for placing on the market using a veterinary medicinal product or premixture where the concentration of veterinary medicinal product in the feedingstuffs is 2 kg per tonne or more: | 320 |
| 5 | Establishment manufacturing feedingstuffs using premixtures or specified feed additive complementary feedingstuffs containing specified feed additives when the feedingstuffs are to be placed on the market: | 170 |
| 6 | Establishment manufacturing feedingstuffs for the manufacturers own use using a veterinary medicinal product or premixture where the concentration of veterinary medicinal product in the feedingstuffs is 2 kg per tonne or more: | 131 |
| 7 | Establishment manufacturing feedingstuffs using premixtures containing specified feed additives when the feedingstuffs are to be used by the person manufacturing the feedingstuffs: | 110 |
| 8 | Establishment distributing specified feed additives, premixtures or feedingstuffs containing specified feed additives, or premixtures or feedingstuffs containing veterinary medicinal products: | 70 |
Extent Information
E303This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Fees relating to premises for supply by suitably qualified personsE+W+S
46.—(1) The fee [F538for an application for the authorisation] of premises for the retail supply of veterinary medicinal products by suitably qualified persons is—
F540(b). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
[F541(1A) The fees for the inspection of sites authorised for the retail supply of veterinary medicinal products by suitably qualified persons are set out in the following table—
Inspection Fees
| Type of sites inspected | Fee payable (£) |
|---|---|
| Sites authorised to supply companion animal medicines | 285 |
| Sites authorised to supply equine medicines | 285 |
| Sites authorised to supply livestock medicines | 338 |
| Sites authorised to supply avian medicines | 285. |
(1B) Where a site is inspected in relation to a single authorisation, and falls within more than one of the categories in the table, only one fee (the highest) is payable.]
(2) The subsequent annual fee is—
F543(b). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
[F544(3) The application fee for authorisation of sites for supply is payable with the application.]
Extent Information
E149This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F538Words in Sch. 7 para. 46(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 192(a)(i)
F539Sum in Sch. 7 para. 46(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 192(a)(ii)
F540Sch. 7 para. 46(1)(b) and word omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 192(a)(iii)
F541Sch. 7 para. 46(1A)(1B) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 192(b)
F542Sum in Sch. 7 para. 46(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 192(c)(i)
F543Sch. 7 para. 46(2)(b) and word omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 192(c)(ii)
F544Sch. 7 para. 46(3) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 192(d)
Fees relating to premises for supply by suitably qualified personsN.I.
46.—(1) The fee to approve of premises for the retail supply of veterinary medicinal products by suitably qualified persons is—
(a)£265; or
(b)if the premises are only authorised to supply veterinary medicinal products for the treatment of—
(i)horses (or horses and companion animals) £145; or
(ii)companion animals £110.
(2) The subsequent annual fee is—
(a)£185; or
(b)if the premises are only authorised to supply veterinary medicinal products for the treatment of—
(i)horses (or horses and companion animals) £95; or
(ii)companion animals £70.
Extent Information
E304This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
PART 6U.K.General
Testing samplesU.K.
47. The fee for testing a sample required to be submitted by the Secretary of State is the full economic cost of the test.
Animal test certificatesE+W+S
48.—(1) The fee for an animal test certificate is [F545£1,170].
(2) The fee for an animal test certificate to administer medicinal products in a small scale trial to test them for clinical safety or efficacy is [F546£40].
F547(3) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
[F548(4) The fee for an application for the variation of the certificate is—
(a)in the case of a small scale trial, £40; and
(b)in the case of any other trial, £390.]
[F549(5) The fee for an application to renew a certificate is—
(a)in the case of a small scale trial, £40; and
(b)in the case of any other trial, £190.]
(6) The Secretary of State may waive the fee if satisfied that the application is in relation to developing a veterinary medicinal product for a limited market (for example, for a minor species, a minor use, or for a disease with restricted regional distribution).
Extent Information
E150This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F545Sum in Sch. 7 para. 48(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 193(a)
F546Sum in Sch. 7 para. 48(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 193(b)
F547Sch. 7 para. 48(3) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(p)(ii); 2020 c. 1, Sch. 5 para. 1(1)
F548 Sch. 7 para. 48(4) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 193(c)
F549Sch. 7 para. 48(5) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 193(d)
Animal test certificatesN.I.
48.—(1) The fee for an animal test certificate is £345 in the case of—
(a)an immunological veterinary medicinal product that has been authorised in another member State for the species on which the proposed test will be conducted;
(b)a pharmaceutical veterinary medicinal product that has been authorised in another member State for use with a food-producing species on which the proposed test will be conducted where the same or similar dosage regime and method of administration is to be used in the medicinal test as is authorised; or
(c)a pharmaceutical veterinary medicinal product authorised in another member State for human or animal use where the test is to be conducted on companion animals only.
(2) The fee for an animal test certificate to administer medicinal products in a small scale trial to test them for clinical safety or efficacy is £30.
(3) In any other case the fee is £815.
(4) The fee for an application for a variation of the certificate is £265 for each change.
(5) The fee for an application to renew a certificate is £130.
(6) The Secretary of State may waive the fee if satisfied that the application is in relation to developing a veterinary medicinal product for a limited market (for example, for a minor species, a minor use, or for a disease with restricted regional distribution).
Extent Information
E305This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Importation of a veterinary medicinal product for treatment under the cascadeE+W+S
49.—(1) The fee for a certificate to import (if necessary) and be in possession of and administer a veterinary medicinal product under the cascade is—
F550(a). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(b)£30 if the veterinary medicinal product is authorised in [F551another] country.
(2) The fee is payable in respect of each animal treated, but in the case of administration to and treatment of a discrete group of animals, the Secretary of State may notify the applicant in writing that a fee for only one animal is payable.
(3) There is no fee if the application is made using the website of the Veterinary Medicines Directorate.
Extent Information
E151This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F550Sch. 7 para. 49(1)(a) omitted (E.W.S.) (31.12.2020) by virtue of The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(q)(i); 2020 c. 1, Sch. 5 para. 1(1)
F551Word in Sch. 7 para. 49(1)(b) substituted (E.W.S.) (31.12.2020) by The Food and Drink, Veterinary Medicines and Residues (Amendment etc.) (EU Exit) Regulations 2019 (S.I. 2019/865), regs. 1(2), 17(4)(q)(ii); 2020 c. 1, Sch. 5 para. 1(1)
Importation of a veterinary medicinal product for treatment under the cascadeN.I.
49.—(1) The fee for a certificate to import (if necessary) and be in possession of and administer a veterinary medicinal product under the cascade is—
(a)£15 if the veterinary medicinal product is authorised in another member State;
(b)£30 if the veterinary medicinal product is authorised in a third country.
(2) The fee is payable in respect of each animal treated, but in the case of administration to and treatment of a discrete group of animals, the Secretary of State may notify the applicant in writing that a fee for only one animal is payable.
(3) There is no fee if the application is made using the website of the Veterinary Medicines Directorate.
Extent Information
E306This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Wholesale dealer’s import certificateU.K.
50.—(1) The fee payable by the holder of a wholesale dealer’s authorisation for a certificate to import and store a veterinary medicinal product not authorised in the United Kingdom to enable it to be supplied for administration under Schedule 4 is [F552£760] .
(2) The fee is only payable if, in the twelve month period immediately before the application, the applicant has supplied the veterinary medicinal product to which the certificate relates in accordance with at least 100 certificates.
Textual Amendments
F552Word in Sch. 7 para. 50 substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 4(3)
Specific batch controlU.K.
[F55351. The fee for an authorisation to release a veterinary medicinal product under specific batch control is—
(a)£560; and
(b)£100 for each additional batch affected by the same issue where the specific batch control application is made at the same time.]
Textual Amendments
F553Sch. 7 para. 51 substituted (14.4.2014) by The Veterinary Medicines (Amendment) Regulations 2014 (S.I. 2014/599), regs. 1, 4(4)
Submission of control tests of an immunological productU.K.
52. The fee for the submission of the results of tests carried out on a batch of immunological products other than autogenous vaccines prior to release is £80.
Export certificatesE+W+S
53. The fee for an application for an export certificate is [F554£54] F555....
Extent Information
E152This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F554Sum in Sch. 7 para. 53 substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 194(a)
F555Words in Sch. 7 para. 53 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 194(b)
Export certificatesN.I.
53. The fee for an application for an export certificate is £30, and £15 for each certified copy.
Extent Information
E307This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
Provision of adviceU.K.
54. The fee for an application for written advice from the Secretary of State as to whether or not a product requires a marketing authorisation is £885.
[F556Provision of scientific adviceE+W+S
54A. The fee for an application for written advice from the Secretary of State in relation to scientific matters is £4,487.]
Textual Amendments
F556Sch. 7 para. 54A inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 195
Appeals to the Veterinary Products CommitteeU.K.
55. The fee for an appeal to the Veterinary Products Committee is £1,500.
Fee relating to an appointed personU.K.
56. The appellant is liable for the full economic cost of a referral to an appointed person subject to a maximum of £5,000.
Fees relating to a veterinary F557... practice premisesE+W+S
57.—[F558(1) The fees for the inspection of a veterinary practice premises are set out in the following table—
| Type of premises inspected | Fee payable (£) |
|---|---|
| Sites registered to supply companion animal medicines | 536 |
| Sites registered to supply equine medicines | 536 |
| Sites registered to supply livestock medicines | 536 |
| Mixed practice premises | 698 |
| Any other type of practice | 451] |
(2) The initial registration and annual fee for the registration of veterinary practice premises with the Royal College of Veterinary Surgeons to supply veterinary medicinal products is [F559£38].
(3) Notwithstanding paragraph 2 of this Schedule, this is payable to the Royal College of Veterinary Surgeons.
[F560(4) For the purposes of sub-paragraph (1) “mixed practice” means premises supplying veterinary medicinal products to livestock in addition to any other category mentioned in that provision.]
Extent Information
E153This version of this provision extends to England and Wales and Scotland only; a separate version has been created for Northern Ireland only
Textual Amendments
F557Word in Sch. 7 para. 57 heading omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 196(d)
F558Sch. 7 para. 57(1) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 196(a)
F559Sum in Sch. 7 para. 57(2) substituted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 196(b)
F560Sch. 7 para. 57(4) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 196(c)
Fees relating to a veterinary surgeon’s practice premisesN.I.
57.—(1) The fee for the inspection of a veterinary surgeon’s practice premises is £350.
(2) The initial registration and annual fee for the registration of veterinary practice premises with the Royal College of Veterinary Surgeons to supply veterinary medicinal products is £34.
(3) Notwithstanding paragraph 2 of this Schedule, this is payable to the Royal College of Veterinary Surgeons.
Extent Information
E308This version of this provision extends to Northern Ireland only; a separate version has been created for England and Wales and Scotland only
[F561Fee in relation to verifying destruction of controlled drugE+W+S
57A. The fee for verifying the destruction of a controlled drug listed in Schedule 2, 3 or 4 to the Misuse of Drugs Regulations 2001 is—
(a)£142; or
(b)£31 (where the verification takes place during the course of an inspection for other purposes).]
Textual Amendments
F561Sch. 7 paras. 57A, 57B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 197
[F561Pharmacovigilance inspectionsE+W+S
57B.—(1) In relation to a pharmacovigilance inspection the fee is—
(a)£3,600 in the case of a large marketing authorisation holder; and
(b)£1,650 in the case of a small marketing authorisation holder.
(2) In sub-paragraph (1)—
“large marketing authorisation holder” means a marketing authorisation holder who holds 30 or more marketing authorisations;
“small marketing authorisation holder” means a marketing authorisation holder who holds fewer than 30 marketing authorisations.]
Textual Amendments
F561Sch. 7 paras. 57A, 57B inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 197
Refund of fees relating to the Veterinary Products Committee or appointed personsU.K.
58. The Secretary of State must refund the fee payable in relation to an appeal to the Veterinary Products Committee or to an appointed person if, as a result of the appeal, the Secretary of State changes the decision that was the subject of the appeal.
Fees relating to an improvement noticeU.K.
59. If an improvement notice is served under these Regulations, the fee for any subsequent inspection necessary as a result of the notice is the full economic cost of the inspection, payable by the person on whom the notice was served.
Non-payment of feesU.K.
60. Where any fee [F562(other than any fee relating to a manufacturing authorisation or wholesale dealer’s authorisation)] is not paid, the Secretary of State may, after giving one month’s written warning, suspend the processing of any application from the person [F563or any authorisation held by the person] who has not paid the fee.
Textual Amendments
F562Words in Sch. 7 para. 60 omitted (E.W.S.) (17.5.2024) by virtue of The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 198(a)
F563Words in Sch. 7 para. 60 inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 198(b)
Waiver or reduction of feesU.K.
61.—(1) If the Secretary of State is satisfied that for reasons of human or animal health or the protection of the environment it is desirable that a product should be authorised for veterinary use or that an authorised product should remain on the market the Secretary of State may waive or reduce any fees payable under these Regulations.
[F564(1A) If the Secretary of State is satisfied that exceptional circumstances exist the Secretary of State may waive or reduce an inspection fee payable under these Regulations.]
(2) An applicant or the holder of a marketing authorisation must provide full written justification for any waiver or reduction.
Textual Amendments
F564Sch. 7 para. 61(1A) inserted (E.W.S.) (17.5.2024) by The Veterinary Medicines (Amendment etc.) Regulations 2024 (S.I. 2024/567), regs. 1(1), 199
Reduction of fees when an application is withdrawnU.K.
62.—(1) Where an application for a marketing authorisation, or any variation referred to in paragraph 17 or 18 as a Type II variation, an extension, an extension-led grouped variation or a Type II led grouped variation is withdrawn before determination, the fee is reduced in accordance with this paragraph.
(2) If no assessment (veterinary, scientific or pharmaceutical) has begun, the reduction is 90%.
(3) If assessment has begun but the Secretary of State has not yet requested further data, the reduction is 50%.
(4) If the Secretary of State has requested further information but it has not yet been provided, the reduction is 25%.
(5) If the further information requested has been supplied but has not yet been fully assessed or the application has not been referred to the Veterinary Products Committee, the reduction is 10%
(6) Once the further information has been fully assessed, or the application has been referred to the Veterinary Products Committee, there is no reduction.
(1)
OJ No L 211, 28.11.2001, p. 1 as last amended by Regulation (EC) No 470/2009 of the European Parliament and of the Council (OJ No L152, 16.6.2009, p. 11). Annex I was inserted by Commission Directive 2009/9/EC (OJ No L 44, 14.2.2009, p. 10).
(2)
OJ No L334, 12.12.2008, p. 7.
(3)
ISBN 9287145873.
(4)
OJ No L 228, 17.8.91, p. 70.
(5)
S. I. 2001/3998; relevant amending instruments are S. I. 2003/1432 and 2005/1653.
(6)
Published at: http://www.vmd.defra.gov.uk/registers/sqpregister.aspx.
(7)
OJ No C 63, 1.3.94, p. 4.
(8)
The number of days of the withdrawal period is calculated by dividing 500 by the mean temperature of the water in degrees Celsius.
(9)
OJ No L42, 13.2.2013, p. 1.
(10)
Published at http://www.rcvs.org.uk/advice-and-guidance/code-of-professional-conduct-for-veterinary-surgeons/.
(11)
OJ No L 92, 7.4.1990, p. 42.
(12)
OJ No L 35, 8.2.2005, p. 1.
(13)
OJ No L86, 6.4.1979, p. 30.
(14)
OJ No L268, 18.10.2003, p. 29. Regulation (EC) no 1831/2003 was last amended by Article 29 of Regulation (EC) No 767/2009 (OJ No L229, 1.9.2009, p. 1.)
(15)
OJ No L 92, 7.4.90, p. 42.
Options/Help
Print Options
PrintThe Whole Instrument
PrintThe Schedules only
You have chosen to open the Whole Instrument
The Whole Instrument you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
You have chosen to open Schedules only
The Schedules you have selected contains over 200 provisions and might take some time to download. You may also experience some issues with your browser, such as an alert box that a script is taking a long time to run.
Would you like to continue?
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As Enacted or Made): The original version of the legislation as it stood when it was enacted or made. No changes have been applied to the text.
See additional information alongside the content
Geographical Extent: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Show Timeline of Changes: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Opening Options
Different options to open legislation in order to view more content on screen at once
Explanatory Memorandum
Explanatory Memorandum sets out a brief statement of the purpose of a Statutory Instrument and provides information about its policy objective and policy implications. They aim to make the Statutory Instrument accessible to readers who are not legally qualified and accompany any Statutory Instrument or Draft Statutory Instrument laid before Parliament from June 2004 onwards.
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as enacted version that was used for the print copy
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
Impact Assessments
Impact Assessments generally accompany all UK Government interventions of a regulatory nature that affect the private sector, civil society organisations and public services. They apply regardless of whether the regulation originates from a domestic or international source and can accompany primary (Acts etc) and secondary legislation (SIs). An Impact Assessment allows those with an interest in the policy area to understand:
- Why the government is proposing to intervene;
- The main options the government is considering, and which one is preferred;
- How and to what extent new policies may impact on them; and,
- The estimated costs and benefits of proposed measures.
Timeline of Changes
This timeline shows the different points in time where a change occurred. The dates will coincide with the earliest date on which the change (e.g an insertion, a repeal or a substitution) that was applied came into force. The first date in the timeline will usually be the earliest date when the provision came into force. In some cases the first date is 01/02/1991 (or for Northern Ireland legislation 01/01/2006). This date is our basedate. No versions before this date are available. For further information see the Editorial Practice Guide and Glossary under Help.
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as made version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.