- Latest available (Revised)
- Original (As adopted by EU)
Commission Implementing Regulation (EU) 2019/1604Show full title
Commission Implementing Regulation (EU) 2019/1604 of 27 September 2019 amending Regulation (EEC) No 2568/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
You are here:
- Regulations originating from the EU
- 2019 No. 1604
- Whole Regulation
- Previous
- Next

What Version
More Resources
Legislation originating from the EU
When the UK left the EU, legislation.gov.uk published EU legislation that had been published by the EU up to IP completion day (31 December 2020 11.00 p.m.). On legislation.gov.uk, these items of legislation are kept up-to-date with any amendments made by the UK since then.


This item of legislation originated from the EU
Legislation.gov.uk publishes the UK version. EUR-Lex publishes the EU version. The EU Exit Web Archive holds a snapshot of EUR-Lex’s version from IP completion day (31 December 2020 11.00 p.m.).
Status:
This is the original version as it was originally adopted in the EU.
This legislation may since have been updated - see the latest available (revised) version
Commission Implementing Regulation (EU) 2019/1604
of 27 September 2019
amending Regulation (EEC) No 2568/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
THE EUROPEAN COMMISSION,
Having regard to the Treaty on the Functioning of the European Union,
Having regard to Regulation (EU) No 1308/2013 of the European Parliament and of the Council of 17 December 2013 establishing a common organisation of the markets in agricultural products and repealing Council Regulations (EEC) No 922/72, (EEC) No 234/79, (EC) No 1037/2001 and (EC) No 1234/2007(1), and in particular point (d) of the first paragraph of Article 91 thereof,
Whereas:
(1) Commission Regulation (EEC) No 2568/91(2) defines the physico-chemical and organoleptic characteristics of olive oil and olive-pomace oil and lays down methods for assessing those characteristics.
(2) The methods and the limit values for the characteristics of oils are regularly updated on the basis of the opinion of chemical experts and in line with the work carried out within the International Olive Council (IOC).
(3) In order to ensure the implementation at Union level of the most recent international standards established by the IOC, certain methods of analysis set out in Regulation (EEC) No 2568/91 should be updated.
(4) The IOC Trade Standard was amended as regards the expression of the limit of the free acidity, peroxide value, organoleptic evaluation (median of the defect and median of the fruity attribute) and the difference between ECN42 (HPLC) and ECN42 (theoretical calculation) for consistency with the precision values of the analytical method.
(5) In accordance with Article 2a(5) of Regulation (EEC) No 2568/91 Member States are to verify whether an olive oil sample is consistent with the category declared by checking the characteristics set out in Annex I to that Regulation either in any order or following the order set out in a decision tree set out in Annex Ib thereto.
(6) In view of recent developments, it is appropriate to update the tables in Annex Ib to Regulation (EEC) No 2568/91 and its appendix as appropriate. It also appears that the term ‘flowchart’ is more appropriate than the term ‘decision tree’ in view of the content of that Annex Ib.
(7) Point 9.4 of Annex XII to Regulation (EEC) No 2568/91 defines the median of the defects as the median of the defect perceived with the greatest intensity. In the context of counter-assessments and given that different panels have to assess the conformity of the oil, it should be clarified that the decision relating to the conformity of the characteristics of an oil with the declared category is solely related to the value of the median of the main defect, irrespective of its nature.
(8) Regulation (EEC) No 2568/91 should therefore be amended accordingly.
(9) The measures provided for in this Regulation are in accordance with the opinion of the Committee for the Common Organisation of the Agricultural Markets,
HAS ADOPTED THIS REGULATION:
Article 1
Regulation (EEC) No 2568/91 is amended as follows:
(1)
Article 2 is amended as follows:
(a)
point (l) of paragraph (1) is replaced by the following:
‘(l)for the determination of the composition and content of sterols and for the determination of alcoholic compounds, by capillary column gas chromatography, the method set out in Annex XIX’;
(b)
the third subparagraph of paragraph (2) is replaced by the following:
‘Should the panel not confirm the category declared as regards the organoleptic characteristics, at the interested party's request, the national authorities or their representatives shall have two counter-assessments by other approved panels carried out without delay. At least one of the panels shall be a panel approved by the producer Member State concerned. The characteristics concerned shall be deemed consonant with the characteristics declared if the two counter-assessments confirm the declared grade. If this is not the case, regardless of the type of defects determined during the counter-assessments, the grading shall be declared inconsistent with the characteristics and the interested party shall be responsible for the cost of the counter-assessments.’;
(2)
point (b) of Article 2a(5) is replaced by the following:
‘(b)following the order set out in Annex Ib on the flowchart, until one of the decisions appearing in the flowchart is reached.’;
(3)
the table ‘ANNEXES Summary’ is replaced by the table in Annex I to this Regulation;
(4)
Annex I is replaced by the text in Annex II to this Regulation;
(5)
point 2.1 of Annex Ia, is replaced by the following:
‘2.1.Each primary sample must be subdivided into laboratory samples, in accordance with point 2.5 of standard EN ISO 5555, and analysed according to the order shown in the flowchart set out in Annex Ib or in any other random order.’;
(6)
Annex Ib is replaced by the text in Annex III to this Regulation;
(7)
Annex V is deleted;
(8)
point 4.2 of Annex VII is replaced by the following:
‘4.2.n-hexane (chromatography grade). Hexane may be replaced by iso-octane (2,2,4- trimethylpentane in chromatography grade), provided that comparable precision values are achieved.’;
(9)
Annex XII is amended in accordance with Annex IV to this Regulation;
(10)
Annex XVII is amended in accordance with Annex V to this Regulation;
(11)
Annex XVIII is amended in accordance with Annex VI to this Regulation;
(12)
Annex XIX is replaced by the text in Annex VII to this Regulation;
(13)
point 4.2 of Annex XX is replaced by the following:
‘4.2.n-hexane, chromatography grade or residue grade. Hexane may be replaced by iso-octane (2,2,4-trimethyl pentane in chromatography grade), provided that comparable precision values are achieved. Solvents with higher boiling point than n-hexane take longer to evaporate. However, they are preferred due to the toxicity of hexane. The purity must by checked; for example, the residue after evaporation of 100 ml of solvent may be controlled.
WARNING — Fumes may ignite. Keep away from sources of heat, sparks or naked flames. Make sure the bottles are always properly closed. Ensure proper ventilation during usage. Avoid build-up of fumes and remove any possible fire risk, such as heaters or electric apparatus not manufactured from non-inflammable material. Pernicious if inhaled, because it may cause nerve cell damage. Avoid breathing in the fumes. Use a suitable respiratory apparatus if necessary. Avoid contact with eyes and skin.
Iso-octane is a flammable liquid that presents a fire hazard. Explosion limits in air are 1,1 % to 6,0 % (volume fraction). It is toxic by ingestion and inhalation. Use a ventilated hood in good operating condition to work with this solvent.’.
Article 2
This Regulation shall enter into force on the twentieth day following that of its publication in the Official Journal of the European Union.
This Regulation shall be binding in its entirety and directly applicable in all Member States.
Done at Brussels, 27 September 2019.
For the Commission
The President
Jean-Claude Juncker
ANNEX I
‘ANNEXES SUMMARY
| Annex I | Olive oil characteristics |
| Annex Ia | Sampling of olive oil or olive-pomace oil delivered in immediate packaging |
| Annex Ib | Flow-chart for verifying whether an olive oil sample is consistent with the category declared |
| Annex II | Determination of free fatty acids, cold method |
| Annex III | Determination of peroxide value |
| Annex IV | Determination of wax content by capillary column gas chromatography |
| Annex VII | Determination of the percentage of 2-glyceryl monopalmitate |
| Annex IX | Spectrophotometric investigation in the ultraviolet |
| Annex X | Determination of fatty acid methyl esters by gas chromatography |
| Annex XI | Determination of the volatile halogenated solvents of olive oil |
| Annex XII | The international olive council's method for the organoleptic assessment of virgin olive oil |
| Annex XV | Oil content of olive residue |
| Annex XVI | Determination of iodine value |
| Annex XVII | Method for the determination of stigmastadienes in vegetable oils |
| Annex XVIII | Determination of the difference between actual and theoretical content of triacylglycerols with ECN 42 |
| Annex XIX | Determination of the sterol composition and content and alcoholic compounds by capillary gas chromatography |
| Annex XX | Method for the determination of the content of waxes, fatty acid methyl esters and fatty acid ethyl esters by capillary gas chromatography |
| Annex XXI | Results of conformity checks carried out on olive oils referred to in Article 8(2)’ |
ANNEX II
‘ANNEX I OLIVE OIL CHARACTERISTICS
Quality characteristics
| a The median of defect may be less than or equal to 3,5 when the fruity median is equal to 0,0. | |||||||||
| Fatty acid ethyl esters(mg/kg) | ≤ 35 | — | — | — | — | — | — | — | |
|---|---|---|---|---|---|---|---|---|---|
| Organoleptic evaluation | Fruity median (Mf) | Mf > 0,0 | Mf > 0,0 | — | — | — | — | — | — |
| Median of defect (Md) (*) | Md = 0,0 | Md ≤ 3,5 | Md> 3,5a | ||||||
| Delta-K | ≤ 0,01 | ≤ 0,01 | — | ≤ 0,16 | ≤ 0,15 | — | ≤ 0,20 | ≤ 0,18 | |
| K268 or K270 | ≤ 0,22 | ≤ 0,25 | — | ≤ 1,25 | ≤ 1,15 | — | ≤ 2,00 | ≤ 1,70 | |
| K232 | ≤ 2,50 | ≤ 2,60 | — | — | — | — | — | — | |
| Peroxide value(mEq O2/kg) | ≤ 20,0 | ≤ 20,0 | — | ≤ 5,0 | ≤ 15,0 | — | ≤ 5,0 | ≤ 15,0 | |
| Acidity(%) (*) | ≤ 0,80 | ≤ 2,0 | > 2,0 | ≤ 0,30 | ≤ 1,00 | — | ≤ 0,30 | ≤ 1,00 | |
| Category | 1.Extra virgin olive oil | 2.Virgin olive oil | 3.Lampante olive oil | 4.Refined olive oil | 5.Olive oil composed of refined olive oil and virgin olive oils | 6.Crude olive-pomace oil | 7.Refined olive-pomace oil | 8.Olive-pomace oil | |
Purity characteristics
| a Other fatty acids content (%): palmitic: 7,50-20,00; palmitoleic: 0,30-3,50; heptadecanoic: ≤ 0,40; heptadecenoic ≤ 0,60; stearic: 0,50-5,00; oleic: 55,00- 83,00; linoleic: 2,50-21,00. | ||||||||||||||
| b Total isomers which could (or could not) be separated by capillary column. | ||||||||||||||
| 2-glyceryl monopalmitate(%) | ≤ 0,9 if total palmitic acid % ≤ 14,00 % | ≤ 1,0 if total palmitic acid % > 14,00 % | ≤ 0,9 if total palmitic acid % ≤ 14,00 % | ≤ 1,0 if total palmitic acid % > 14,00 % | ≤ 0,9 if total palmitic acid % ≤ 14,00 % | ≤ 1,1 if total palmitic acid % > 14,00 % | ≤ 0,9 if total palmitic acid % ≤ 14,00 % | ≤ 1,1 if total palmitic acid % > 14,00 % | ≤ 0,9 if total palmitic acid % ≤ 14,00 % | ≤ 1,0 if total palmitic acid % > 14,00 % | ≤ 1,4 | ≤ 1,4 | ≤ 1,2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Difference: ECN42 (HPLC) and ECN42(theoretical calculation) | ≤ |0,20| | ≤ |0,20| | ≤ |0,30| | ≤ |0,30| | ≤|0,30| | ≤ |0,60| | ≤ |0,50| | ≤ |0,50| | ||||||
| Stigmasta- dienes(mg/kg)b | ≤ 0,05 | ≤ 0,05 | ≤ 0,50 | — | — | — | — | — | ||||||
| Total trans- linoleic + translinolenic isomers(%) | ≤ 0,05 | ≤ 0,05 | ≤ 0,10 | ≤ 0,30 | ≤ 0,30 | ≤ 0,10 | ≤ 0,35 | ≤ 0,35 | ||||||
| Total transoleic isomers(%) | ≤ 0,05 | ≤ 0,05 | ≤ 0,10 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,40 | ≤ 0,40 | ||||||
| Fatty acid compositiona | Lignoceric(%) | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | |||||
| Behenic(%) | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,20 | ≤ 0,30 | ≤ 0,30 | ≤ 0,30 | ||||||
| Eicosenoic(%) | ≤ 0,50 | ≤ 0,50 | ≤ 0,50 | ≤ 0,50 | ≤ 0,50 | ≤ 0,50 | ≤ 0,50 | ≤ 0,50 | ||||||
| Arachidic(%) | ≤ 0,60 | ≤ 0,60 | ≤ 0,60 | ≤ 0,60 | ≤ 0,60 | ≤ 0,60 | ≤ 0,60 | ≤ 0,60 | ||||||
| Linolenic(%) | ≤ 1,00 | ≤ 1,00 | ≤ 1,00 | ≤ 1,00 | ≤ 1,00 | ≤ 1,00 | ≤ 1,00 | ≤ 1,00 | ||||||
| Myristic(%) | ≤ 0,03 | ≤ 0,03 | ≤ 0,03 | ≤ 0,03 | ≤ 0,03 | ≤ 0,03 | ≤ 0,03 | ≤ 0,03 | ||||||
| Category | 1.Extra virgin olive oil | 2.Virgin olive oil | 3.Lampante olive oil | 4.Refined olive oil | 5.Olive oil composed of refined olive oil and virgin olive oils | 6.Crude olive-pomace oil | 7.Refined olive-pomace oil | 8.Olive-pomace oil | ||||||
| a See the Appendix to this Annex. | |||||||||
| b App β-sitosterol: Delta-5,23-stigmastadienol+clerosterol+beta-sitosterol+sitostanol+delta-5-avenasterol+delta-5,24-stigmastadienol. | |||||||||
| c Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be lampante olive oil if the total aliphatic alcohol content is less than or equal to 350 mg/kg or if the erythrodiol and uvaol content is less than or equal to 3,5 %. | |||||||||
| d Oils with a wax content of between 300 mg/kg and 350 mg/kg are considered to be crude olive-pomace oil if the total aliphatic alcohol content is above 350 mg/kg and if the erythrodiol and uvaol content is greater than 3,5 %. | |||||||||
| Waxes (mg/kg)(**) | C42 + C44 + C46 ≤ 150 | C42 + C44 + C46 ≤ 150 | C40 + C42 + C44 + C46 ≤ 300c | C40 + C42 + C44 + C46 ≤ 350 | C40 + C42 + C44 + C46 ≤ 350 | C40 + C42 + C44 + C46 > 350d | C40 + C42 + C44 + C46 > 350 | C40 + C42 + C44 + C46 > 350 | |
|---|---|---|---|---|---|---|---|---|---|
| Erythrodiol and uvaol(%) (**) | ≤ 4,5 | ≤ 4,5 | ≤ 4,5c | ≤ 4,5 | ≤ 4,5 | > 4,5d | > 4,5 | > 4,5 | |
| Total sterols(mg/kg) | ≥ 1 000 | ≥ 1 000 | ≥ 1 000 | ≥ 1 000 | ≥ 1 000 | ≥ 2 500 | ≥ 1 800 | ≥ 1 600 | |
| Sterols composition | Delta-7-stigmastenola(%) | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 |
| App β–sitosterolb(%) | ≥ 93,0 | ≥ 93,0 | ≥ 93,0 | ≥ 93,0 | ≥ 93,0 | ≥ 93,0 | ≥ 93,0 | ≥ 93,0 | |
| Stigmasterol(%) | < Camp. | < Camp. | — | < Camp. | < Camp. | — | < Camp. | < Camp. | |
| Campesterola(%) | ≤ 4,0 | ≤ 4,0 | ≤ 4,0 | ≤ 4,0 | ≤ 4,0 | ≤ 4,0 | ≤ 4,0 | ≤ 4,0 | |
| Brassicasterol(%) | ≤ 0,1 | ≤ 0,1 | ≤ 0,1 | ≤ 0,1 | ≤ 0,1 | ≤ 0,2 | ≤ 0,2 | ≤ 0,2 | |
| Cholesterol(%) | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | ≤ 0,5 | |
| Category | 1.Extra virgin olive oil | 2.Virgin olive oil | 3.Lampante olive oil | 4.Refined olive oil | 5.Olive oil composed of refined olive oil and virgin olive oils | 6.Crude olive-pomace oil | 7.Refined olive-pomace oil | 8.Olive-pomace oil | |
Notes:
(a)The results of the analyses must be expressed to the same number of decimal places as used for each characteristic. The last digit must be increased by one unit if the following digit is greater than 4.
(b)If just a single characteristic does not match the values stated, the category of an oil can be changed or the oil is declared non-compliant for the purposes of this Regulation.
(c)For lampante olive oil, both quality characteristics marked with an asterisk (*) may differ simultaneously from the limits established for that category.
(d)If a characteristic is marked with two asterisks (**), this means that for crude olive-pomace oil, it is possible for both the relevant limits to be different from the stated values at the same time. For olive-pomace oil and refined olive-pomace oil one of the relevant limits may be different from the stated values.
Appendix Decision trees
Campesterol decision tree for virgin and extra virgin olive oils:

The other parameters shall comply with the limits fixed in this Regulation.
Delta-7-stigmastenol decision tree for:
Extra virgin and virgin olive oils
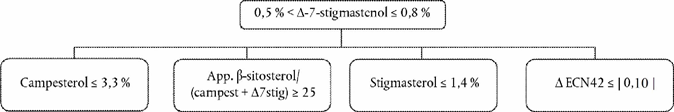
The other parameters shall comply with the limits fixed in this Regulation.
Olive-pomace oils (crude and refined)

The other parameters shall comply with the limits fixed in this Regulation.’
ANNEX III
‘ANNEX Ib FLOW-CHART FOR VERIFYING WHETHER AN OLIVE OIL SAMPLE IS CONSISTENT WITH THE CATEGORY DECLARED
General table

Table 1 — Extra Virgin Olive Oil — Quality criteria
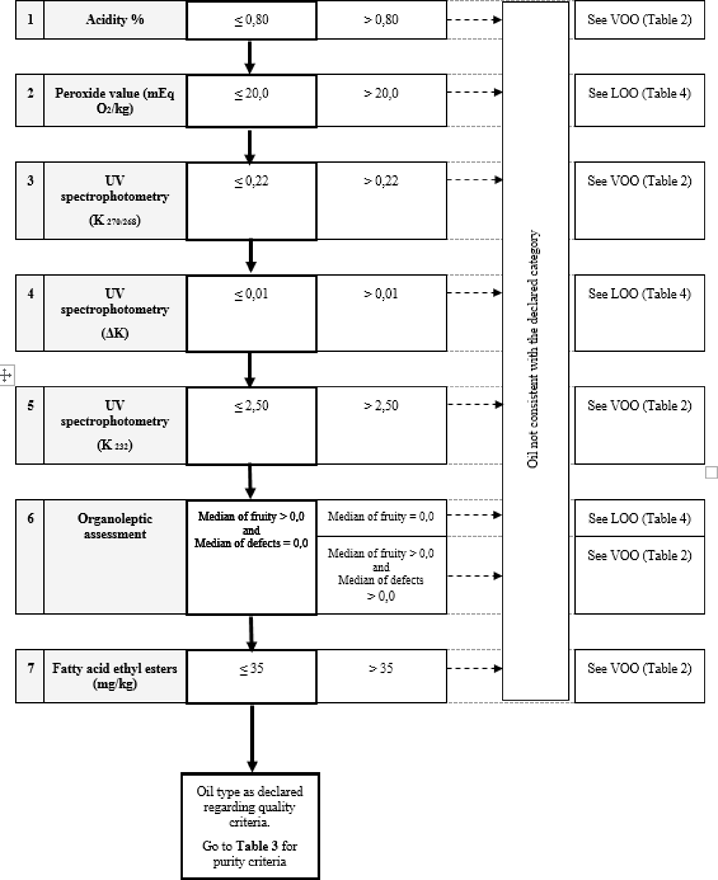
Table 2 — Virgin Olive Oil — Quality criteria

Table 3 — Extra Virgin Olive Oil and Virgin Olive Oil — Purity criteria

Table 4 — Lampante Olive Oil — Purity criteria
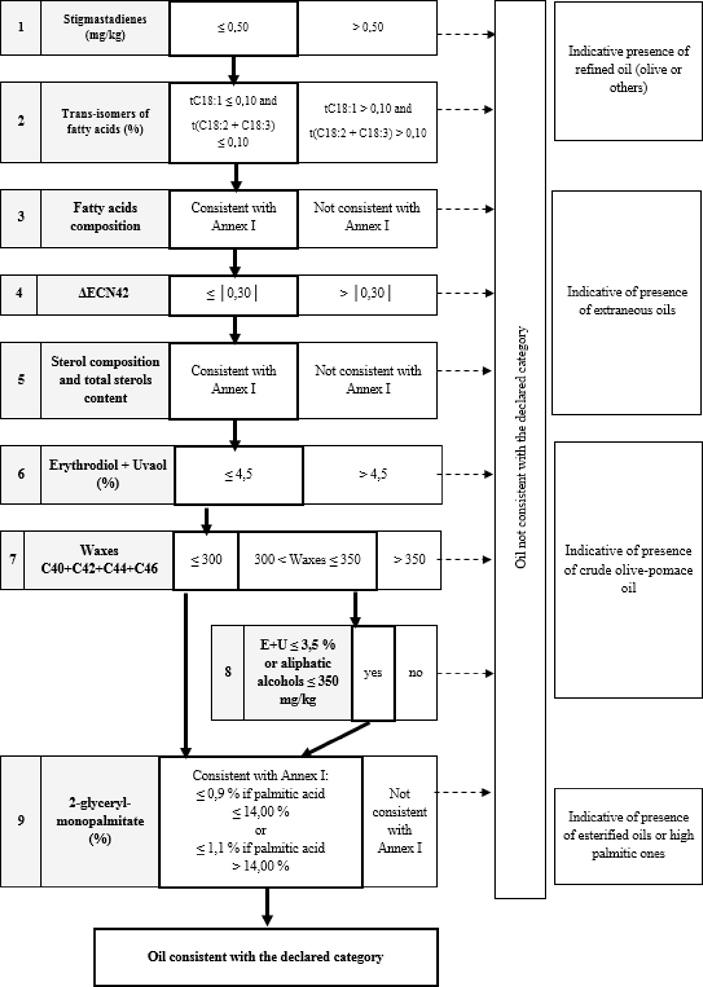
Table 5 — Refined Olive Oil — Quality criteria
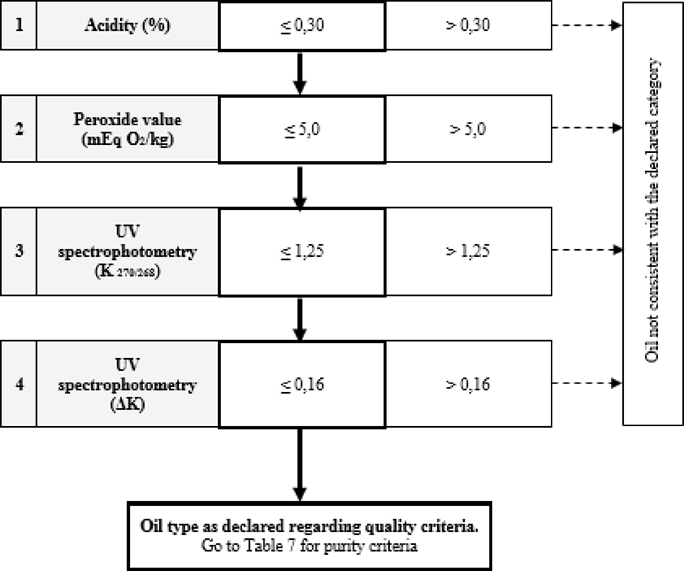
Table 6 — Olive Oil (composed of refined olive oil and virgin olive oils) — Quality criteria

Table 7 — Refined Olive Oil and olive oil composed of refined olive oil and Virgin Olive Oils — Purity criteria

Table 8 — Crude Olive-Pomace Oil — Purity criteria
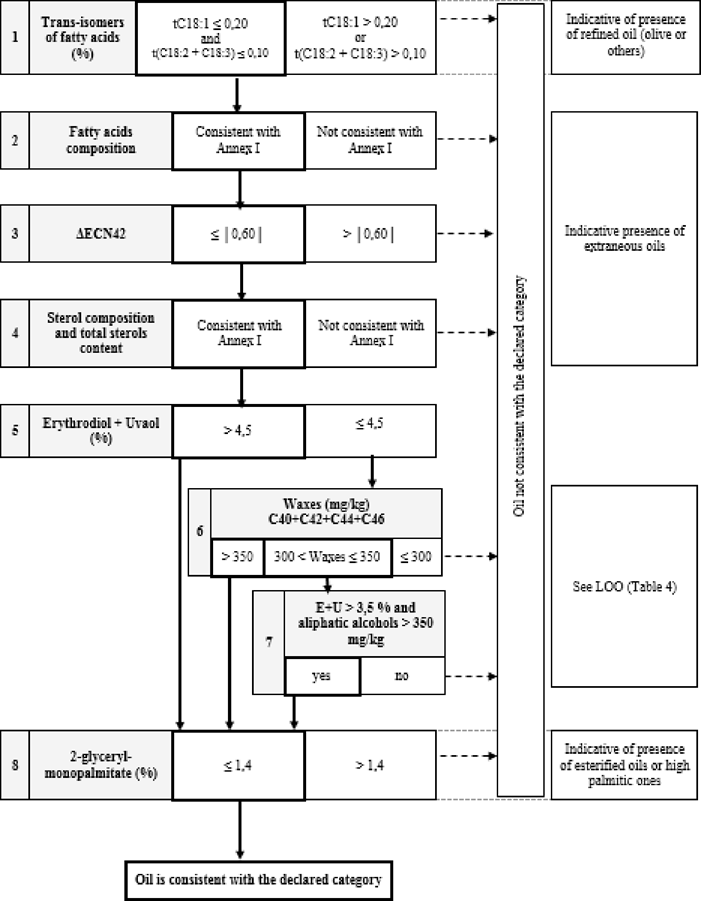
Table 9 — Refined Olive-Pomace Oil — Quality criteria

Table 10 — Olive Pomace Oil — Quality criteria

Table 11 — Refined Olive-Pomace Oil and Olive-Pomace Oil — Purity criteria

’’;
ANNEX IV
Annex XII is amended as follows:
(1)
point 3.3 is replaced by the following:
‘3.3. Optional terminology for labelling purposes
Upon request, the panel leader may certify that the oils which have been assessed comply with the definitions and ranges corresponding solely to the following terms according to the intensity and perception of the attributes.
Positive attributes (fruity, bitter and pungent): According to the intensity of perception:
Robust, when the median of the attribute is more than 6,0;
Medium, when the median of the attribute is more than 3,0 and less or equal to 6,0;
Delicate, when the median of the attribute is less or equal to 3,0.
| Fruitiness | Set of olfactory sensations characteristic of the oil which depends on the variety of olive and comes from sound, fresh olives in which neither green nor ripe fruitiness predominates. It is perceived directly and/or through the back of the nose. |
| Green fruitiness | Set of olfactory sensations characteristic of the oil which is reminiscent of green fruit, depends on the variety of olive and comes from green, sound, fresh olives. It is perceived directly and/or through the back of the nose. |
| Ripe fruitiness | Set of olfactory sensations characteristic of the oil which is reminiscent of ripe fruit, depends on the variety of olive and comes from sound, fresh olives. It is perceived directly and/or through the back of the nose. |
| Well balanced | Oil which does not display a lack of balance, by which is meant the olfactory-gustatory and tactile sensation where the median of the bitter attribute and the median of the pungent attribute are not more than 2,0 points above the median of the fruitiness. |
| Mild oil | Oil for which the median of the bitter and pungent attributes is 2,0 or less. |
List of terms according to the intensity of perception:
| Terms subject to production of an organoleptic test certificate | Median of the attribute |
|---|---|
| Fruitiness | — |
| Ripe fruitiness | — |
| Green fruitiness | — |
| Delicate fruitiness | ≤ 3,0 |
| Medium fruitiness | 3,0 < Me ≤ 6,0 |
| Robust fruitiness | > 6,0 |
| Delicate ripe fruitiness | ≤ 3,0 |
| Medium ripe fruitiness | 3,0 < Me ≤ 6,0 |
| Robust ripe fruitiness | > 6,0 |
| Delicate green fruitiness | ≤ 3,0 |
| Medium green fruitiness | 3,0 < Me ≤ 6,0 |
| Robust green fruitiness | > 6,0 |
| Delicate bitterness | ≤ 3,0 |
| Medium bitterness | 3,0 < Me ≤ 6,0 |
| Robust bitterness | > 6,0 |
| Delicate pungency | ≤ 3,0 |
| Medium pungency | 3,0 < Me ≤ 6,0 |
| Robust pungency | > 6,0 |
| Well balanced oil | The median of the bitter attribute and the median of the pungent attribute are not more than 2,0 points above the median of the fruitiness. |
| Mild oil | The median of the bitter attribute and the median of the pungent attribute are 2,0 or less.’ |
(2)
point 9.4 is replaced by the following:
‘9.4. Classification of the oil
The oil is graded as follows in line with the median of the defects and the median for the fruity attribute. The median of the defects is defined as the median of the defect perceived with the greatest intensity. The median of the defects and the median of the fruity attribute are expressed to one decimal place.
The oil is graded by comparing the median value of the defects and the median of the fruity attribute with the reference ranges given below. The error of the method has been taken into account when establishing the limits of these ranges, which are therefore considered to be absolute. The software packages allow the grading to be displayed as a table of statistics or a graph.
(a)
Extra virgin olive oil: the median of the defects is 0,0 and the median of the fruity attribute is above 0,0;
(b)
Virgin olive oil: the median of the defects is above 0,0 but not more than 3,5 and the median of the fruity attribute is above 0,0;
(c)
Lampante virgin olive oil: the median of the defects is above 3,5 or the median of the defects is less than or equal to 3,5 and the fruity median is equal to 0,0.
Note 1: When the median of the bitter and/or pungent attribute is more than 5,0, the panel leader shall state so on the test certificate.
For assessments intended to monitor compliance, one test shall be carried out. In the case of counter assessments, the analysis must be carried out in duplicate in different tasting sessions. The results of the duplicate analysis must be statistically homogenous (see point 9.5). If not, the sample must be reanalysed twice again. The final value of the median of the classification attributes will be calculated using the average of both medians.’.
ANNEX V
Annex XVII is amended as follows:
(1)
point 5.1 is replaced by the following:
‘5.1.Hexane or mixture of alkanes of b.p. interval 65 to 70 °C, distilled with rectifying column. Hexane may be replaced by iso-octane (2,2,4-trimethyl pentane in chromatography grade), provided that comparable precision values are achieved. The residue after evaporation of 100 ml of solvent may be controlled. Solvents with higher boiling point than n-hexane take longer to evaporate. However, they are preferred due to the toxicity of hexane.’;
(2)
in point 6.3.3, the following text is added:
‘Note 10. When stigmastadienes appear in concentrations of more than 4 mg/kg, if quantifying is required, the method of the International Olive Council for determination of sterenes in refined oil must be applied.’.
ANNEX VI
Annex XVIII is amended as follows:
(1)
point 4.2.1 is replaced by the following:
‘4.2.1.Petroleum ether 40-60 °C chromatographic grade or hexane. Hexane may be replaced by iso-octane (2,2,4-trimethyl pentane in chromatography grade), provided that comparable precision values are achieved. Solvents with higher boiling point than n-hexane take longer to evaporate. However, they are preferred due to the toxicity of hexane.’;
ANNEX VII
‘ANNEX XIX
DETERMINATION OF THE STEROL COMPOSITION AND CONTENT AND ALCOHOLIC COMPOUNDS BY CAPILLARY GAS CHROMATOGRAPHY
1.SCOPE
The method describes a procedure for determining the individual and total alcoholic compound content of olive oils and olive pomace oils as well as of blends of these two oils.
The alcoholic compounds in olive and olive pomace oils comprise aliphatic alcohols, sterols and triterpenic dialcohols.
2.PRINCIPLE
The oils, with added α-cholestanol and 1-eicosanol as internal standards, are saponified with potassium hydroxide in ethanolic solution and the unsaponifiable matter is then extracted with ethyl ether.
The different alcoholic compounds fractions are separated from the unsaponifiable matter either by thin-layer chromatography on a basic silica gel plate (reference method) or by HPLC with a silica gel column. The fraction recovered from the silica gel separation is transformed into trimethylsilyl ethers and then analysed by capillary column gas chromatography.
PART 1 PREPARATION OF THE UNSAPONIFIABLE MATTER
1.SCOPE
This Part describes the preparation and extraction of the unsaponifiable matter. It includes the preparation and extraction of the unsaponifiable matter from olive and olive-pomace oils.
2.PRINCIPLE
A test portion is saponified by boiling under reflux with an ethanolic potassium hydroxide solution. The unsaponifiable matter is extracted with diethyl ether.
3.APPARATUS
The usual laboratory equipment and in particular the following:
3.1.
Round bottomed flask fitted with a reflux condenser with ground-glass joints, 250 mL.
3.2.
Separating funnel, 500 mL.
3.3.
Flasks, 250 mL.
3.4.
Microsyringes, 100 μL and 500 μL.
3.5.
Cylindrical filter funnel with a G3 porous septum (porosity 15-40 μm) of diameter approximately 2 cm and a depth of 5 cm, suitable for filtration under vacuum with male ground-glass joint.
3.6.
Conical flask with ground-glass female joint, 50 mL, which can be fitted to the filter funnel (3.5).
3.7.
Test tube with a tapering bottom and a sealing glass stopper, 10 mL.
3.8.
Calcium dichloride desiccator.
4.REAGENTS
4.1.
Potassium hydroxide minimum titre 85 %.
4.2.
Potassium hydroxide ethanolic solution, approximately 2 M.
Dissolve 130 g of potassium hydroxide (4.1) with cooling in 200 ml of distilled water and then make up to one litre with ethanol (4.7). Keep the solution in well-stoppered dark glass bottles and stored maximum 2 days.
4.3.
Ethyl ether, for analysis quality.
4.4.
Anhydrous sodium sulphate, for analysis quality.
4.5.
Acetone, for chromatography quality.
4.6.
Ethyl ether, for chromatography quality.
4.7.
Ethanol of analytical quality.
4.8.
Ethyl acetate of analytical quality.
4.9.
Internal standard, α-cholestanol, purity more than 99 % (purity must be checked by GC analysis).
4.10.
Internal standard solution of α-cholestanol, 0,2 solution (m/V) in ethyl acetate (4.8).
4.11.
Phenolphthalein solution, 10 g/L in ethanol (4.7).
4.12.
A 0,1 % (m/v) solution of 1-eicosanol in ethyl acetate (internal standard).
5.PROCEDURE
Using a 500 μL micro-syringe (3.4) introduce into the 250 mL flask (3.1) a volume of the α-cholestanol internal standard solution (4.10) and a volume of 1-eicosanol (4.12) containing an amount of cholestanol and eicosanol corresponding to approximately 10 % of the sterol and alcohol content of the sample. For example, for 5 g of olive oil sample add 500 μL of the α-cholestanol solution (4.10) and 250 μL of 1-eicosanol solution (4.12). For pomace olive oils add 1500 μL of both α-cholestanol solution (4.10) and 1-eicosanol (4.12). Evaporate until dryness with a gentle current of nitrogen in a warm water bath. After cooling the flask, weigh 5,00 ± 0,01 g of the dry filtered sample into the same flask.
Note 1: Animal or vegetable oils and fats containing appreciable quantities of cholesterol may show a peak having a retention time identical to cholestanol. If this case occurs, the sterol fraction will have to be analysed in duplicate with and without internal standard.
Add 50 mL of 2M ethanolic potassium hydroxide solution (4.2) and some pumice, fit the reflux condenser and heat to gentle boiling until saponification takes place (the solution becomes clear). Continue heating for a further 20 minutes, then add 50 mL of distilled water from the top of the condenser, detach the condenser and cool the flask to approximately 30 °C.
Transfer the contents of the flask quantitatively into a 500 mL separating funnel (3.2) using several portions of distilled water (50 mL). Add approximately 80 ml of ethyl ether (4.6), shake vigorously for approximately 60 seconds, periodically releasing the pressure by inverting the separating funnel and opening the stopcock. Allow standing until there is complete separation of the two phases (Note 2). Then draw off the soap solution as completely as possible into a second separating funnel. Perform two further extractions on the water-alcohol phase in the same way using 60 to 70 mL of ethyl ether (4.6).
Note 2: Any emulsion can be destroyed by adding small quantities of ethanol (4.7).
Combine the three ether extracts in one separating funnel containing 50 mL of water. Continue to wash with water (50 mL) until the wash water no longer gives a pink colour on the addition of a drop of phenolphthalein solution (4.11). When the wash water has been removed, filter on anhydrous sodium sulphate (4.4) into a previously weighed 250 mL flask, washing the funnel and filter with small quantities of ethyl ether (4.6).
Evaporate the solvent by distillation in a rotary evaporator at 30 °C under vacuum. Add 5mL of acetone (4.5) and remove the volatile solvent completely in a gentle current of nitrogen. Dry the residue in the oven at 103 ± 2 °C for 15 min. Cool in desiccators and weigh to the nearest 0,1 mg.
PART 2 SEPARATION OF THE ALCOHOLIC COMPOUNDS FRACTIONS
1.SCOPE
The unsaponifiable matter prepared in Part 1 is fractionated in the different alcoholic compounds, aliphatic alcohols, sterols and triterpenic dialcohols (erythrodiol and uvaol).
2.PRINCIPLE
The unsaponifiable matter can be fractionated using basic thin layer chromatography (reference method), revealed and the corresponding bands scratched and extracted. As an alternative method of separation, HPLC using a silica gel column and UV detector and the different fractions collected. The aliphatic and triterpenic alcohols as well as the sterol and triterpenic dialcohols are isolated together.
3.APPARATUS
The usual laboratory equipment and in particular the following:
3.1.
Complete apparatus for analysis by thin-layer chromatography using 20 × 20 cm glass plates.
3.2.
Ultraviolet lamp with a wavelength of 366 or 254 nm.
3.3.
Microsyringes, 100 μL and 500 μL.
3.4.
Cylindrical filter funnel with a G3 porous septum (porosity 15-40 μm) of diameter approximately 2 cm and a depth of 5 cm, suitable for filtration under vacuum with male ground-glass joint.
3.5.
Conical flask with ground-glass female joint, 50 mL which can be fitted to the filter funnel (3.4).
3.6.
Test tube with a tapering bottom and a sealing glass stopper, 10 mL.
3.7.
Calcium dichloride desiccator.
3.8.
HPLC system, consisting of:
3.8.1.
Binary pump.
3.8.2.
Manual or automatic injector equipped with 200 μL injection loop.
3.8.3.
In-line degasser.
3.8.4.
UV-VIS or IR detector.
3.9.
HPLC column (25 cm × 4 mm i.d.) with silica gel 60 (5 μm particle size).
3.10.
Syringe filter, 0,45 μm.
3.11.
Conical flask 25 mL.
4.REAGENTS
4.1.
Potassium hydroxide minimum titre 85 %.
4.2.
Potassium hydroxide ethanolic solution, approximately 2 M.
Dissolve 130 g of potassium hydroxide (4.1) with cooling in 200 ml of distilled water and then make up to one litre with ethanol (4.9). Keep the solution in well-stoppered dark glass bottles and stored maximum 2 days.
4.3.
Ethyl ether, for analysis quality.
4.4.
Potassium hydroxide ethanolic solution, approximately 0,2 M.
Dissolve 13 g of potassium hydroxide (4.1) in 20 ml of distilled water and make up to one litre with ethanol (4.9).
4.5.
Glass 20x20 plates coated with silica gel, without fluorescence indicator, thickness 0,25 mm (commercially available ready for use).
4.6.
Acetone, for chromatography quality.
4.7.
n-Hexane, for chromatography quality.
4.8.
Ethyl ether, for chromatography quality.
4.9.
Ethanol of analytical quality.
4.10.
Ethyl acetate of analytical quality.
4.11.
Reference solution for thin-layer chromatography: cholesterol, phytosterols, alcohols and Erythrodiol 5 % solution in Ethyl acetate (4.10).
4.12.
Solution of 2,7-dichlorofluorescein, 0,2 % in ethanolic solution. Make slightly basic by adding a few drops of 2 M alcoholic potassium hydroxide solution (4.2).
4.13.
n-Hexane (4.7)/ethyl ether (4.8) mixture 65:35 (V/V).
4.14.
HPLC mobile phase n-hexane (4.7)/ethyl ether (4.8) (1:1) (V/V).
5.REFERENCE METHOD: SEPARATION OF THE ALCOHOLIC COMPOUNDS BY BASIC THIN-LAYER CHROMATOGRAPHY (TLC) PLATE
Preparation of the basic thin layer chromatography plates. Immerse or dip the silica gel plates (4.5) about 4 cm in the 0,2 M ethanolic potassium hydroxide solution (4.4) for 10 seconds, then allow to dry in a fume cupboard for two hours and finally place in an oven at 100 °C for one hour.
Remove from the oven and keep in a calcium chloride desiccator (3.7) until required for use (plates treated in this way must be used within 15 days).
Place hexane/ethyl ether mixture (4.13) (Note 3) into the development chamber, to a depth of approximately 1 cm. Close the chamber with the appropriate cover and leave thus for at least half an hour, in a cool place, so that liquid-vapour equilibrium is established. Strips of filter paper dipping into the eluent may be placed on the internal surfaces of the chamber. This reduces developing time by approximately one-third and brings about more uniform and regular elution of the components.
Note 3: The developing mixture should be replaced for every test, in order to achieve perfectly reproducible elution conditions. Alternative solvent 50:50 (V/V) n-hexane/ethyl ether may be used.
Prepare an approximately 5 % solution of the unsaponifiable prepared in Part 1 in ethyl acetate (4.10) and, using the 100 μL microsyringe (3.3), depose 0,3 ml of the solution on a narrow and uniform streak on the lower end (2 cm) of the chromatographic plate (4.5). In line with the streak, place 2 to 3 μL of the material reference solution (4.11), so that the sterol, triterpene dialcohols and alcohols bands can be identified after developing.
Place the plate in the developing chamber (3.1). The ambient temperature should be maintained between 15 and 20 °C (Note 4). Immediately close the chamber with the cover and allow eluting until the solvent front reaches approximately 1 cm from the upper edge of the plate. Remove the plate from the developing chamber and evaporate the solvent in a flow of hot air or by leaving the plate for a short while, under a hood.
Note 4: Higher temperature could worsen the separation.
Spray the plate lightly and uniformly with the 2,7-dichlorofluorescein solution (4.12) and then leave to dry. When the plate is observed under ultraviolet lamp (3.2), the sterols, triterpene dialcohols and alcohols bands can be identified through being aligned with the spots obtained from the reference solution (4.11). Mark the limits of the bands along the edges of the fluorescence with a black pencil (see TLC plate in Figure 1).
By using a metal spatula, scrape off the silica gel of the marked area. Place the finely comminuted material removed into the filter funnel (3.4). Add 10 mL of hot ethyl acetate (4.10), mix carefully with the metal spatula and filter (under vacuum if necessary), collecting the filtrate in the conical flask (3.5.) attached to the filter funnel.
Wash the residue in the flask three times with ethyl ether (4.3) (approximately 10 mL each time), collecting the filtrate in the same flask attached to the funnel, evaporate the filtrate to a volume of 4 to 5 mL, transfer the residual solution to the previously weighed 10 mL test tube (3.6), evaporate to dryness by mild heating, in a gentle flow of nitrogen, make up again using a few drops of acetone (4.6), evaporate again to dryness. The residue contained in the test tube consists of the sterol and triterpene dialcohols or the alcohols and triterpenic alcohols fractions.
6.SEPARATION OF THE ALCOHOLIC FRACTION BY HPLC
The unsaponifiable obtained from Part 1 is dissolved in 3 mL of the mobile phase (4.14), filter the solution with a syringe filter (3.10) and reserve.
Inject 200 μL of the filtered unsaponifiable solution in the HPLC (3.8).
Run the HPLC separation at 0,8 mL/min, discard the first 5 min. and collect in 25 mL conical flasks (3.11) between the 5 and 10 min. for aliphatic and triterpenic alcohols and between 11 and 25 min for sterols and erythrodiol and uvaol (Note 5).
The separation can be monitored with an UV detector at 210 nm wavelengths or a refractive index detector (see Figure 6).
The fractions are evaporated until dryness and prepared for chromatographic analysis.
Note 5: Carefully control the pressure of the HPLC pump, the ethyl ether can increase the pressure, adjust the flow to keep the pressure under control.
PART 3 GAS CHROMATOGRAPHIC ANALYSIS OF THE ALCOHOLIC COMPOUNDS FRACTIONS
1.SCOPE
This part gives general guidance for the application of capillary column gas chromatography to determine the qualitative and quantitative composition of the alcoholic compounds isolated in accordance with the method specified in Part 2 of this method.
2.PRINCIPLE
The fractions collected from the unsaponifiable matter using TLC or HPLC are derivatized into trimethylsilyl ethers and analysed by capillary column gas chromatography with split injection and flame ionization detector.
3.APPARATUS
The usual laboratory equipment and in particular the following:
3.1.
Test tube with a tapering bottom and a sealing glass stopper, 10 mL.
3.2.
Gas chromatograph suitable for use with a capillary column with split injection system, consisting of:
3.2.1.
A thermostatic chamber for columns capable of maintaining the desired temperature with an accuracy of ± 1 °C;
3.2.2.
A temperature-adjustable injection unit with a persilanised glass vaporising element and split system;
3.2.3.
A flame ionisation detector (FID);
3.2.4.
Data acquisition system suitable for use with the FID detector (3.10.3.), capable of manual integration.
3.3.
Fused-silica capillary column of length 20 to 30 m, internal diameter 0,25 to 0,32 mm, coated with 5 % Diphenyl - 95 % Dimethylpolysiloxane (SE-52 or SE-54 stationary phase or equivalent), to a uniform thickness between 0,10 and 0,30 μm.
3.4.
Microsyringe, of 10 μL capacity, for gas chromatography, with cemented needle suitable for split injection.
4.REAGENTS
4.1.
Anhydrous pyridine, for chromatography quality.
4.2.
Hexamethyl disilazane of analytical quality.
4.3.
Trimethylchlorosilane of analytical quality.
4.4.
Sample solutions of sterol trimethylsilyl ethers. To be prepared at the time of use from sterols and erythrodiol obtained from oils containing them.
4.5.
Standard solutions of trimethylsilyl ethers of aliphatic alcohols from C20 to C28. They may be prepared from mixtures of pure alcohols at the time they are required for use.
4.6.
Carrier gas: hydrogen or helium, gas-chromatographic purity.
4.7.
Auxiliary gases: hydrogen, helium, nitrogen and air, of gas-chromatographic purity.
4.8.
Silylation reagent, consisting of a 9:3:1 (V/V/V) mixture of pyridine/hexamethyl disilazane/trimethylchlorosilane.
4.9.
n-Hexane, for chromatography quality.
5.PREPARATION OF THE TRIMETHYLSILYL ETHERS
Add the silylation reagent (4.8) (Note 6), in the ratio of 50 μl for every milligram of alcoholic compound, in the test tube (3.1) containing the alcoholic compound fraction, avoiding any uptake of moisture (Note 7).
Note 6: Ready for use solutions are available commercially. Other silylation reagents, such as, for example, bistrimethylsilyl trifluor acetamide + 1 % trimethylchlorosilane, which has to be diluted with an equal volume of anhydrous pyridine, are also available. Pyridine can be replaced by the same amount of acetonitrile.
Note 7: The slight opalescence, which may form, is normal and does not cause any anomaly. The formation of a white flock or the appearance of a pink colour is indicative of the presence of moisture or deterioration of the reagent. If these occur the test must be repeated (only if hexamethyldisilazane/trimethylchlorosilane is used).
Stopper the test tube (3.1), shake carefully (without overturning) until the compounds are completely dissolved. Leave to stand for at least 15 minutes at ambient temperature and then centrifuge for a few minutes. The clear solution is ready for gas chromatographic analysis.
6.GAS CHROMATOGRAPHIC ANALYSIS
6.1.Preliminary operations, capillary column conditioning
Fit the column (3.3) in the gas chromatograph, by attaching the inlet end to the split injector and the outlet end to the detector.
Carry out general checks on the gas chromatograph unit (leaks from the gas circuits, detector efficiency, efficiency of the splitting system and recording system, etc.).
If the column is being used for the first time, it is recommended that it should be subjected to conditioning: passing a gentle flow of gas through the column itself, then switch on the gas chromatography unit and begin a gradual heating, up to a temperature of at least 20 °C above the operating temperature (Note 8). Hold this temperature for at least two hours, then place the entire unit in operating mode (adjustment of gas flows and splitting, ignition of the flame, connection with the computing system, adjustment of the column, detector and injector temperature, etc.) and then record the signal with a sensitivity at least two times greater than that one intended for the analysis. The course of the base line must be linear, without peaks of any kind, and must not show drift. A negative straight-line drift indicates leakage from the column connections; a positive drift indicates inadequate conditioning of the column.
Note 8: The conditioning temperature must always be at least 20 °C less than the maximum temperature specified for the stationary phase used.
6.2.Operating conditions
Optimize the temperature programme and the carrier gas flow so that chromatograms similar to Figures 3 to 6 are obtained.
The following parameters were tested and found useful:
6.2.1.Aliphatic alcohols
| Oven Program | 180 °C (8 min.) → 260 °C (at 5 °C/min) → 260 °C (15 min) |
|---|---|
| Injector Temperature | 280 °C |
| Detector Temperature | 290 °C |
| Linear Velocity of Carrier gas | Helium (20 to 30 cm/s); Hydrogen (30 to 50 cm/s) |
| Split Ratio | 1:50 to 1:100 |
| Volume Injected | 0,5 to 1 μL of TMSE solution |
6.2.2.Sterol and triterpenic dialcohols
| Oven Program | 260 ± 5 °C Isothermal |
|---|---|
| Injector Temperature | 280 – 300 °C |
| Detector Temperature | 280 – 300 °C |
| Linear Velocity of Carrier gas | Helium (20 to 30 cm/s); Hydrogen (30 to 50 cm/s) |
| Split Ratio | 1:50 to 1:100 |
| Volume Injected | 0,5 to 1 μL of TMSE solution |
These conditions may be changed according to the characteristics of the column and gas chromatograph, so as to obtain chromatograms, which meet the following requirements:
Alcohol C26 retention time shall be 18 ± 5 minutes.
Alcohol C22 peak shall be 80 ± 20 % of the full-scale value for olive oil and 40 ± 20 % of the full-scale value for olive-pomace oil.
The retention time for the β-sitosterol peak should be at 20 ± 5 min.
The campesterol peak should be: for olive oil (mean content 3 %) 20 ± 5 % of full scale.
All the present sterols must be separated. In addition to being separated, the peaks must also be completely resolved, i.e. the peak trace should return to the base line before leaving for the next peak. Incomplete resolution is, however, tolerated, provided that the peak at RRT 1,02 (Sitostanol) can be quantified using the perpendicular.
6.3.Analytical procedure
By using the 10 μl microsyringe (3.4), take 1 μl of hexane, draw in 0,5 μl of air and then 0,5 to 1 μl of the sample solution. Raise the plunger of the syringe further, so the needle is emptied. Push the needle through the membrane of the injector and after one to two seconds, inject rapidly, and then slowly remove the needle after around five seconds. An automatic injector can be used as well.
Carry out the recording until the TMSE of the corresponding alcoholic compounds present are completely eluted. The base line must continue to meet the requirements of the corresponding operating conditions (6.2.1 or 6.2.2).
6.4.Peak identification
Identify individual peaks on the basis of retention times and by comparison with the mixture of the aliphatic and triterpenic alcohols or the sterol and triterpene dialcohols TMSE, analysed under the same conditions. A chromatogram of the aliphatic and triterpenic alcohols fraction is showed in Figure 3 and the corresponding chromatograms for sterols and triterpenic dialcohols are showed in Figure 2.
The aliphatic alcohols are eluted in the following order: C20-ol (I.S.), C22-ol, C23-ol, C24-ol, C25-ol, C26-ol, C27-ol and C28-ol.
The sterols and triterpene dialcohols are eluted in the following order: cholesterol, brassicasterol, ergosterol, 24-methylen-cholesterol, campesterol, campestanol, stigmasterol, Δ7-campesterol, Δ5,23-stigmastadienol, clerosterol, β-sistosterol, sitostanol, Δ5-avenasterol, Δ5,24-stigmastadienol, Δ7-stigmastenol, Δ7-avenasterol, erythrodiol and uvaol.
6.5.Quantitative evaluation
The peak areas of 1-eicosanol and of the aliphatic alcohols C22, C24, C26, C28 are calculated by a data acquisition system. The response factor for 1-eicosanol should be considered equal to 1.
Calculate the areas of the α-cholestanol and the sterol and triterpene dialcohols peaks by using the computing system. Ignore peaks for any compound, which are not included (ergosterol must not be calculated) among those listed in Table 1. The response factor for α-cholestanol should be considered equal to 1.
Calculate the concentration of each individual alcoholic compound, in mg/kg of fatty material, as follows:

where:
Ax
=
Peak area for alcoholic compound x, in computing system counts.
As
=
Area of the 1-eicosanol/α-cholestanol peak, in computing system counts.
ms
=
Mass of added 1-eicosanol/α-cholestanol, in milligrams.
m
=
Mass of the sample used for determination, in grams.
7.EXPRESSION OF THE RESULTS
Report individual aliphatic and triterpenic alcohols concentrations as mg/kg of fatty material and their sum as 'total aliphatic alcohol content'. The total content is the sum of C22, C24, C26 and C28.
The composition of each of the individual alcoholic compounds should be expressed to one decimal point.
Total sterol concentration should be expressed without any decimal point.
Calculate the percentage of each individual sterol from the ratio of the relevant peak area to the total peak area for sterols:
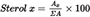
where:
Ax
=
Peak area for sterol x.
ΣA
=
Total peak area for sterols.
Apparent β-sitosterol: Δ5,23-stigmastadienol + clerosterol + β-sitosterol + sitostanol + Δ5-avenasterol + Δ5,24-stigmastadienol.
Calculate the percentage of erythrodiol and uvaol:

where:
AEr
=
Area of Erythrodiol in computing system counts.
AUv
=
Area of Uvaol in computing system counts.
Σ AT
=
Sum area for sterol + erythrodiol + uvaol in computing system counts.
Besides the calculation of relative percentage of single sterols and triterpenic dialcohols and the total concentration of sterols, the concentration of erythrodiol and of uvaol and their sum, in mg/kg of fatty material must be calculated, according the following expressions:


where:
AEr
=
Peak area of Erythrodiol, in computing system counts.
AUv
=
Area of Uvaol in computing system counts.
As
=
Area of the α-cholestanol peak, in computing system counts.
ms
=
Mass of added α-cholestanol, in milligrams.
m
=
Mass of the sample used for determination, in grams.
Appendix

Figure 1 — TLC of the unsaponifiable fraction from olive pomace oil eluted twice with hexane:diethyl ether (65:35), developed with SO4H2 (50 %) and heated. The bands that should be scrapped are the ones contained within the rectangle, 1 are the bands for aliphatic alcohols and 2 for the sterols and triterpenic dialcohols.
Table I — Relative retention times for sterols
| Peak | Identification | Relative retention time | ||
|---|---|---|---|---|
| SE 54 column | SE 52 column | |||
| 1 | Cholesterol | Δ-5-cholesten-3β-ol | 0,67 | 0,63 |
| 2 | Cholestanol | 5α-cholestan-3β -ol | 0,68 | 0,64 |
| 3 | Brassicasterol | [24S]-24-methyl-Δ-5,22-cholestadien-3β -ol | 0,73 | 0,71 |
| * | Ergosterol | [24S]-24-methyl-Δ-5,7,22 cholestatrien-3β -ol | 0,78 | 0,76 |
| 4 | 24-methylene-cholesterol | 24-methylene-Δ-5,24-cholestadien-3β -o1 | 0,82 | 0,80 |
| 5 | Campesterol | (24R)-24-methyl-Δ-5-cholesten-3β -ol | 0,83 | 0,81 |
| 6 | Campestanol | (24R)-24-methyl-cholestan-3β -ol | 0,85 | 0,82 |
| 7 | Stigmasterol | (24S)-24-ethyl-Δ-5,22-cholestadien-3β -ol | 0,88 | 0,87 |
| 8 | Δ-7-campesterol | (24R)-24-methyl-Δ-7-cholesten-3β -ol | 0,93 | 0,92 |
| 9 | Δ-5,23-stigmastadienol | (24R,S)-24-ethyl-Δ-5,23-choIestadien-3β -ol | 0,95 | 0,95 |
| 10 | Clerosterol | (24S)-24-ethyl-Δ-5,25-cholestadien-3β -ol | 0,96 | 0,96 |
| 11 | ß-sitosterol | (24R)-24-ethyl-Δ-5-cholesten-3β -ol | 1,00 | 1,00 |
| 12 | Sitostanol | 24-ethyl-cholestan-3β -ol | 1,02 | 1,02 |
| 13 | Δ-5-avenasterol | (24Z)-24-ethylidene-Δ-cholesten-3β -ol | 1,03 | 1,03 |
| 14 | Δ-5,24-stigmastadienol | (24R,S)-24-ethyl-Δ-5,24-cholestadien-3β -ol | 1,08 | 1,08 |
| 15 | Δ-7-stigmastenol | (24R,S)-24-ethyl-Δ-7-cholesten-3β -ol | 1,12 | 1,12 |
| 16 | Δ-7-avenasterol | (24Z)-24-ethylidene-Δ-7-cholesten-3β -ol | 1,16 | 1,16 |
| 17 | Erythrodiol | 5α-olean-12-en-3β,28-diol | 1,41 | 1,41 |
| 18 | Uvaol | Δ12-ursen-3β,28-diol | 1,52 | 1,52 |

Figure 2 — GC-FID chromatographic profile of the sterol and triterpenic dialcohols from refined olive oil. (1) Cholesterol, (2) α-cholestanol (I.S.), (3) 24-methylencholesterol, (4) campesterol, (5) campestanol, (6) stigmasterol, (7) Δ5,23-stigmastadienol, (8) clerosterol, (9) β-sitosterol, (10) sitostanol, (11) Δ5-avenasterol, (12) Δ5,24-stigmastadienol, (13) Δ7-stigmastenol, (14) Δ7-avenasterol, (15) erythrodiol, (16) uvaol.

Figure 3 — GC-FID chromatographic profile of the sterol and triterpenic dialcohols from a lampante olive oil. (1) Cholesterol, (2) α-cholestanol, (3) brassicasterol, (4) 24-methylencholesterol, (5) campesterol, (6) campestanol, (7) stigmasterol, (8) Δ7-campesterol, (9) Δ5,23-stigmastadienol, (10) clerosterol, (11) β-sitosterol, (12) sitostanol, (13) Δ5-avenasterol, (14) Δ5,24-stigmastadienol, (15) Δ7-stigmastenol, (16) Δ7-avenasterol, (17) erythrodiol, (18) uvaol.

Figure 4 — GC-FID chromatographic profile of aliphatic alcohols and triterpenic alcohols of olive oil. (I.S.) C20-ol, (1) C22-ol, (2) C24-ol, (3) C26-ol, (4) C28-ol, (5) triterpenic alcohols.

Figure 5 — GC-FID chromatographic profile of aliphatic alcohols and triterpenic alcohols of a refined olive oil and a second centrifugation olive oil. (I.S.) C20-ol, (1) C22-ol, (2) C24-ol, (3) C26-ol, (4) C28-ol, (5) triterpenic alcohols.

Figure 6 — HPLC Chromatogram of an olive oil unsaponifiable separated by HPLC using a UV detector. (1) Aliphatic and triperpenic alcohols; (2) Sterols and triterpenic dialcohols’
(2)
Commission Regulation (EEC) No 2568/91 of 11 July 1991 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis (OJ L 248, 5.9.1991, p. 1).
Options/Help
Print Options
PrintThe Whole Regulation
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As adopted by EU): The original version of the legislation as it stood when it was first adopted in the EU. No changes have been applied to the text.
Opening Options
Different options to open legislation in order to view more content on screen at once
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the EU Official Journal
- lists of changes made by and/or affecting this legislation item
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as adopted version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
The data on this page is available in the alternative data formats listed:
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.