- Latest available (Revised)
- Point in Time (01/04/2009)
- Original (As made)
The Blood Safety and Quality Regulations 2005
You are here:
- UK Statutory Instruments
- 2005 No. 50
- Whole Instrument
- Previous
- Next

What Version
Advanced Features
- Show Geographical Extent(e.g. England, Wales, Scotland and Northern Ireland)
- Show Timeline of Changes
More Resources
Changes over time for: The Blood Safety and Quality Regulations 2005
Version Superseded: 16/12/2009
Alternative versions:
- 08/02/2005- Amendment
- 08/04/2005- Amendment
- 01/10/2005- Amendment
- 08/11/2005- Amendment
- 31/08/2006- Amendment
- 01/04/2007- Amendment
- 01/04/2008- Amendment
- 01/05/2008- Amendment
- 01/04/2009- Amendment
- 01/04/2009
Point in time - 16/12/2009- Amendment
- 01/04/2010- Amendment
- 01/10/2010- Amendment
- 28/10/2011- Amendment
- 01/04/2013- Amendment
- 18/07/2016- Amendment
- 15/02/2018- Amendment
- 01/04/2018- Amendment
- 31/12/2020- Amendment
- 27/07/2021- Amendment
- 01/04/2023- Amendment
- 21/03/2024- Amendment
Status:
Point in time view as at 01/04/2009.
Changes to legislation:
There are currently no known outstanding effects for the The Blood Safety and Quality Regulations 2005.

Changes to Legislation
Revised legislation carried on this site may not be fully up to date. At the current time any known changes or effects made by subsequent legislation have been applied to the text of the legislation you are viewing by the editorial team. Please see ‘Frequently Asked Questions’ for details regarding the timescales for which new effects are identified and recorded on this site.
Statutory Instruments
2005 No. 50
HEALTH AND SAFETY
The Blood Safety and Quality Regulations 2005
Made
13th January 2005
Laid before Parliament
18th January 2005
Coming into force
For all purposes other than regulation 25(1)
8th February 2005
For the purposes of regulation 25(1)
8th November 2005
The Secretary of State for Health, being a Minister designated M1 for the purposes of section 2(2) of the European Communities Act 1972 M2 in relation to health protection measures regulating the use of material of human origin, in exercise of the powers conferred on him by the said section 2(2) and, with the consent of the Treasury, of the powers conferred by section 56(1) and (2) of the Finance Act 1973 M3, hereby makes the following Regulations:—
Marginal Citations
M21972 c. 68. Under section 57(1) of the Scotland Act 1998 (c. 46), despite the transfer to Scottish Ministers of functions in relation to implementing obligations under Community law in relation to devolved matters, the functions of the Secretary of State in relation to implementing these obligations continues to be exercisable by him as regards Scotland.
Citation, commencement and interpretationU.K.
1.—(1) These Regulations may be cited as the Blood Safety and Quality Regulations 2005.
(2) Except for regulation 25(1), which shall come into force on 8th November 2005, these Regulations shall come into force on 8th February 2005.
(3) In these Regulations—
“autologous transfusion” means a transfusion in which the donor and the recipient are the same person and in which pre-deposited blood or blood components are used;
[F1“biomedical research institution” means any body which carries out biomedical research;]
“blood” means whole human blood collected from a donor and processed either for transfusion or for further manufacturing;
“blood component” means a therapeutic constituent of human blood (red cells, white cells, platelets and plasma) that can be prepared by various methods;
“blood component release” means a process which enables a blood component to be released from a quarantine status by the use of systems and procedures to ensure that the finished product meets its release specification;
[F2“blood establishment” means any person who carries out any of the activities specified in regulation 3(2) which require an authorisation by virtue of that regulation;]
“blood product” means any therapeutic product derived from human blood or plasma;
[F1“care home”—
(a)
in England and Wales, has the same meaning as in section 3 of the Care Standards Act 2000,
(b)
in Scotland, has the same meaning as in section 2 of the Regulation of Care (Scotland) Act 2001, and
(c)
in Northern Ireland, has the same meaning as in article 2 of the Health and Personal Social Services (Quality, Improvement and Regulation)(Northern Ireland) Order 2003;]
“Commission” means the European Commission;
[F1“Commission Directive 2005/62/EC” means Commission Directive 2005/62/EC of 30th September 2005 implementing Directive 2002/98/EC of the European Parliament and of the Council as regards Community standards and specifications relating to a quality system for blood establishmentsF3;]
“deferral” means suspension of the eligibility of an individual to donate blood or blood components, such suspension being either permanent or temporary;
“the Directive” means Directive 2002/98/EC of the European Parliament and of the Council of 27 January 2003 setting standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components M4;
“distribution” means the act of delivery of blood and blood components to other blood establishments, hospital blood banks and manufacturers of blood products, other than the issuing of blood or blood components for transfusion;
“doctor” means a registered medical practitioner;
“donor carer” means a person who has passed both the written and practical examinations of the [F4NHS Blood and Transplant (Gwaed a Thrawsblaniadau'r GIG)], the Scottish National Blood Transfusion Service M5, the Northern Ireland Blood Transfusion Service M6 or the Welsh Blood Service M7 in the care of blood donors and who holds a current certificate of competence, awarded by that body, in the care of blood donors;
[F1“facility” means—
(a)
a hospital,
(b)
any other facility or service owned or managed by a health service body,
(c)
a care home,
(d)
an independent clinic,
(e)
a manufacturer, or
(f)
a biomedical research institute;]
[F5“health service hospital” means a hospital owned or managed by a health service body;]
“haemovigilance” means a set of organised surveillance procedures relating to serious adverse or unexpected events or reactions in donors or recipients, and the epidemiological follow-up of donors;
“health service body” means—
(a)
a Strategic Health Authority, Special Health Authority, Primary Care Trust or Local Health Board established under the National Health Service Act 1977,
(b)
a Health Board or Special Health Board established under the National Health Service (Scotland) Act 1978,
(c)
a Health and Social Services Board established under the Health and Personal Social Services (Northern Ireland) Order 1972 M8,
(d)
a special health and social services agency established under the Health and Personal Social Services (Special Agencies) (Northern Ireland) Order 1990 M9,
(e)
the Common Services Agency for the Scottish Health Service established under the National Health Service (Scotland) Act 1978,
(f)
the Northern Ireland Central Services Agency for the Health and Social Services established under the Health and Personal Social Services (Northern Ireland) Order 1972,
(g)
a National Health Service trust established under the National Health Service and Community Care Act 1990 M10, or the National Health Service (Scotland) Act 1978,
(h)
an NHS foundation trust within the meaning of section 1(1) of the Health and Social Care (Community Health and Standards) Act 2003 M11, or
(i)
a Health and Social Services trust established under the Health and Personal Social Services (Northern Ireland) Order 1991 M12;
“hospital” means a health service hospital or an independent hospital;
“hospital blood bank” means any unit within a hospital which stores and distributes, and may perform compatibility tests on, blood and blood components exclusively for use within hospital facilities, including hospital based transfusion activities;
[F1“imputability” means the likelihood that a serious adverse reaction in a recipient can be attributed to the blood or blood component transfused, or that a serious adverse reaction in a donor can be attributed to the donation process;]
[F1“independent clinic”—
(a)
in England and Wales, has the same meaning as in section 2 of the Care Standards Act 2000,
(b)
in Scotland, has the same meaning as in section 2 of the Regulation of Care (Scotland) Act 2001, and
(c)
in Northern Ireland, has the same meaning as in article 2 of the Health and Personal Social Services (Quality, Improvement and Regulations)(Northern Ireland) Order 2003;]
[F6“independent hospital”—
(a)
in England and Wales, has the same meaning as in section 2 of the Care Standards Act 2000,
(b)
in Scotland, has the same meaning as in section 2 of the Regulation of Care (Scotland) Act 2001, and
(c)
in Northern Ireland, has the same meaning as in article 2 of the Health and Personal Social Services (Quality, Improvement and Regulation)(Northern Ireland) Order 2003]
“inspection” means formal and objective control to identify problems in accordance with standards adopted to assess compliance with these Regulations;
“inspector” means a person appointed by the Secretary of State to carry out inspections pursuant to regulation 15(10);
[F1“issue” means the provision of blood or blood components by a blood establishment or a hospital blood bank for transfusion to a recipient;]
[F1“manufacturer” means a person who—
(a)
holds a licence under section 8(2) of the Medicines Act 1968 to manufacture medicinal products;
(b)
holds an authorisation to manufacture an investigational medicinal product granted pursuant to regulation 36 of the Medicines for Human Use (Clinical Trials) Regulations 2004; or
(c)
falls within the definition of “manufacturer” in paragraph (1) of regulation 2 of the Medical Devices Regulations 2002;]
“nurse” means a registered nurse or registered midwife;
[F1“person responsible for the management of a facility” means—
(a)
in the case of a hospital, facility or service which is owned or managed by an NHS body, that body,
(b)
in the case of an independent hospital, an independent clinic or a care home, the registered person,
(c)
in the case of a manufacturer or a biomedical research institution, the manufacturer or biomedical research institution;]
“person responsible for management of a hospital blood bank” means—
(a)
in the case of hospital blood bank located in a hospital managed by a health service body, that body, and
(b)
in the case of an independent hospital, the registered person;
[F1“person responsible for the management of a reporting establishment” means a blood establishment, the person responsible for the management of a facility or the person responsible for the management of a hospital blood bank;]
“qualified health professional” means—
(a)
a doctor;
(b)
a nurse, or
(c)
a donor carer;
[F1“recipient” means a person who has been transfused with blood or blood components;]
[F7“registered person” means the person registered as the manager of an independent hospital, a care home or an independent clinic following an application to be registered as such pursuant to—
(a)
section 12(3) of the Care Standards Act 2000,
(b)
section 7(1) of the Regulation of Care (Scotland) Act 2001, or
(c)
article 13(1) of the Health and Personal Social Services (Quality, Improvement and Regulation)(Northern Ireland) Order 2003]
[F1“reporting establishment” means the blood establishment, the hospital blood bank or the facility where the transfusion takes place;]
“reporting year” means the period of twelve months ending on 31st March;
“responsible person” in relation to a blood establishment means the person who has been designated pursuant to regulation 6 as the responsible person for that blood establishment,
“serious adverse event” means any untoward occurrence associated with the collection, testing, processing, storage and distribution of blood or blood components that might lead to death or life-threatening, disabling or incapacitating conditions for patients or which results in, or prolongs, hospitalisation or morbidity;
“serious adverse reaction” means an unintended response in a donor or in a patient associated with the collection or transfusion of blood or blood components that is fatal, life-threatening, disabling or incapacitating, or which results in or prolongs hospitalisation or morbidity;
“site”, in relation to a blood establishment, means any premises at which the blood establishment carries out any of the activities listed in regulation 3(2), but shall not include any premises not owned or managed by the blood establishment at which blood is collected, or any mobile blood collection unit;
[F1“third country” means any country other than a Member State; and]
[F1“traceability” means the ability to trace each individual unit of blood or blood component from the donor to its final destination (whether this is a recipient, a manufacturer of medicinal products or disposal) and from its final destination back to the donor;]
“validation” means the establishment of documented and objective evidence that the particular requirements for a specific intended use can be consistently fulfilled.
Textual Amendments
F1Words in reg. 1(3) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 2(d)
F2Words in reg. 1(3) substituted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 2
F3O.J. L 256 1.10.2005 p 14.
F4Words in reg. 1 substituted (E.W.) (1.10.2005) by The National Blood Authority and United Kingdom Transplant (Abolition) Order 2005 (S.I. 2005/2532), art. 1(1), Sch. 2 para. 7
F5Words in reg. 1(3) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 2(a)
F6Words in reg. 1(3) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 2(b)
F7Words in reg. 1(3) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 2(c)
Marginal Citations
M4O.J. No. L33, 8.2.2003, p.30.
M5The Scottish National Blood Transfusion Service is managed by the Common Services Agency established by section 10 of, and Schedule 5 to, the National Health Service (Scotland) Act 1978 (c. 29). The Common Services Agency was designated for this purpose by the NHS (Functions of the Common Services Agency)(Scotland) Order (S.I. 1974/467).
M6The Northern Ireland Blood Transfusion Service was established under Article 10(1)(d) of the Health and Personal Social Services (Northern Ireland) Order (S.I. 1972/1265) (N.I. 14).
M7The Welsh Blood Service is provided and managed by the Velindre National Health Service Trust. The Velindre NHS Trust was established, and designated for this purpose by the Velindre National Health Service Trust (Establishment) Order (1993/2838), as amended by S.I. 1999/826 and 2002/442 and 2199.
Designation of the competent authority and scope of the RegulationsU.K.
2.—(1) The Secretary of State is designated the competent authority for the purpose of the Directive.
(2) Subject to the following paragraphs, the requirements of these Regulations apply to the collection and testing of blood and blood components, whatever their intended purpose, and to their processing, storage, and distribution when they are intended to be used for transfusion.
(3) These Regulations apply without prejudice to the Medical Devices Regulations 2002 M13.
(4) These Regulations do not apply to blood stem cells.
Marginal Citations
Requirement for authorisationU.K.
3.—(1) Subject to paragraph (3), no person may carry on any of the activities listed in paragraph (2) otherwise than in accordance with an authorisation granted under regulation 4.
(2) The activities referred to in paragraph (1) are—
(a)the collection and testing of blood or blood components, whatever their intended purpose; F8...
(b)the processing, storage and distribution of blood and blood components when they are intended to be used for transfusion [F9; and]
[F10(c)the import of blood or blood components from a third country;.]
(3) The restriction in paragraph (1) shall not apply to—
(a)the storage and distribution of, and the performance of compatibility tests on, blood and blood components exclusively for use within hospital facilities, including transfusion activities where such activities are performed by a hospital blood bank; F11...
(b)any person carrying out any of the activities referred to in paragraph (2), where that person carries out that activity on behalf of, [F12or pursuant] to a contractual arrangement with—
(i)a blood establishment which is authorised under these regulations to carry out the activity in question; or
(ii)a person responsible for management of a hospital blood bank [F13; and]
[F14(c)the import of blood and blood components from a third country when undertaken by—
(i)a manufacturer, or
(ii)a person acting on behalf of and pursuant to a contractual arrangement with a manufacturer,
for the purposes of manufacturing a medicinal product within the meaning of the Medicines act 1968 or the Medical Devices Regulations 2002;]
Textual Amendments
F8Word in reg. 3(2)(a) omitted (31.8.2006) by virtue of The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 3(2)(a)
F9Word in reg. 3(2)(b) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 3(2)(b)
F10Reg. 3(2)(c) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 3(2)(c)
F11Word in reg. 3(3)(a) omitted (31.8.2006) by virtue of The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 3(3)(a)
F12Words in reg. 3(3)(b) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 3(3)(b)(i)
F13Word in reg. 3(3)(b) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 3(3)(b)(ii)
F14Reg. 3(3)(c) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 3(3)(c)
Authorisation of a blood establishmentU.K.
4.—(1) The Secretary of State may grant an authorisation to a blood establishment to carry out any of the activities referred to in regulation 3(2).
(2) An application for authorisation under paragraph (1) shall be made to the Secretary of State.
(3) An application must—
(a)include the information set out in paragraph (4); and
(b)be accompanied by a fee of the amount prescribed in regulation 22(2)(a).
(4) The information referred to in [F15paragraph (3)] is—
(a)the name and address of the blood establishment and general information about its activities which shall include—
(i)details of each site at which it wishes to carry out any of the activities referred to in regulation 3(2),
(ii)a description of the activities which it wishes to carry out at each site,
(iii)where it has or intends to enter into a contractual arrangement with any person to carry out any of the services in respect [F16of which] it is seeking authorisation, the name and address of that person and of the services which he will carry out,
(iv)the name, qualifications and contact details of the responsible person for the establishment,
(v)the list of hospital blood banks which it supplies; and
(b)a description of the quality system in place at each site for each activity in respect of which the application for authorisation is made, which shall include the following information—
(i)documentation, such as an organisation chart, setting out the responsibilities of responsible persons and reporting relationships,
(ii)documentation, such as a site master file or quality manual, describing the quality system and explaining how it meets the requirements of Part 5 of the Schedule,
(iii)details of the number and qualifications of personnel,
(iv)details of hygiene provisions,
(v)details of premises and equipment, and
(vi)a list of standard operating procedures for—
(aa)recruitment, retention and assessment of donors,
(bb)processing, testing, distribution and recall of blood and blood components, and
(cc)the reporting and recording of serious adverse reactions and events.
(5) The Secretary of State may—
(a)grant or refuse any application for authorisation made under paragraph (3); and
(b)grant such application—
(i)in respect of particular sites or activities only, and
(ii)subject to conditions.
(6) Where the Secretary of State grants an application for authorisation, he shall give notice in writing to the blood establishment specifying—
(a)the activities which the blood establishment may undertake under these Regulations at each site in respect of which authorisation is granted; and
(b)the conditions which apply to the undertaking of those activities.
(7) Subject to the requirements of paragraph (8), the Secretary of State may at any time remove or vary any of the conditions referred to in paragraph (5)(b)(ii), or may impose additional conditions.
(8) Where the Secretary of State removes or varies any condition or imposes any additional condition pursuant to paragraph (7), he shall serve a notice on the blood establishment in question which shall—
(a)give details of the conditions which he proposes to remove, or of the variation which he proposes to make to any existing conditions, or of any additional condition which he proposes to impose;
(b)give the reasons for his decision; and
(c)specify the date, which shall be not less than 14 days from the date on which the notice is served, from which the removal or variation of any condition, or the imposition of any additional condition shall apply.
(9) A blood establishment may not make any substantial change in the activities which it undertakes without the prior written approval of the Secretary of State.
(10) Any application for approval to make a substantial change in its activities shall be made in writing to the Secretary of State, and shall be accompanied by a fee of the amount prescribed in regulation 22(2)(b).
(11) For the purpose of this regulation, a substantial change in a blood establishment's activities is any change—
(a)to the sites from which the blood establishment operates or to the activities to be carried out at each site;
(b)which would result in breach of these regulations or of any condition specified by the Secretary of State pursuant to this regulation; or
(c)to the quality system which is likely to have a substantial impact on the conduct of, or might compromise the safety of, any of the activities which the blood establishment has been authorised to undertake pursuant to this regulation.
Textual Amendments
F15Words in reg. 4(4) substituted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 2(a)
F16Words in reg. 4(4)(a)(iii) substituted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 2(b)
Suspension or revocation of authorisationU.K.
5.—(1) The Secretary of State may suspend or revoke the authorisation of a blood establishment on one or more of the following grounds—
(a)that the blood establishment has failed, in any material respect, to comply with the requirements of these regulations;
(b)that the collection, testing, processing, storage or distribution of blood or blood components by the establishment cannot be carried out safely;
(c)that any blood or blood components cannot be supplied to hospital blood banks in such a state that they could be safely administered for transfusion; or
(d)that the information given by the blood establishment pursuant to regulation 4(3) was false or incomplete in any material respect.
(2) Subject to paragraph (3), before suspending or revoking the authorisation of a blood establishment, the Secretary of State shall serve a notice on the blood establishment stating that he intends to suspend or revoke its authorisation with effect from the date specified in the notice, which date shall be not less than 7 days from the date on which the notice is served.
(3) Where the Secretary of State considers that it is necessary in the interests of safety, he may, by a notice served on a blood establishment, suspend or revoke its authorisation with immediate effect.
(4) Where—
(a)the blood establishment has failed, in any material respect, to comply with the requirements of these regulations; or
(b)the information given by the blood establishment pursuant to [F17regulation 4(3)] was false or incomplete in any material respect,
and the Secretary of State considers that the failure in question is not sufficiently serious to warrant suspension or revocation of the authorisation of the blood establishment in the first instance, he may serve a notice on the responsible person of the blood establishment in accordance with paragraph (5).
(5) A notice served under this paragraph shall—
(a)identify the requirements of the regulations of which the blood establishment is in breach or, in the case of false and incomplete information, the further information which is required;
(b)identify the action which the blood establishment is required to take; and
(c)give the timescale within which the blood establishment shall take the action identified in sub-paragraph (b).
(6) If the blood establishment fails to comply with the requirements set out in the notice within the specified timescale, the Secretary of State may, by a notice served on the blood establishment, suspend or revoke the authorisation of the blood establishment.
(7) A suspension or revocation pursuant to paragraph (6) shall take effect—
(a)in a case where the Secretary of State considers that it is necessary in the interests of safety, immediately; or
(b)in all other cases, from a date specified in the notice.
(8) Any suspension pursuant to paragraphs (1) or (6) shall be for such period as the Secretary of State shall consider necessary having regard to the reasons for the suspension.
(9) The suspension or revocation of an authorisation under paragraph (1) or paragraph (6) may be total, or may be limited to a particular activity or to one or more activities carried out at a particular site or sites, or to a particular blood component.
Textual Amendments
F17Words in reg. 5(4)(b) substituted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 3
The responsible person for a blood establishmentU.K.
6.—(1) A blood establishment shall designate a person who is responsible for the following tasks—
(a)ensuring that every unit of blood or blood component that has been collected or tested for any purpose has been collected and tested in accordance with the requirements of these Regulations;
(b)ensuring that every unit of blood or blood components intended for transfusion has been processed, stored and distributed in accordance with the requirements of these Regulations;
(c)providing information to the Secretary of State relating to the authorisation of the blood establishment for the purposes of regulation 4; and
(d)the implementation in the blood establishment of the requirements of regulations 7, 8 and 14.
(2) A blood establishment shall not designate a person under paragraph (1) unless that person has—
(a)a diploma, certificate or other evidence of formal qualification in the field of medical or biological sciences awarded on completion of—
(i)a university course of study, or
(ii)a course recognised as an equivalent course by the Secretary of State; and
(b)practical post-graduate experience in areas of work relevant to the responsibilities of the responsible person under these Regulations for at least 2 years, in an establishment (or more than one establishment) authorised in any Member State in to undertake activities related to the collection or testing (or both) of blood and blood components, or to their preparation, storage and distribution.
(3) The Secretary of State shall from time to time publish details of courses recognised by him for the purpose of paragraph (2)(a)(ii).
(4) The responsible person may delegate any of the tasks specified in paragraph (1) to other persons who shall be qualified by training and experience to perform them.
(5) Blood establishments shall notify the Secretary of State of the name of any persons to whom tasks have been delegated by the responsible person under paragraph (4), and the specific tasks which have been delegated to such persons.
(6) Where the responsible person or a person to whom tasks have been delegated under paragraph (4) is permanently or temporarily replaced, the blood establishment shall without delay provide the Secretary of State with the name of the replacement, details of his qualifications and the date on which the replacement began his duties.
(7) If the Secretary of State considers that the responsible person does not meet the requirements of paragraph (2) [F18or that he is failing to carry out the tasks specified in paragraph (1) adequately or at all], he may serve a notice to that effect on the blood establishment.
(8) If, within 14 days of receiving a notice in accordance with paragraph (7), a blood establishment is not able to demonstrate to the reasonable satisfaction of the Secretary of State that the responsible person does meet the requirements of paragraph (2) [F19or that he is carrying out the tasks specified in paragraph (1) adequately], it shall, without delay—
(a)relieve him of the duties of responsible person in respect of the establishment;
(b)appoint a new responsible person in his place; and
(c)notify the Secretary of State that it has appointed a new responsible person and provide details of the name and qualifications of the person appointed.
Textual Amendments
F18Words in reg. 6(7) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 3(a)
F19Words in reg. 6(8) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 3(b)
Blood establishment requirementsU.K.
7.—(1) A blood establishment shall—
(a)ensure that the personnel directly involved in the collection, testing, processing, storage and distribution of human blood and blood components for the blood establishment are qualified to perform those tasks and are provided with timely, relevant and regularly updated training;
(b)establish and maintain a quality system for blood establishments based on the principles of [F20good practice, which complies with the Community standards and requirements set out in the Annex to Commission Directive 2005/62/ECF21;]
(c)ensure that all testing and processes of the blood establishment which are referred to in Parts 2 to 5 of the Schedule are validated;
(d)maintain documentation on operational procedures, guidelines, training and reference manuals and reporting forms so that they are readily available for inspection under regulation 15;
F22(e). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
(f)establish and maintain a procedure, which is accurate, efficient and verifiable, for the withdrawal from distribution of blood or blood components associated with any notification referred to in paragraph (e) [F23; and]
[F24(g)retain, for a period of at least 15 years, a record of any serious adverse events which may affect the quality or safety of blood and blood components.]
(2) A blood establishment shall, in relation to the donation of blood—
(a)give all prospective donors of blood or blood components information in accordance with Part A of Part 2 of the Schedule;
(b)obtain from all persons who are willing to provide blood or blood components, information in accordance with Part B of Part 2 of the Schedule;
(c)put and keep in place procedures for the evaluation of donors;
(d)apply eligibility criteria for all donors of blood and blood components in accordance with Part 3 of the Schedule;
(e)maintain records of the results of donor evaluations and report to donors any relevant abnormal findings from the evaluations;
(f)ensure that—
(i)an examination of the donor, including an interview, is carried out before any donation of blood or blood components,
(ii)a qualified health professional is responsible for giving to and gathering from donors the information which is necessary to assess their eligibility to donate, and
(iii)on the basis of that information, a qualified health professional assesses the eligibility of all donors to donate; and
(g)encourage voluntary and unpaid blood donations with a view to ensuring that blood and blood components are, in so far as possible, provided from such donations, in particular, by—
(i)disseminating information about blood donation, and
(ii)advertising for blood donors.
(3) A blood establishment shall ensure that, in relation to the blood and blood components which it collects, processes, stores or distributes—
(a)each donation of blood and blood components (including blood and blood components which are imported into the European Community) is tested in conformity with—
(i)the basic testing requirements for whole blood and apheresis donations, specified in paragraph (7), and
(ii)any additional tests which may be necessary for specific components, types of donors or epidemiological situations;
(b)the storage, transport and distribution conditions of blood and blood components comply with the requirements of Part 4 of the Schedule; and
(c)quality and safety requirements for blood and blood components meet the standards specified in Part 5 of the Schedule.
(4) A blood establishment shall, in relation to the activities specified in regulation 3(2) for which it is responsible, maintain records, for a minimum period of 15 years, of—
(a)the information specified in paragraphs (5) and (6),
(b)the conduct of the tests referred to in paragraph (3)(a).
(5) The information specified in this paragraph is—
(a)the total number of donors who give blood and blood components;
(b)the total number of donations;
(c)an updated list of the hospital blood banks which it supplies;
(d)the total number of whole donations not used;
(e)the number of each component produced and distributed;
(f)the incidence and prevalence of transfusion transmissible infectious markers in donors of blood and blood components;
(g)the number of product recalls; and
(h)the number of serious adverse events and serious reactions reported;
(6) The information specified in this paragraph is—
(a)information provided to donors by the blood establishment in accordance with paragraph (2)(a);
(b)information obtained from donors by the blood establishment in accordance with paragraph (2)(b); and
(c)information relating to the suitability of blood and plasma donors in accordance with the eligibility criteria specified in Part 3 of the Schedule.
(7) The basic testing requirements with which blood establishments must ensure compliance pursuant to paragraph (3)(a)(i) are—
(a)testing to establish ABO Group, except in respect of plasma intended only for fractionation;
(b)testing to establish Rh D Group, except in respect of plasma intended only for fractionation; and
(c)testing for the following infections of donors—
(i)Hepatitis B (HBs-Ag);
(ii)Hepatitis C (Anti-HCV);
(iii)HIV 1 and 2 (Anti-HIV 1 and 2).
(8) The Secretary of State may issue guidance as to the additional tests referred to in paragraph (3)(a)(ii) which are necessary in relation to specific components, types of donor or epidemiological situations and blood establishments shall have regard to such guidance.
(9) As soon as practicable after the end of the reporting year, each blood establishment shall provide to the Secretary of State a report specifying—
(a)the information referred to in paragraph (3) for that year; and
(b)details of the steps it has taken during that year to comply with paragraph (2)(g).
Textual Amendments
F20Words in reg. 7(1)(b) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 4(1)(a)
F21 OJ L 256, 1.10.2005, p 41.
F22Reg. 7(1)(e) omitted (31.8.2006) by virtue of The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 4(1)(b)
F23Word in reg. 7(1)(f) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 4(1)(c)
F24Reg. 7(1)(g) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 4(1)(d)
Labelling of blood and blood components and traceabilityU.K.
8.—(1) A blood establishment shall ensure that the label on each unit of blood or blood component supplied by it, or imported by it from outside the European Community, shall contain the following information—
(a)the official name of the component;
(b)the volume or weight or number of cells in the component, as appropriate;
(c)a unique numeric or alphanumeric donation indication;
(d)the name of the producing blood establishment;
(e)the ABO Group, except in the case of plasma intended only for fractionation;
(f)the Rh D Group, either Rh D positive or Rh D negative, except in the case of plasma intended only for fractionation;
(g)the date or time of expiry, as appropriate;
(h)the temperature of storage;
(i)the name, composition and volume of any anticoagulant and any additive solution.
[F25(2) A blood establishment shall maintain, in relation to all blood and blood components collected or prepared by it (including blood and blood components which are imported by it into the European Community)—
(a)records of the information referred to in paragraph (1) above;
(b)the records referred to in Part A of Part 6 to the Schedule; and
(c)such other records as are necessary to ensure full traceability of blood and blood components and identification of each single donation, unit and component.]
[F26(3) The records referred to in sub-paragraph (a) [F27of paragraph (2)] shall be maintained—
(a)in an appropriate and readable storage medium, and
(b)for a period of not less than 30 years.
(4) A blood establishment shall ensure that the traceability system in place in the blood establishment enables the tracing of blood and blood components to their location and processing stage.
(5) A blood establishment shall have in place a system to uniquely identify each donor, each blood unit collected and each blood component prepared, whatever its intended purpose, and the facilities to which a given unit of blood or blood component has been delivered.”.
(6) A blood establishment shall ensure, when it issues a unit of blood or blood components for transfusion, that the facility to which the unit of blood is issued has in place a procedure to verify that each unit of blood issued has been transfused to the intended recipient or, if not transfused, to verify its subsequent disposition.]
Textual Amendments
F25Reg. 8(2) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 5(2)
F26Reg. 8(3)-(6) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 5(3)
F27Words in reg. 8(3) inserted (1.5.2008) by The Medicines for Human Use (Clinical Trials) and Blood Safety and Quality (Amendment) Regulations 2008 (S.I. 2008/941), regs. 1(1), 7
Hospital blood bank requirementsU.K.
9.—(1) The person responsible for the management of a hospital blood bank shall—
(a)ensure that personnel directly involved in the testing, storage and distribution of human blood and blood components for the hospital blood bank are qualified to perform those tasks and are provided with timely, relevant and regularly updated training;
(b)establish and maintain a quality system for the hospital blood bank which is based on the principles of [F28good practice, which complies with the Community standards and requirements set out the Annex to Commission Directive 2005/62/EC insofar as these are applicable to hospital blood banks;]
(c)ensure that all processes referred to in Part 4 of the Schedule which are applicable to activities carried out by the hospital blood bank, are validated;
(d)maintain documentation on operational procedures, guidelines, training and reference manuals and reporting forms so that they are readily available for inspection under regulation 15;
[F29(e)maintain in an appropriate and readable storage medium and for a period of not less than 30 years—
(i)the data set out in Part 6 of the Schedule (insofar as those data are applicable to the activities carried out by the hospital blood bank), and
(ii)such other data as are needed to ensure full traceability of blood and blood components and the unique identification of each unit of blood and each blood component from the point of receipt of the blood or blood components by the hospital blood bank;]
[F30(f)retain, for a period of at least 15 years, a record of any serious adverse events which may affect the quality or safety of blood and blood components;]
(g)establish and maintain a procedure, which is accurate, efficient and verifiable, for the withdrawal from distribution of blood or blood components associated with any notification referred to in paragraph (f); F31...
(h)ensure that the storage, transport and distribution conditions of blood and blood components by the hospital blood bank comply with the requirements of Part 4 of the Schedule [F32; and]
[F33(i)ensure that the traceability system in place in the hospital blood bank enables the tracing of blood components to their final destination; and
(j)where it delivers blood or blood components for transfusion at another facility, have in place a system to uniquely identify the facility to which a given unit of blood or blood component has been delivered.]
[F34(2) A person responsible for management of a hospital blood bank shall ensure that when a hospital blood bank issues a unit of blood for transfusion, that it has in place a procedure to verify that each unit of blood issued has been transfused to the intended recipient, or if not transfused, to verify its subsequent disposition.]
Textual Amendments
F28Words in reg. 9(1)(b) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 6(2)(a)
F29Reg. 9(1)(e) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 6(2)(b)
F30Reg. 9(1)(f) substituted (1.5.2008) by The Medicines for Human Use (Clinical Trials) and Blood Safety and Quality (Amendment) Regulations 2008 (S.I. 2008/941), regs. 1(1), 8
F31Word in reg. 9(1)(g) omitted (31.8.2006) by virtue of The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 6(2)(d)
F32Word in reg. 9(1)(h) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 6(2)(e)
F33Reg. 9(1)(i)(j) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 6(2)(f)
F34Reg. 9(2) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 6(3)
Requirement for hospital blood banks to provide information to the Secretary of StateU.K.
10.—(1) [F35On or before the date specified in paragraph (1A)], the person responsible for management of a hospital blood bank shall submit [F36a report] to the Secretary of State, which shall—
(a)include a declaration that the hospital blood bank has in place appropriate systems to ensure compliance with the requirements of these Regulations; and
(b)provide details of the systems which it has in place to ensure such compliance.
[F37(1A) The date referred to in paragraph (1) is—
(a)in relation to the reporting year ending on 31st March 2006, 31st December 2005; and
(b)in relation to each subsequent reporting year, 30th April following the end of that year.]
(2) The person responsible for management of a hospital blood bank shall without delay notify the Secretary of State of any changes to the matters in respect of which evidence has been supplied pursuant to paragraph (1) which might affect compliance with the requirements of these Regulations.
Textual Amendments
F35Words in reg. 10(1) substituted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 4(a)(i)
F36Words in reg. 10(1) substituted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 4(a)(ii)
F37Reg. 10(1A) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 4(b)
Service of notices relating to hospital blood banksU.K.
11.—(1) If the Secretary of State is of the opinion that—
(a)the person responsible for management of a hospital blood bank has failed, in any material respect, to comply with the requirements of these regulations; or
(b)the testing, storage or distribution of blood or blood components by the hospital blood bank is such that any blood or blood components cannot be safely administered for transfusion; or
(c)the information given by the person responsible for management of a hospital blood bank pursuant to regulation 10 was false or incomplete in any material respect,
he may serve a notice on the person responsible for management of the hospital bank requiring that the hospital ceases to conduct any of the activities specified in the notice, or refrains from administering to patients any blood or blood components specified in the notice, until the requirements of paragraph (4) are met.
(2) Subject to paragraph (3), any notice served by the Secretary of State pursuant to paragraph (1) shall specify the date from which the prohibition specified in the notice shall take effect, which shall be not less than 7 days from the date on which the notice is served.
(3) Where the Secretary of State considers that it is necessary in the interests of safety, he may specify in the notice that the prohibition takes immediate effect.
(4) The requirements of this paragraph are, as may be applicable in each case, that—
(a)that the person responsible for management of the hospital blood bank is no longer in breach of the requirements of these regulations;
(b)that the hospital blood bank is able to show that the activity or product referred to in the notice given pursuant to paragraph (1)(b) may be safely carried out or, as the case may be, administered; or
(c)that all necessary information has been supplied to the Secretary of State.
Objections to suspensions, revocations etcU.K.
12.—(1) A blood establishment or a person responsible for the management of a hospital blood bank who—
(a)objects to any suspension or revocation of authorisation, or to any notice served pursuant to regulation 4(8), 5 or 11; or
(b)objects to the refusal of authorisation or the imposition of any condition pursuant to regulation 4(5),
may notify the Secretary of State of its desire to make written representations to, [F38or to] appear before and be heard by, a person appointed by the Secretary of State for that purpose.
(2) Any notification of an objection pursuant to paragraph 1 shall be made within 14 days of service on the blood establishment or the person responsible for the management of the hospital blood bank of the notice to which the notification pursuant to paragraph (1) relates.
(3) Where the Secretary of State receives a notification pursuant to paragraph (1), he shall appoint a person to consider the matter.
(4) The person appointed pursuant to paragraph (3) shall determine the procedure to be followed with respect to the consideration of any objection.
(5) The person appointed pursuant to paragraph (3) shall consider any written or oral objections made by the blood establishment or the person responsible for management of the hospital blood bank in support of its objection, and shall make a recommendation to the Secretary of State.
(6) A recommendation made pursuant to paragraph (5) shall be made in writing to the Secretary of State, and a copy of it shall be sent to the blood establishment or the person responsible for the management of the hospital blood bank concerned, or to its nominated representative.
(7) The Secretary of State shall take into account any recommendation made pursuant to paragraph (5).
(8) Within 14 days of receipt of any recommendation made pursuant to paragraph (5), the Secretary of State shall inform the blood establishment or the person responsible for the management of the hospital blood bank whether he accepts the recommendation and, if he does not accept it, of the reasons for his decision.
(9) [F39Subject to paragraph (11),] where the Secretary of State is notified of an objection pursuant to paragraph (1)(a) before the date upon which the suspension or revocation or the notice is due to take effect, the suspension or revocation or notice in respect of which the objection is made shall not take effect until—
(a)the person appointed pursuant to regulation (3) has considered the matter in accordance with the provisions of this regulation and made a recommendation; and
(b)the Secretary of State has informed the blood establishment or the person responsible for the management of the hospital blood bank concerned of his decision with regard to the recommendation pursuant to paragraph (8),.
(10) Subject to paragraph (11), where the Secretary of State is notified of an objection pursuant to paragraph (1)(a), within the period specified in paragraph (2), to a suspension, revocation or other notice which has already taken effect on the date the notification was made, the suspension, revocation or notice in respect of which the objection is made shall cease to have effect until—
(a)the person appointed pursuant to regulation (3) has considered the matter in accordance with the provisions of this regulation and made a recommendation; and
(b)the Secretary of State has informed the blood establishment or the person responsible for the management of the hospital blood bank concerned of his decision with regard to the recommendation pursuant to paragraph (8).
(11) [F40Paragraphs (9) and (10)] shall not apply—
(a)in relation to a suspension or revocation, or a notice served pursuant to regulation 11, which takes immediate effect in accordance with regulation 5(3) or 11(3); or
(b)in any other case, where the Secretary of State determines that it is necessary in the interests of public safety for the suspension, revocation or notice to take effect on the date originally specified, and serves a notice in writing to that effect on the blood establishment or person responsible for management of the hospital blood bank concerned.
Textual Amendments
F38Words in reg. 12(1) substituted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 4(a)
F39Words in reg. 12(9) inserted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 4(b)
F40Words in reg. 12(11) substituted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 4(c)
[F41Requirement that facilities retain certain dataU.K.
12A.—(1) A person responsible for management of a facility shall ensure that the facility—
(a)retains the data set out in Section B of Part 6 of the Schedule, in an appropriate and readable storage medium, for a period of at least 30 years; and
(b)has in place a system in place to record each unit of blood or blood component received, whether or not locally used, and the final destination of that received unit whether transfused, used in the manufacture of medicinal products, discarded or returned to the blood establishment or hospital blood bank.
Textual Amendments
F41Regs. 12A, 12B inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 7
Requirement to report serious adverse reactions and eventsU.K.
12B.—(1) A person responsible for management of a reporting establishment shall ensure that the reporting establishment—
(a)has in place procedures to retain the record of transfusions for a period of at least 30 years;
(b)notifies blood establishments without delay of any serious adverse reactions observed in recipients during or after transfusion which may be attributable to the quality or safety of blood or blood components; and
(c)notifies the Secretary of State as soon as is known all relevant information about suspected serious adverse reactions using the notification formats set out in Section A and Section C of Part 7 of the Schedule.
(2) A person responsible for management of a reporting establishment shall ensure that the reporting establishment—
(a)notifies the Secretary of State of all relevant information about serious adverse reactions of imputability level 2 and 3 as referred to in Section B of Part 7 of the Schedule, which may be attributable to the quality and safety of blood or blood components;
(b)notifies the Secretary of State, as soon as is known, of any case of transmission of infectious agents by blood or blood components;
(c)as part of the notification referred to in paragraph (a), describes the actions taken with respect to other implicated blood or blood components that have been distributed for transfusion or for plasma fractionation;
(d)as soon as is reasonably practicable after each suspected serious adverse reaction, evaluates that reaction according to the imputability levels set out in Section B of Part 7 of the Schedule;
(e)completes the serious adverse reaction notification, upon conclusion of the investigation, using the format set out in Section C of Part 7 to the Schedule; and
(f)submits a complete report to the Secretary of State on serious adverse reactions in any calendar year by no later than 1st April in the following calendar year, using the format set out in Section D of Part 7 to the Schedule.
(3) A person responsible for management of a reporting establishment shall ensure that the reporting establishment notifies the Secretary of State as soon as is known, using the notification formats set out in Section A of Part 8 of the Schedule, of all relevant information about serious adverse events which may put in danger donors or recipients other than those directly involved in the event concerned.
(4) A person responsible for management of a reporting establishment shall ensure that the reporting establishment—
(a)as soon as is reasonably practicable after each serious adverse event, evaluates that serious adverse event to identify preventable causes within the process;
(b)upon completion of the investigation, completes the serious adverse event notification, using the format set out in Section B of Part 8 of the Schedule; and
[F42(c)submits a complete report to the Secretary of State on serious adverse events in any calendar year by no later than 1st April in the following calendar year, using the format set out in Section C of Part 8 of the Schedule.]
(5) Provided that either the condition set out in paragraph (6)(a), or the conditions set out in paragraph (6)(b) and (c) are satisfied, a facility may make arrangements with a hospital blood bank for the hospital blood bank to submit to the Secretary of State or the blood establishment the reports required by paragraphs (1)(b) and (c), (2)(a),(b),(e) and (f) and [F43(4)(b)] and (c) on the facility’s behalf.
(6) The conditions referred to in paragraph (5) are that—
(a)the person responsible for management of the hospital blood bank is the same person as the person responsible for management of the facility with which the arrangement is made; or
(b)the arrangements referred to in paragraph (5) must be—
(i)evidenced by a written agreement, and
(ii)made with the person responsible for management of the hospital blood bank who supplied the blood or blood components to the facility for transfusion; and
(c)the facility must supply the information necessary to enable the hospital blood bank to make the reports within the timescale specified by this regulation in relation to that report .]
Textual Amendments
F41Regs. 12A, 12B inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 7
F42Reg. 12B(4)(c) substituted (1.4.2007) by The Blood Safety and Quality (Amendment) Regulations 2007 (S.I. 2007/604), regs. 1(1), 2
F43Word in reg. 12B(5) substituted (1.5.2008) by The Medicines for Human Use (Clinical Trials) and Blood Safety and Quality (Amendment) Regulations 2008 (S.I. 2008/941), regs. 1(1), 9
Import of blood and blood components into the United KingdomU.K.
[F4413. Any person who imports blood or blood components into the United kingdom from a third country must ensure that each unit of blood and each blood components which he imports—
(a)has been prepared in accordance with standards equivalent to the Community standards and requirements set out in the Annex to Commission Directive 2005/62/EC; and
(b)meets standards of quality and safety equivalent to those laid down in Part 5 of the Schedule.]
Textual Amendments
F44Reg. 13 substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 8
Disclosure of information by blood establishments and hospital blood banksU.K.
14.—(1) A blood establishment and the person responsible for management of a hospital blood bank shall ensure that all information which is collected for the purposes of these Regulations is held securely so that it is—
(a)available for the purpose of, tracing donations;
(b)not disclosed except—
(i)in accordance with one or more of the requirements of paragraph (2), or
(ii)where they have been rendered anonymous so that donors are no longer identifiable;
(c)subject to safeguards against unauthorised additions, deletions or modifications.
(2) The requirements of this paragraph are—
(a)the disclosure is made in accordance with an order of a court or is otherwise required by law;
(b)the disclosure is to an inspector appointed by the Secretary of State in accordance with regulation 15(10); or
(c)the disclosure is for the purpose of tracing a donation from donor to recipient or recipient to donor.
(3) Where a disclosure is made to an inspector pursuant to paragraph (2)(b), the inspector shall not further disclose the information received unless—
(a)the disclosure is made in accordance with an order of a court or is otherwise required by law;
(b)the disclosure is to another officer of the Secretary of State where this is necessary for the proper performance of the inspector or officer's duties; or
(c)the information has been rendered anonymous so that that donors are no longer identifiable.
(4) Where a disclosure is made by an inspector to another officer of the Secretary of State pursuant to paragraph (3), that person shall not further disclose the information he receives other than in accordance with the requirements of that paragraph.
(5) The responsible person of the blood establishment and the person responsible for management of the hospital blood bank shall ensure that they put in place a procedure to ensure that any discrepancies relating to data which are brought to their attention are resolved without delay.
Inspections, etc.U.K.
15.—(1) The Secretary of State shall conduct a regular inspection of each site of a blood establishment, not less than once every two years, for the purpose of ensuring that—
(a)blood establishments comply with the requirements of these Regulations; and
(b)problems relating to compliance with those requirements are identified.
(2) The Secretary of State may conduct such additional inspections of blood establishments sites as he considers necessary for the purpose of ensuring compliance with the requirements of these Regulations.
(3) The Secretary of State may also serve a notice on a blood establishment requiring that it furnish him with such information concerning its compliance with these Regulations as shall be specified in the notice within such period as shall be specified in the notice.
(4) Any blood establishment which receives a request or information in accordance with paragraph (3) shall provide the information requested within the period specified in the notice.
(5) The Secretary of State may inspect hospital blood banks [F45and facilities] with a view to ensuring that—
(a)hospital blood banks [F45and facilities] and persons responsible for the management of such blood banks [F45and facilities] comply with the requirements of these Regulations; and
(b)problems relating to compliance with those requirements are identified.
(6) The Secretary of State may also serve a notice on the person responsible for managing a hospital blood bank [F46or a facility] requiring that he furnish him with such information concerning the compliance of the blood bank [F46or a facility] with these Regulations as shall be specified in the notice within such period shall be specified in the notice.
(7) Any person responsible for management of a hospital blood bank [F47or a facility] who receives a request for information in accordance with paragraph (6) shall provide the information requested within the period specified in the notice.
(8) In the event of any serious adverse event or any serious adverse reaction or suspicion thereof, the Secretary of State shall request such information or conduct such inspections in accordance with this regulation as he shall consider appropriate.
(9) Any reference to an inspection of a site which the Secretary of State is required or empowered to conduct by virtue of this regulation, shall be construed so as to include an inspection of premises within the UK at which any of the activities listed in regulation 3(2) are carried out by any person on behalf of, and pursuant to a contractual arrangement with, a blood establishment or, as the case may be, a person responsible for management of a hospital blood bank.
(10) The Secretary of State may appoint such persons to be inspectors as he thinks necessary for the proper discharge by them of his functions set out in these Regulations, and he may appoint such persons upon such terms and conditions (including conditions as to remuneration, benefits, allowances and reimbursement for expenses) as he thinks fit.
Textual Amendments
F45Words in reg. 15(5) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 9(a)
F46Words in reg. 15(6) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 9(b)
F47Words in reg. 15(7) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 9(c)
Records to be kept by the Secretary of StateU.K.
16.—(1) The Secretary of State shall keep such records of information which he receives from, or relating to, blood establishments as he considers appropriate and shall, in particular, keep records relating to—
(a)authorisations under regulation 4;
(b)the designation of responsible persons under regulation 6;
(c)notification of serious adverse events and serious adverse reactions by such establishments pursuant to [F48regulation 12B];
(d)inspections or requests for information under regulation 15;
(e)the operation, during the period from [F498th February 2005] to 7th November 2005, of blood establishments licensed under section 8 of the Medicines Act 1968.
(2) The Secretary of State shall keep such records of information which he receives from persons responsible for management of hospital blood banks [F50and facilities], or otherwise F51... relating to hospital blood banks [F52or facilities], as he considers appropriate and shall, in particular keep records relating to—
(a)notification of serious adverse events and serious adverse reactions pursuant to [F53regulation 12B];
(b)the information supplied by hospital blood banks pursuant to regulation 10;
(c)inspections or requests for information under regulation 15.
Textual Amendments
F48Words in reg. 16(1)(c) substituted (1.4.2007) by The Blood Safety and Quality (Amendment) Regulations 2007 (S.I. 2007/604), regs. 1(1), 3(a)
F49Words in reg. 16(1)(e) substituted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 5
F50Words in reg. 16(2) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 10(a)
F51Word in reg. 16(2) omitted (31.8.2006) by virtue of The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 10(b)
F52Words in reg. 16(2) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 10(c)
F53Words in reg. 16(2)(a) substituted (1.4.2007) by The Blood Safety and Quality (Amendment) Regulations 2007 (S.I. 2007/604), regs. 1(1), 3(b)
[F54Requirement that the Secretary of State communicate certain information to other competent authoritiesU.K.
16A. The Secretary of State shall communicate to the competent authorities of other Member States such information as is appropriate with regard to serious adverse reactions and events in order to guarantee that blood or blood components known or suspected to be defective are withdrawn from use and discarded.]
Textual Amendments
F54Reg. 16A inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 11
Powers of entry, etc.U.K.
17.—(1) For the purposes of enforcing compliance with these Regulations or conducting inspections pursuant to regulation 15, an inspector appointed in accordance with regulation 15(10) shall, upon production of evidence that he is so authorised, have the right—
(a)at any reasonable hour to enter any premises, other than premises used only as a private dwelling house, which he has reason to believe it is necessary for him to visit, including—
(i)any premises owned or managed by a blood establishment or person responsible for management of a hospital blood bank, or at which the blood establishment or person responsible for management of a hospital blood bank carries out any of the activities referred to in regulation 3;
(ii)any premises of any person who carries out any of the activities referred to in regulation 3(2) on behalf of, [F55or] pursuant to a contractual arrangement with, a blood establishment or a person responsible for management of a hospital blood bank; F56...
(iii)where any facilities for donor evaluation and testing are in the premises of any person other than a blood establishment or hospital blood bank, those facilities in that person's premises; [F57and]
[F58(iv)any premises where transfusion of blood or blood components takes place, or which are owned or managed by a person responsible for management of a facility to which blood or blood components have been delivered.]
(b)to carry out at those premises during that visit inspections, examinations, tests and analyses as he considers necessary;
(c)to require the production of, and inspect any article or substance at, the premises;
(d)to require the production of, inspect and take copies of, or extracts from, any book, document, data or record (in whatever form it is held) at, or (in the case of computer data or records) accessible at the premises;
(e)F59... to take possession of any samples for examination and analysis and any other article, substance, book, document, data, record (in whatever form they are held) at, or (in the case of computer data or records) accessible at, the premises;
(f)to question any person whom he finds at the premises and whom he has reasonable cause to believe is able to give him relevant information;
(g)to require any person to afford him such assistance as he considers necessary with respect to any matter within that person's control, or in relation to which that person has responsibilities;
(h)to require, as he considers necessary, any person to afford him such facilities as he may reasonably require that person to afford him;
but nothing in this paragraph shall be taken to compel the production by any person of a document of which he would on grounds of legal professional privilege be entitled to withhold production on an order for disclosure in an action in the High Court or, as the case may be, on an order for production of documents in an action in the Court of Session.
(2) If a justice of the peace is satisfied by any written information on oath that there are reasonable grounds for entry into any premises, other than premises used only as a private dwelling house, for any purpose mentioned in paragraph (1), and—
(a)admission to the premises has been refused or is likely to be refused and notice of intention to apply for a warrant under this sub-paragraph has been given to the occupier;
(b)an application for admission, or the giving of such notice, would defeat the object of the entry; or
(c)the premises are unoccupied or the occupier is temporarily absent and it might defeat the object of the entry to await his return,
the justice may, by warrant signed by him, which shall continue in force for a period of one month, authorise an inspector to enter the premises, if need be by force.
(3) An inspector entering premises by virtue of paragraph (1) or of a warrant under paragraph (2) may take with him when he enters those premises such equipment as may appear to him necessary and any person who is authorised by the Secretary of State to accompany him on that visit.
(4) On leaving any premises which an inspector is authorised to enter by a warrant under paragraph (2), he shall, if the premises are unoccupied, or the occupier is temporarily absent, leave the premises as effectively secured against trespassers as he found them.
(5) Where, pursuant to paragraph (1)(e), an inspector takes possession of any article, substance, book, document, data or record, he shall leave at the premises with a responsible person, or if there is no such person present on the premises, leave in the premises in a prominent position, a statement giving particulars of the article, substance, book, document, data or record sufficient to identify it and stating that he has taken possession of it.
(6) Where, pursuant to paragraph (1)(e) an inspector takes a sample for analysis, the Secretary of State may, subject to the requirements of paragraph (7), make such arrangements for analysis of that sample as he considers appropriate.
(7) The requirements of this paragraph are—
(a)that the Secretary of State shall inform the responsible person of the blood establishment or person responsible for the management of the hospital blood bank from which the sample was taken that he intends to make arrangements for analysis of the sample, and of the tests which he intends should be made; and
(b)that if the responsible person or person responsible for the management of the hospital blood bank so requests, the Secretary of State shall divide the sample of which an analysis is to be made into three equal parts and deal with those parts in accordance with the requirements of paragraph (8).
(8) The requirements of this paragraph are—
(a)that the Secretary of State shall make arrangements for the testing of one part of the sample;
(b)that one part of the sample shall be sent to the responsible person of the blood establishment or person responsible for the management of the hospital blood bank; and
(c)that one part of the sample shall be retained by the Secretary of State for a reasonable period in case of dispute.
Textual Amendments
F55Word in reg. 17(1)(a)(ii) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 12(a)(i)
F56Word in reg. 17(1)(a)(ii) omitted (31.8.2006) by virtue of The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 12(a)(ii)
F57Word in reg. 17(1)(a)(iii) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 12(b)
F58Reg. 17(1)(a)(iv) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 12(c)
F59Words in reg. 17(1)(e) omitted (8.4.2005) by virtue of The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 6
Criminal offencesU.K.
18.—(1) Any person who contravenes any of the following provisions—
(a)regulation 3(1)
(b)regulation 7;
(c)regulation 9;
(d)regulation 13;
(e)regulation 23(2),
[F60(f)regulation 12A, and
(g)regulation 12B,]
shall be guilty of an offence.
(2) Any person who contravenes any of the following provisions—
(a)regulation 4(9);
(b)regulation 6, other than regulation 6(3);
(c)regulation 8;
(d)regulation 10;
(e)regulation 15(4) and (7),
shall be guilty of an offence
(3) Any person who fails to comply with a notice of suspension or revocation of his authorisation served pursuant to regulation 5, save where the operation of that notice has been suspended pursuant to regulation 12, or has been withdrawn or revoked by the Secretary of State, shall be guilty of an offence.
(4) Any person who knowingly sells or supplies blood or any blood component which is not labelled in accordance with the requirements of regulation 8, shall be guilty of an offence.
(5) Any person who contravenes the requirements of any notice served by the Secretary of State under regulation 11(1), shall be guilty of an offence.
(6) Any person who—
(a)contravenes regulation 14; or
(b)discloses any information referred to in regulation 14(1) to which they have access by virtue of these regulations, otherwise than in accordance one or more of the requirements specified in regulation 14(2) and (3),
shall be guilty of an offence.
(7) Subject to [F61paragraph (8)] —
(a)any person who—
(i)intentionally obstructs an inspector, or;
(ii)without reasonable cause fails to comply with any requirements made of him by an inspector, in circumstances where that inspector is acting in pursuance of any of his functions under these Regulations; or
(iii)any person who, in purported compliance with any such requirement as is mentioned in sub-paragraph (a)(ii), intentionally or recklessly furnishes information which is false or misleading in a material respect,
shall be guilty of an offence.
(8) Nothing in paragraph (7)(a)(ii) shall be construed as requiring any person to answer any question or give any information if to do so might incriminate him or, in the case of a person who is [F62married or a civil partner, his spouse or civil partner].
Textual Amendments
F60Reg. 18(1)(f)(g) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 13
F61Words in reg. 18(7) substituted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 7
F62Words in reg. 18(8) substituted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 5
PenaltiesU.K.
19.—(1) A person guilty of an offence under regulation 18(1), (3), (5) or (7) shall be liable—
(a)on summary conviction to a fine not exceeding the statutory maximum or to imprisonment for a term not exceeding [F633 months], or to both; or
(b)on conviction on indictment, to a fine, or to imprisonment for a term not exceeding 2 years, or to both.
(2) A person guilty of an offence under regulation 18(2), (4) or (6) shall be liable on summary conviction to a fine not exceeding level 5 on the standard scale, or to imprisonment for a term not exceeding [F633 months], or to both.
Textual Amendments
F63Words in reg. 19 substituted (8.4.2005) by The Blood Safety and Quality (Amendment) Regulations 2005 (S.I. 2005/1098), regs. 1(1), 8
Defence of due diligenceU.K.
20.—(1) In any proceedings for an offence under any of the preceding provisions of these Regulations, it shall be a defence for the person charged to prove that he took all reasonable precautions and exercised all due diligence to avoid commission of the offence.
(2) Where evidence is adduced which is sufficient to raise an issue with respect to that defence, the court or jury shall assume that the defence is satisfied unless the prosecution proves beyond all reasonable doubt that it is not.
Offences by bodies corporate and Scottish partnershipsU.K.
21. Where an offence under these Regulations is committed by a body corporate or a Scottish partnership and is proved to have been committed with the consent or connivance of, or to be attributable to, any neglect on the part of—
(a)any director, manager, secretary, partner or similar officer of the body corporate or Scottish partnership; or
(b)any person who was purporting to act in any such capacity,
he, as well as the body corporate or Scottish partnership, shall be deemed to be guilty of that offence and he shall be liable to be proceeded against and punished accordingly.
FeesU.K.
22.—(1) Blood establishments shall pay to the Secretary of State such fees as are payable in accordance with paragraphs (2) and (3).
(2) The fees payable pursuant to paragraph (1) by blood establishments in relation to authorisation under regulation 3 are—
(a)in respect of an application for authorisation pursuant to regulation 3, the sum of [F64£3,044];
(b)in respect of an application for approval of a substantial change pursuant to regulation 4(10), the sum of [F65£513]; and
[F66(bb)in respect of the assessment by the Secretary of State of serious adverse events and serious adverse reactions notified by blood establishments, an annual haemovigilance fee calculated in accordance with paragraph (2A); and]
(c)in connection with the holding of an authorisation under regulation 3, an annual fee of the sum of [F67£458].
[F68(2A) The fee payable under paragraph (2)(bb) shall be—
(a)in respect of the reporting year ending on 31st March 2006, £156; and
(b)in respect of each subsequent reporting year, [F69£487].]
(3) Where the Secretary of State carries out an inspection at a site of a blood establishment he may charge the establishment and that establishment shall, if so charged, pay to the Secretary of State a fee calculated in accordance with the following sub-paragraphs—
[F70(a)for an inspection where the time taken to carry out the inspection at the site is not more than 7 hours, the sum of [F71£2,557]; and
(b)for an inspection where the time taken to carry out the inspection at the site is more than 7 hours, the sum in sub-paragraph (a) and thereafter at the rate of [F72£1,279], for each additional period of 3 hours and 30 minutes or less taken to make the inspection.]
[F73(3A) In respect of each reporting year in which a hospital blood bank has operated, the person who is responsible for management of that hospital blood bank shall pay to the Secretary of State a fee of [F74£676].
(3B) Subject to [F75paragraphs (3D) and (3E)], in respect of the assessment by the Secretary of State of serious adverse events and serious adverse reactions notified by hospital blood banks [F76or facilities], the person who is responsible for management of a hospital blood bank [F77or facility] shall pay to the Secretary of State an annual haemovigilance fee calculated in accordance with paragraph (3C).
(3C) The fee payable under paragraph (3C) shall be—
(b)in respect of the reporting year ending on 31st March 2006, £156; and
(c)in any other case, [F78£487].
(3D) No fee shall be payable under paragraph (3B) by a person responsible for the management of a hospital blood bank if that person is authorised as a blood establishment under these Regulations.]
[F79(3E) No fee shall be payable under paragraph (3B) by a person responsible for the management of a facility where the facility makes arrangements with a hospital blood bank, pursuant to regulation 12B(5) that the hospital blood bank will report serious adverse reactions and events to the Secretary of State on behalf of the facility.]
(4) Where the Secretary of State carries out an inspection of a hospital blood bank [F80or a facility] he may charge the person responsible for management of the hospital blood bank [F81or the facility] and that person shall, if so charged, pay to the Secretary of State a fee calculated in accordance with paragraph (5).
(5) The fees payable by hospital blood banks [F82or facilities] in respect of inspections are—
[F83(a)for an inspection where the time taken to carry out the inspection at the site is not more than 7 hours, the sum of [F84£2,557]; and
(b)for an inspection where the time taken to carry out the inspection at the site is more than 7 hours, the sum in sub-paragraph (a) and thereafter at the rate of [F85£1,279], for each additional period of 3 hours and 30 minutes or less taken to make the inspection.]
[F86(5A) Where the Secretary of State carries out an inspection of a contract laboratory, he may charge the person having control of that laboratory and that person shall, if so charged, pay to the Secretary of State a fee calculated in accordance with paragraph (5B).
(5B) Subject to paragraph (5C), the fee payable under paragraph (5A) shall be—
[F87(a)for an inspection where the time taken to carry out the inspection at the site is not more than 7 hours, the sum of [F88£2,557]; and
(b)for an inspection where the time taken to carry out the inspection at the site is more than 7 hours, the sum in sub-paragraph (a) and thereafter at the rate of [F89£1,279], for each additional period of 3 hours and 30 minutes or less taken to make the inspection.]
(5C) Where an inspection referred to in paragraph (5A) takes place at the same time as an inspection by a person appointed by the Good Laboratory Practice Monitoring Authority under regulation 3(4) of the Good Laboratory Practice Regulations 1999, for the purposes of ascertaining whether the contract laboratory complies with the principles of good laboratory practice, the fee payable under paragraph (5A) shall be—
[F90(a)for an inspection where the time taken to carry out the inspection at the site is not more than 7 hours, the sum of [F91£2,557]; and
(b)for an inspection where the time taken to carry out the inspection at the site is more than 7 hours, the sum in sub-paragraph (a) and thereafter at the rate of [F92£1,279], for each additional period of 3 hours and 30 minutes or less taken to make the inspection.]
F93(5D) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .]
(6) In this regulation—
[F94“contract laboratory” means a laboratory carrying out testing of blood or blood components on behalf of, and pursuant to a contractual arrangement with—
(a)
a blood establishment which is authorised under these Regulations; or
(b)
a person responsible for management of a hospital blood bank;]
F95...
F95...
F95...
F95...
[F96(6A) For the purposes of this regulation if an inspection is carried out by more than one inspector, the time taken by the Secretary of State to carry out the inspection is the aggregate of times taken by each inspector in carrying out the inspection at the site.]
(7) [F97Subject to paragraph (7A),] any fee payable under this regulation shall be payable at the following times—
(i)the fee payable pursuant to paragraph (2)(a) in respect of an application for authorisation to operate a blood establishment, and the fee payable pursuant to paragraph (2)(b) in respect of an application to make a substantial change, shall, in each case, be payable at the time the application is made;
(ii)the periodic fee payable pursuant to paragraph (2)(c) shall be payable on the first anniversary of the grant by the Secretary of State of authorisation to operate as a blood establishment, and whilst the blood establishment continues to be authorised to operate as such pursuant to these Regulations, annually thereafter;
[F98(iia)any fee payable pursuant to paragraph (2)(bb) or (3B) shall be payable—
(aa)if it is payable in respect of the reporting year ending on 31st March 2006, on 31st December 2005, and
(bb)if it is payable in respect of any subsequent reporting year, on 30th April during that year;
(iib)the fee payable pursuant to paragraph (3A) shall be payable—
(aa)if it is payable in respect of the reporting year ending on 31st March 2006, on 31st December 2005, and
(bb)if it is payable in respect of any subsequent reporting year, on 30th April following the end of that year;]
(iii)any other fee payable under this regulation shall be payable within fourteen days following written notice from the Secretary of State requiring payment of the fee.
[F99(7A) In the case of a blood establishment granted an authorisation under regulation 4 before 8th November 2005, the periodic fee payable pursuant to paragraph (2)(c) shall be payable on 8th November 2006 and, while the blood establishment continues to be authorised to operate as such pursuant to these Regulations, annually thereafter.]
(8) All unpaid sums due by way of, or on account of, any fees payable under this regulation shall be recoverable as debts due to the Crown.
(9) The Secretary of State may in exceptional circumstances where it appears to him to be in the interests of safety or otherwise appropriate to do so—
(a)waive any fee or reduce any fee or part of a fee otherwise payable under this regulation; or
(b)refund the whole or part of any fee paid pursuant to this regulation.
Textual Amendments
F64Word in reg. 22(2)(a) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(2)(a)
F65Word in reg. 22(2)(b) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(2)(b)
F66Reg. 22(2)(bb) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 6(2)
F67Word in reg. 22(2)(c) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(2)(c)
F68Reg. 22(2A) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 6(3)
F69Word in reg. 22(2A)(b) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(3)
F70Reg. 22(3)(a)(b) substituted for reg. 22(3)(a)-(f) (1.4.2008) by The Blood Safety and Quality (Fees Amendment) Regulations 2008 (S.I. 2008/525), regs. 1(1), 2(4)
F71Word in reg. 22(3)(a) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(4)(a)
F72Word in reg. 22(3)(b) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(4)(b)
F73Reg. 22(3A)-(3D) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 6(4)
F74Word in reg. 22(3A) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(5)
F75Words in reg. 22(3B) substituted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 14(a)(i)
F76Words in reg. 22(3B) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 14(a)(ii)
F77Words in reg. 22(3B) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 14(a)(iii)
F78Word in reg. 22(3C)(c) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(6)
F79Reg. 22(3E) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 14(b)
F80Words in reg. 22(4) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 14(c)(i)
F81Words in reg. 22(4) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 14(c)(ii)
F82Words in reg. 22(5) inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 14(d)
F83Reg. 22(5)(a)(b) substituted for reg. 22(5)(a)-(c) (1.4.2008) by The Blood Safety and Quality (Fees Amendment) Regulations 2008 (S.I. 2008/525), regs. 1(1), 2(7)
F84Word in reg. 22(5)(a) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(7)(a)
F85Word in reg. 22(5)(b) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(7)(b)
F86Reg. 22(5A)-(5D) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 6(5)
F87Reg. 22(5B)(a)(b) substituted for reg. 22(5B)(a)-(c) (1.4.2008) by The Blood Safety and Quality (Fees Amendment) Regulations 2008 (S.I. 2008/525), regs. 1(1), 2(8)
F88Word in reg. 22(5B)(a) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(8)(a)
F89Word in reg. 22(5B)(b) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(8)(b)
F90Reg. 22(5C)(a)(b) substituted for reg. 22(5C)(a)-(c) (1.4.2008) by The Blood Safety and Quality (Fees Amendment) Regulations 2008 (S.I. 2008/525), regs. 1(1), 2(9)
F91Word in reg. 22(5C)(a) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(9)(a)
F92Word in reg. 22(5C)(b) substituted (1.4.2009) by The Blood Safety and Quality (Fees Amendment) Regulations 2009 (S.I. 2009/372), regs. 1(1), 2(9)(b)
F93Reg. 22(5D) omitted (1.4.2008) by virtue of The Blood Safety and Quality (Fees Amendment) Regulations 2008 (S.I. 2008/525), regs. 1(1), 2(10)
F94Words in reg. 22(6) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 6(6)
F95Words in reg. 22(6) omitted (1.4.2008) by virtue of The Blood Safety and Quality (Fees Amendment) Regulations 2008 (S.I. 2008/525), regs. 1(1), 2(11)
F96Reg. 22(6A) inserted (1.4.2008) by The Blood Safety and Quality (Fees Amendment) Regulations 2008 (S.I. 2008/525), regs. 1(1), 2(12)
F97Words in reg. 22(7) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 6(7)(a)
F98Reg. 22(7)(iia)(iib) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 6(7)(b)
F99Reg. 22(7A) inserted (8.11.2005) by The Blood Safety and Quality (Amendment) (No. 2) Regulations 2005 (S.I. 2005/2898), regs. 1(1), 6(8)
Specific epidemiological situationsU.K.
23.—(1) Where the Secretary of State is aware of a specific epidemiological situation, such as an outbreak of a disease, which may affect the safety of blood donations, and as a result of which he considers that specific deferral criteria for the collection of blood donations should be adopted, he shall—
(a)notify blood establishments that those criteria must be adopted; and
(b)notify the Commission of—
(i)the epidemiological situation; and
(ii)the additional deferral criteria which blood establishments are required to adopt in relation to it pursuant to sub-paragraph (a).
(2) A blood establishment shall adopt and comply with any criteria for additional tests notified to them by the Secretary of State pursuant to paragraph (1).
Transitional provisionsU.K.
24.—(1) Subject to paragraph (2), these Regulations, other than regulations 13 and 16, shall not apply before 8th November 2005 in relation to—
(a)any blood establishment licensed under section 8 of the Medicines Act 1968 M14;
(b)any hospital blood bank.
(2) From the date these Regulations come into force, a blood establishment licensed under section 8 of the Medicines Act 1968 may apply for, and the Secretary of State may grant, an authorisation under regulation 4 to have effect as from 8th November 2005.
Marginal Citations
Consequential amendmentsU.K.
25.—(1) The Medicines Act 1968 shall be amended as follows—
(a)in section 7 (general provisions as to dealing with medicinal products) omit paragraph (a) of subsection (6A) M15;
(b)in section 8 (provisions as to manufacture and wholesale dealing), omit paragraph (a) of subsection (4) M16;
(c)in section 130 (meaning of “medicinal product” and related expressions) in subsection (5), after paragraph (b), insert the following new paragraph—
“(ba) whole human blood and human blood components;”.
(d)In section 130, after subsection (5A) insert the following new subsection—
“(5B) For the purposes of this section, “human blood component” means any of the following constituents of human blood: red cells, white cells, platelets and plasma.”.
(2) The Medicines (Standard Provisions for Licenses and Certificates) Regulations 1971 shall be amended as follows—
(a)in regulation 2 (interpretation), after the definition of “BCG vaccine” insert the following definitions—
““blood” means whole human blood;
“blood component” means a therapeutic constituent of blood (red cells, white cells, platelets and plasma);”; and
(b)in Schedule 2 insert the following new paragraph—
“5C. The licence holder shall ensure that any blood or blood component imported into the United Kingdom and used by him as a starting material or raw material in the manufacture of a medicinal product, shall meet equivalent standards of quality and safety to those laid down in Commission Directive 2004/33/EC, implementing Directive 2003/98/EC of the European Parliament and of the Council as regards certain technical requirements for blood and blood components.”.
(3) In the Medicines for Human Use (Marketing Authorisations Etc.) Regulations 1994 M17, in regulation 1 (citation, commencement and interpretation), in paragraph (2), in the definition of “the 2001 Directive”, after “as amended by” insert— “ Directive 2002/98/EC of the European Parliament and of the Council setting standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components and amending Directive 2001/83/EC, ”.
Marginal Citations
M15Subsection (6A) was inserted by S.I. 1992/604 and amended by S.I. 1994/276.
M16Subsection (4) was substituted by S.I. 1992/604 and amended by S.I. 1994/276.
M17S.I. 1994/3144; relevant amending instruments are S.I. 2001/795, 2002/236, 2003/2321 and 2004/3224.
Signed by authority of the Secretary of State for Health
Melanie Johnson
Parliamentary Under Secretary of State,
Department of Health
We consent,
Joan Ryan
Nick Ainger
Two of the Lords Commissioners' of Her Majesty's Treasury
Regulations 4(4)(b), 7(1)(c), 2(a), (b) and (d), and (3)(b) and (c), 9(1)(c) and (h) and 13.
SCHEDULEU.K.
PART 1U.K.
DefinitionsU.K.
The following definitions apply for the purposes of this Schedule.
1. “Autologous donation” means blood and blood components collected from an individual and intended solely for subsequent autologous transfusion or other human application to that same individual.U.K.
2. “Allogeneic donation” means blood and blood components collected from an individual and intended for transfusion to another individual, for use in medical devices or as starting material or raw material for manufacturing into medicinal products.U.K.
3. “Whole blood” means a single blood donation.U.K.
4. “Cryopreservation” means prolongation of the storage life of blood components by freezing.U.K.
5. “Plasma” means the liquid portion of the blood in which the cells are suspended. Plasma may be separated from the cellular portion of a whole blood collection for therapeutic use as fresh-frozen plasma or further processed to cryoprecipitate and cryoprecipitate-depleted plasma for transfusion. It may be used for the manufacture of medicinal products derived from human blood and human plasma or used in the preparation of pooled platelets, or pooled, leucocyte-depleted platelets. It may also be used for re-suspension of red cell preparations for exchange transfusion or perinatal transfusion.U.K.
6. “Cryoprecipitate” means a plasma component prepared from plasma, fresh-frozen, by freeze-thaw precipitation of proteins and subsequent concentration and re-suspension of the precipitated proteins in a small volume of the plasma.U.K.
7. “Washed” means a process of removing plasma or storage medium from cellular products by centrifugation, decanting of the supernatant liquid from the cells and addition of an isotonic suspension fluid, which in turn is generally removed and replaced following further centrifugation of the suspension. The centrifugation, decanting, replacing process may be repeated several times.U.K.
8. “Red cells” means the red cells from a single whole blood donation, with a large proportion of the plasma from the donation removed.U.K.
9. “Red cells, buffy coat removed” means the red cells from a single whole blood donation, with a large proportion of the plasma from the donation removed. The buffy coat, containing a large proportion of the platelets and leucocytes in the donated unit, is removed.U.K.
10. “Red cells, leucocyte-depleted” means the red cells from a single whole blood donation, with a large proportion of the plasma from the donation removed, and from which leucocytes are removed.U.K.
11. “Red cells in additive solution” means the red cells from a single whole blood donation, with a large proportion of the plasma from the donation removed. A nutrient or preservative solution is added.U.K.
12. “Additive solution” means a solution specifically formulated to maintain beneficial properties of cellular components during storage.U.K.
13. “Red cells, buffy coat removed, in additive solution” means the red cells from a single whole blood donation, with a large proportion of the plasma from the donation removed. The buffy coat, containing a large proportion of the platelets and leucocytes in the donated unit, is removed. A nutrient or preservative solution is added.U.K.
14. “Buffy coat” means a blood component prepared by centrifugation of a unit of whole blood, and which contains a considerable proportion of the leucocytes and platelets.U.K.
15. “Red cells, leucocyte-depleted, in additive solution” means the red cells from a single whole blood donation, with a large proportion of the plasma from the donation removed, and from which leucocytes are removed. A nutrient or preservative solution is added.U.K.
16. “Red cells, apheresis” means the red cells from an apheresis red cell donation.U.K.
17. “Apheresis” means a method of obtaining one or more blood components by machine processing of whole blood in which the residual components of the blood are returned to the donor during or at the end of the process.U.K.
18. “Platelets, apheresis” means a concentrated suspension of blood platelets obtained by apheresis.U.K.
19. “Platelets, apheresis, leucocyte-depleted” means a concentrated suspension of blood platelets, obtained by apheresis, and from which leucocytes are removed.U.K.
20. “Platelets, recovered, pooled” means a concentrated suspension of blood platelets, obtained by processing of whole blood units and pooling the platelets from the units during or after separation.U.K.
21. “Platelets, recovered, pooled, leucocyte-depleted” means a concentrated suspension of blood platelets, obtained by processing of whole blood units and pooling the platelets from the units during or after separation, and from which leucocytes are removed.U.K.
22. “Platelets, recovered, single unit” means a concentrated suspension of blood platelets, obtained by processing of a single unit of whole blood.U.K.
23. “Platelets, recovered, single unit, leucocyte-depleted” means a concentrated suspension of blood platelets, obtained by processing of a single whole blood unit from which leucocytes are removed.U.K.
24. “Plasma, fresh-frozen” means the supernatant plasma separated from a whole blood donation or plasma collected by apheresis, frozen and stored.U.K.
25. “Plasma, cryoprecipitate-depleted for transfusion” means a plasma component prepared from a unit of plasma, fresh-frozen. It comprises the residual portion after the cryoprecipitate has been removed.U.K.
26. “Granulocytes, apheresis” means a concentrated suspension of granulocytes obtained by apheresis.U.K.
27. “Statistical process control” means a method of quality control of a product or a process that relies on a system of analysis of an adequate sample size without the need to measure every product of the process.U.K.
PART 2U.K.INFORMATION REQUIREMENTS FOR DONORS
Part A – Information to be provided to prospective donors of blood or blood componentsU.K.
1. Accurate educational materials, which are written in terms which can be understood by members of the general public, about the essential nature of blood, the blood donation procedure, the components derived from whole blood and apheresis donations, and the important benefits to patients.U.K.
2. For both allogeneic and autologous donations, the reasons for requiring an examination and health and medical history, and the testing of donations, and the significance of “informed consent”.U.K.
3. For allogeneic donations, the criteria for self-deferral, and temporary and permanent deferral, and the reasons why individuals are not to donate blood or blood components if there could be a risk for the recipient.U.K.
4. For autologous donations, the possibility of deferral and the reasons why the donation procedure would not take place in the presence of a health risk to the individual whether as donor or recipient of the autologous blood or blood components.U.K.
5. Information on the protection of personal data, including confirmation that there will be no disclosure of the identity of the donor, of information concerning the donor's health, and of the results of the tests performed, other than in accordance with the requirements of these Regulations.U.K.
6. The reasons why individuals are not to make donations which may be detrimental to their health.U.K.
7. Specific information on the nature of the procedures involved either in the allogeneic or autologous donation process and their respective associated risks. For autologous donations, the possibility that the autologous blood and blood components may not suffice for the intended transfusion requirements.U.K.
8. Information on the option for donors to change their mind about donating prior to proceeding further, or the possibility of withdrawing or self-deferring at any time during the donation process, without any undue embarrassment or discomfort.U.K.
9. The reasons why it is important that donors inform the blood establishment of any subsequent event that may render any prior donation unsuitable for transfusion.U.K.
10. Information on the responsibility of the blood establishment to inform the donor, through an appropriate mechanism, if test results show any abnormality of significance to the donor's health.U.K.
11. Information as to why unused autologous blood and blood components will be discarded and not transfused to other patients.U.K.
12. Information that test results detecting markers for viruses, such as HIV, HBV, HCV or other relevant blood transmissible microbiologic agents, will result in donor deferral and destruction of the collected unit.U.K.
13. Information on the opportunity for donors to ask questions at any time.U.K.
Part B – Information to be obtained from donors by blood establishments at every donationU.K.
Identification of the donorU.K.
14. Personal data uniquely, and without any risk of mistaken identity, distinguishing the donor, as well as contact details.
Health and medical history of the donorU.K.
15. Health and medical history, provided on a questionnaire and through a personal interview performed by a qualified health professional, that includes relevant factors that may assist in identifying and screening out persons whose donation could present a health risk to others, such as the possibility of transmitting diseases, or health risks to themselves.
Signature of the donorU.K.
16. Signature of the donor, on the donor questionnaire, countersigned by the qualified health professional responsible for obtaining the health history confirming that the donor has—
(a)read and understood the educational materials provided;
(b)had an opportunity to ask questions;
(c)been provided with satisfactory responses to any questions asked;
(d)given informed consent to proceed with the donation process;
(e)been informed, in the case of autologous donations, that the donated blood and blood components may not be sufficient for the intended transfusion requirements; and
(f)acknowledged that all the information provided by the donor is true to the best of his knowledge.
PART 3U.K.ELIGIBILITY CRITERIA FOR DONORS OF WHOLE BLOOD AND BLOOD COMPONENTS
Acceptance criteria for donors of whole blood and blood componentsU.K.
1.
Under exceptional circumstances, individual donations from donors who do not comply with following criteria may be authorised by a qualified healthcare professional in the blood establishment. All such cases must be clearly documented and subject to the quality management provisions in Articles 11, 12 and 13 of Directive 2002/98/EC.
The criteria in this paragraph do not apply to autologous donations.
1.1. Age and body weight of donorsU.K.
| Age | 18 to 65 years | |
|---|---|---|
| 17 years | Where, in the opinion of a qualified health professional, the donor has sufficient knowledge and understanding of what is involved in the process of blood donation to give their informed consent, or otherwise with the written consent of a person with parental responsibility. | |
| First time donors over 60 years | — at the discretion of the doctor in the blood establishment | |
| Over 65 years | — with permission of the doctor in the blood establishment, given annually | |
| Body weight | ≥ 50 kg for donors either of whole blood or apheresis blood components | |
1.2. Haemoglobin levels in donor's bloodU.K.
| Haemoglobin | For females ≥ 125 g/l | For males ≥ 135 g/l | Applicable to allogeneic donors of whole blood and cellular components |
1.3. Protein levels in donor's bloodU.K.
| Protein | ≥ 60 g/l | The protein analysis for apheresis plasma donations must be performed at least annually |
1.4. Platelet levels in donor's bloodU.K.
| Platelets | Platelet number greater than or equal to 150 x 109 /1 | Level required for apheresis platelet donors |
DEFERRAL CRITERIA FOR DONORS OF WHOLE BLOOD AND BLOOD COMPONENTS
Deferral criteria for donors of whole blood and blood componentsU.K.
2.1. Permanent deferral criteria for donors of allogeneic donations
| Cardiovascular disease | Prospective donors with active or past serious cardiovascular disease, except congenital abnormalities with complete cure |
| Central nervous system disease | A history of serious CNS disease |
| Abnormal bleeding tendency | Prospective donors who give a history of a coagulopathy |
| Repeated episodes of syncope, or a history of convulsions | Other than childhood convulsions or where at least three years have elapsed since the date the donor last took anticonvulsant medication without any recurrence of convulsions |
| Gastrointestinal. Genitourinary, haematological, immunological, metabolic, renal, or respiratory system diseases | Prospective donors with serious active, chronic, or relapsing disease |
| Diabetes | If being treated with insulin |
| Infectious diseases | Hepatitis B, except for HBsAg-negative persons who are demonstrated to be immune |
| Hepatitis C | |
| HIV – 1 and 2 | |
| HTLV I/II | |
| Babesiosis (*) | |
| Kala Azar (visceral leishmaniasis) (*) | |
| Trypanosomiasis cruzi (Chagas' disease) (*) | |
| Malignant diseases | Except in situ cancer with complete recovery |
| Transmissible spongiform encephalopathies (TSEs) (e.g. Creutzfeldt Jakob Disease, variant Creutzfeldt Jakob Disease) | Persons who have a family history which places them at risk of developing a TSE, or persons who have received a corneal or dura mater graft, or who have been treated in the past with medicines made from human pituitary glands. For variant Creutzfeldt Jacob disease, further precautionary measures may be recommended. |
| Intravenous (IV) or intramuscular (IM) drug use | Any history of non-prescribed IV or IM drug use, including body-building steroids or hormones |
| Xenotransplant recipients | |
| Sexual behaviour | Persons whose sexual behaviour puts them at high risk of acquiring severe infectious diseases that can be transmitted by blood |
2.2. Temporary deferral criteria for donors of allogeneic donationsU.K.
2.2.1. InfectionsU.K.
Duration of deferral periodU.K.
After an infectious illness, prospective donors shall be deferred for at least two weeks following the date of full clinical recovery.
However, the following deferral periods shall apply for the infections listed in the table:
| Brucellosis (*) | 2 years following the date of full recovery |
| Osteomyelitis | 2 years after confirmed cured |
| Q fever (*) | 2 years following the date of confirmed cure |
| Syphilis (*) | 1 year following the date of confirmed cure |
| Toxoplasmosis (*) | 6 months following the date of clinical recovery |
| Tuberculosis | 2 years following the date of confirmed cure |
| Rheumatic fever | 2 years following the date of cessation of symptoms, unless evidence of chronic heart disease |
Fever >38°C | 2 weeks following the date of cessation of symptoms |
| Flu-like illness | 2 weeks after cessation of symptoms |
Malaria (*) | |
| — individuals who have lived in a malarial area within the first five years of life | 3 years following return from last visit to any endemic area, provided person remains symptom free; may be reduced to 4 months if an immunologic or molecular genomic test is negative at each donation. |
| — individuals with a history of malaria | 3 years following cessation of treatment and absence of symptoms. Donations may be accepted thereafter only if an immunologic or molecular genomic test is negative |
| — asymptomic visitors to endemic areas | 6 months after leaving the endemic area unless an immunologic or molecular genomic test is negative |
| — individuals with a history of undiagnosed febrile illness during or within six months of a visit to an endemic area | 3 years following resolution of symptoms; may be reduced to 4 months if an immunologic or molecular test is negative |
| West Nile Virus (WNV) (*) | 28 days after leaving an area with ongoing transmission of WNV to humans |
2.2.2. Exposure to risk of acquiring a transfusion-transmissible infectionU.K.
— Endoscopic examination using flexible instruments, — mocusal splash with blood or needlestick injury, — transfusion of blood components, — tissue or cell transplant of human origin, — major surgery, — tattoo or body piercing, — acupuncture unless performed by a qualified practitioner and with sterile single-use needles, — persons at risk due to close household contact with persons with hepatitis B. | Defer 6 months, or 4 months provided a NAT test for hepatitis C is negative |
| Persons whose behaviour or activity places them at risk of acquiring infectious diseases that may be transmitted by blood. | Defer after cessation of risk behaviour for a period determined by the disease in question, and by the availability of appropriate tests. |
2.2.3. VaccinationU.K.
| Attenuated viruses or bacteria | 4 weeks |
| Inactivated/killed viruses, bacteria or rickettsiae | No deferral if well |
| Toxoids | No deferral if well |
| Hepatitis A or hepatitis B vaccines | No deferral if well and if no exposure |
| Rabies | No deferral if well and if no exposure If vaccination is given following exposure defer for one year |
| Tick-borne encephalitis vaccines | No deferral if well and if no exposure |
2.2.4. Other temporary deferralsU.K.
| Pregnancy | 6 months after delivery or termination, except in exceptional circumstances and at the discretion of a physician |
| Minor surgery | 1 week |
| Dental treatment | Minor treatment by dentist or dental hygienist – defer until next day (NB: Tooth extraction, root-filling and similar treatment is considered as minor surgery) |
| Medication | Based on the nature of the prescribed medicine, its mode of action an the disease being treated |
2.3. Deferral for particular epidemiological situationsU.K.
| Particular epidemiological situations (e.g. disease outbreaks) | Deferral consistent with the epidemiological situation |
2.4. Deferral criteria for donors of autologous donationsU.K.
| Serious cardiac disease | Depending on the clinical setting of the blood collection |
| Active bacterial infection |
PART 4U.K.STORAGE, TRANSPORT AND DISTRIBUTION CONDITIONS FOR BLOOD AND BLOOD COMPONENTS
1. STORAGEU.K.
1.1. Liquid storageU.K.
| Component | Temperature of storage | Maximum storage time |
|---|---|---|
| Red cell preparations and whole blood (if used for transfusion as whole blood) | +2 to +6°C | 28 to 49 days according to the processes used for collection, processing and storage |
| Platelet preparations | +20 to +24°C | 5 days, may be stored for 7 days in conjunction with detection or reduction of bacterial contamination |
| Granulocytes | +20 to +24°C | 24 hours |
1.2. CryopreservationU.K.
| Component | Storage conditions and duration |
|---|---|
| Red blood cells | Up to 30 years according to processes used for collection, processing and storage |
| Platelets | Up to 24 months according to processes used for collection, processing and storage |
| Plasma and cryoprecipitate | Up to 36 months according to processes used for collection, processing and storage |
| Cryopreserved red blood cells and platelets must be formulated in a suitable medium after thawing. The allowable storage period after thawing to depend on the method used. | |
TRANSPORT AND DISTRIBUTIONU.K.
2. Transport and distribution of blood and blood components at all stages of the transfusion chain must be under conditions that maintain the integrity of the product.
ADDITIONAL REQUIREMENTS FOR AUTOLOGOUS DONATIONSU.K.
3.
3.1. Autologous blood and blood components must be clearly identified as such and stored, transported and distributed separately from allogeneic blood and blood components.U.K.
3.2. Autologous blood and blood components must be labelled as required by regulation 8, and, in addition, the label must include the identification of the donor and the warning “FOR AUTOLOGOUS TRANSFUSION ONLY”.U.K.
PART 5U.K.QUALITY AND SAFETY REQUIREMENTS FOR BLOOD AND BLOOD COMPONENTS
1. THE BLOOD COMPONENTSU.K.
| 1. Red cell preparations | The components listed in points 1.1 to 1.8 may be further processed within blood establishments and must be labelled accordingly |
| 1.1 | Red cells |
| 1.2 | Red cells, buffy coat removed |
| 1.3 | Red cells, leucocyte-depleted |
| 1.4 | Red cells, in additive solution |
| 1.5 | Red cells, buffy coat removed, in additive solution |
| 1.6 | Red cells, leucocyte-depleted, in additive solution |
| 1.7 | Red cells, apheresis |
| 1.8 | Whole blood |
| 2. Platelet preparations | The components listed in points 2.1 to 2.6 may be further processed within blood establishments and must be labelled accordingly |
| 2.1 | Platelets, apheresis |
| 2.2 | Platelets, apheresis, leucocyte-depleted |
| 2.3 | Platelets, recovered, pooled |
| 2.4 | Platelets, recovered, pooled, leucocyte-depleted |
| 2.5 | Platelets, recovered, single unit |
| 2.6 | Platelets, recovered, single unit, leucocyte-depleted |
| 3. Plasma preparations | The components listed in 3.1 to 3.3 may be further processed within blood establishments and must be labelled accordingly |
| 3.1 | Fresh-frozen plasma |
| 3.2 | Fresh-frozen plasma, cryoprecipitate-depleted |
| 3.3 | Cryoprecipitate |
| 4. | Granulocytes, apheresis |
2. QUALITY CONTROL REQUIREMENTS FOR BLOOD AND BLOOD COMPONENTSU.K.
2.1. Blood and blood components must comply with the following technical quality measurements and meet the acceptable results.U.K.
2.2. Appropriate bacteriological control of the collection and manufacturing process must be performed.U.K.
2.3. For autologous donations, the measures marked with an asterisk (*) are recommendations only.U.K.
| Component | Quality measures required | Acceptable results for quality measures |
|---|---|---|
| The required frequency of sampling for all measurements shall be determined using statistical process control | ||
| Red cells | Volume | Valid for storage characteristics to maintain product within specifications for haemoglobin and haemolysis |
| Haemoglobin (*) | Not less than 45g per unit | |
| Haemolysis | Less than 0.8% of red cell mass at end of the shelf life | |
| Red cells, buffy coat removed | Volume | Valid for storage characteristics to maintain product within specifications for haemoglobin and haemolysis |
| Haemoglobin (*) | Not less than 43 g per unit | |
| Haemolysis | Less than 0.8% of red cell mass at the end of the shelf life | |
| Red cells, leucocyte-depleted | Volume | Valid for storage characteristics to maintain product within specifications for haemoglobin and haemolysis |
| Haemoglobin (*) | Not less than 40g per unit | |
| Leucocyte content | Less than 1 x 106 per unit | |
| Haemolysis | Less than 0.8% of red cell mass at the end of the shelf life | |
| Red cells, in additive solution | Volume | Valid for storage characteristics to maintain product within specifications for haemoglobin and haemolysis |
| Haemoglobin (*) | Not less than 45g per unit | |
| Haemolysis | Less than 0.8% of red cell mass at end of the shelf life | |
| Red cells, buffy coat removed, in additive solution | Volume | Valid for storage characteristics to maintain product within specifications for haemoglobin and haemolysis |
| Haemoglobin (*) | Not less than 43g per unit | |
| Haemolysis | Less than 0.8% of red cell mass at the end of the shelf life | |
| Red cells, leucocyte-depleted, in additive solution | Volume | Valid for storage characteristics to maintain product within specifications for haemoglobin and haemolysis |
| Haemoglobin (*) | Not less than 40g per unit | |
| Leucocyte content | Less than 1 x 106 per unit | |
| Haemolysis | Less than 0.8% of red cell mass at the end of the shelf life | |
| Red cells, apheresis | Volume | Valid for storage characteristics to maintain product within specifications for haemoglobin and haemolysis |
| Haemoglobin (*) | Not less than 40g per unit | |
| Haemolysis | Less than 0.8% of red cell mass at the end of the shelf life | |
| Whole blood | Volume | Valid for storage characteristics to maintain product within specifications for haemoglobin and haemolysis 450ml +/- 50ml For paediatric autologous whole blood collections – not to exceed 10.5ml per kg body weight |
| Haemoglobin (*) | Not less than 45g per unit | |
| Haemolysis | Less than 0.8% of red cell mass at the end of the shelf life | |
| Platelets, apheresis | Volume | Valid for storage characteristics to maintain product within specifications for pH |
| Platelet content | Variations in platelet content per single donation are permitted within the limits that comply with validated preparation and preservation conditions | |
| pH | 6.4 -7.4 corrected for 22°C, at the end of the shelf life | |
| Platelets, apheresis, leucocyte-depleted | Volume | Valid for storage characteristics to maintain product within specifications for pH |
| Platelet content | Variations in platelet content per single donation are permitted within the limits that comply with validated preparation and preservation conditions | |
| Leucocyte content | Less than 1 x 106 per unit | |
| pH | 6.4-7.4 corrected for 22°C, at the end of the shelf life | |
| Platelets, recovered, pooled | Volume | Valid for storage characteristics to maintain product within specifications for pH |
| Platelet content | Variations in platelet content per pool are permitted within limits that comply with validated preparation and preservation conditions | |
| Leucocyte content | Less than 0.2 x 109 per single unit (platelet-rich plasma method) Less than 0.05 x 109 per single unit (buffy coat method) | |
| pH | 6.4-7.4 corrected for 22°C, at the end of the shelf life | |
| Platelets, recovered, pooled, leucocyte-depleted | Volume | Valid for storage characteristics to maintain product within specifications for pH |
| Platelet content | Variations in platelet content per pool are permitted within limits that comply with validated preparation and preservation conditions | |
| Leucocyte content | Less than 1 x 106 per pool | |
| pH | 6.4-7.4 corrected for 22°C, at the end of the shelf life | |
| Platelets, recovered, single unit | Volume | Valid for storage characteristics to maintain product within specifications for pH |
| Platelet content | Variations in platelet content per single unit are permitted within limits that comply with validated preparation and preservation conditions | |
| Leucocyte content | Less than 0.2 x 109 per single unit (platelet-rich plasma method) Less than 0.05 x 109 per single unit (buffy coat method) | |
| pH | 6.4-7.4 corrected for 22°C, at the end of the shelf life | |
| Platelets, recovered, single unit, leucocyte-depleted | Volume | Valid for storage characteristics to maintain product within specifications for pH |
| Platelet content | Variations in platelet content per single unit are permitted within limits that comply with validated preparation and preservation conditions | |
| Leucocyte content | Less than 1 x 106 per unit | |
| pH | 6.4-7.4 corrected for 22°C, at the end of the shelf life | |
| Plasma, fresh-frozen | Volume | Stated volume +/- 10% |
| Factor VIIIc(*) | Average (after freezing and thawing): 70% or more of the value of the freshly collected plasma unit | |
| Total protein | Not less than 50g/l | |
| Residual cellular content(*) | Red cells: less than 6.0 x 109/l Leucocytes: less than 0.1 x 109/l Platelets: less than 50 x 109/l | |
| Plasma, fresh-frozen, cryoprecipitate-depleted | Volume | Stated volume +/-10% |
| Residual cellular content(*) | Red cells: less than 6.0 x 109/l Leucocytes: less than 0.1 x 109/l Platelets: less than 50 x 109/l | |
| Cryoprecipitate | Fribrinogen content(*) | Greater than or equal to 140mg per unit |
| Fractor VIIIc content (*) | Greater than or equal to 70 international units per unit | |
| Granulocytes, apheresis | Volume | Less than 500ml |
| Granulocyte content | Greater than 1 x 1010 granulocytes per unit |
[F100PART 6U.K.RECORD OF DATA ON TRACEABILITY
Textual Amendments
F100Sch. Pts. 6-8 inserted (31.8.2006) by The Blood Safety and Quality (Amendment) Regulations 2006 (S.I. 2006/2013), regs. 1(1), 15
A. BY BLOOD ESTABLISHMENTSU.K.
1. Blood establishment identificationU.K.
2. Blood donor identificationU.K.
3. Blood unit identificationU.K.
4. Individual blood component identificationU.K.
5. Date of collection (year/month/day)U.K.
6. Facilities to which blood units or blood components are distributed, or subsequent disposition.U.K.
B. BY FACILITIESU.K.
1. Blood component supplier identificationU.K.
2. Issued blood component identificationU.K.
3. Transfused recipient identificationU.K.
4. For blood units not transfused, confirmation of subsequent dispositionU.K.
5. Date of transfusion or disposition (year/month/day)U.K.
6. Lot number of the component, if relevant.U.K.
PART 7U.K.NOTIFICATION OF SERIOUS ADVERSE REACTIONS
SECTION AU.K.Rapid notification format for suspected serious adverse reactions

SECTION BU.K.Serious adverse reactions – imputability levels
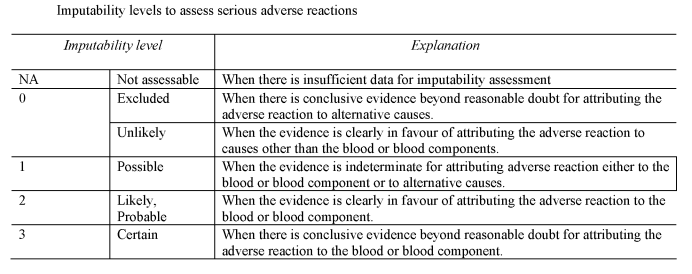
SECTION CU.K.Confirmation format for serious adverse reactions

SECTION DU.K.Annual notification format for serious adverse reactions
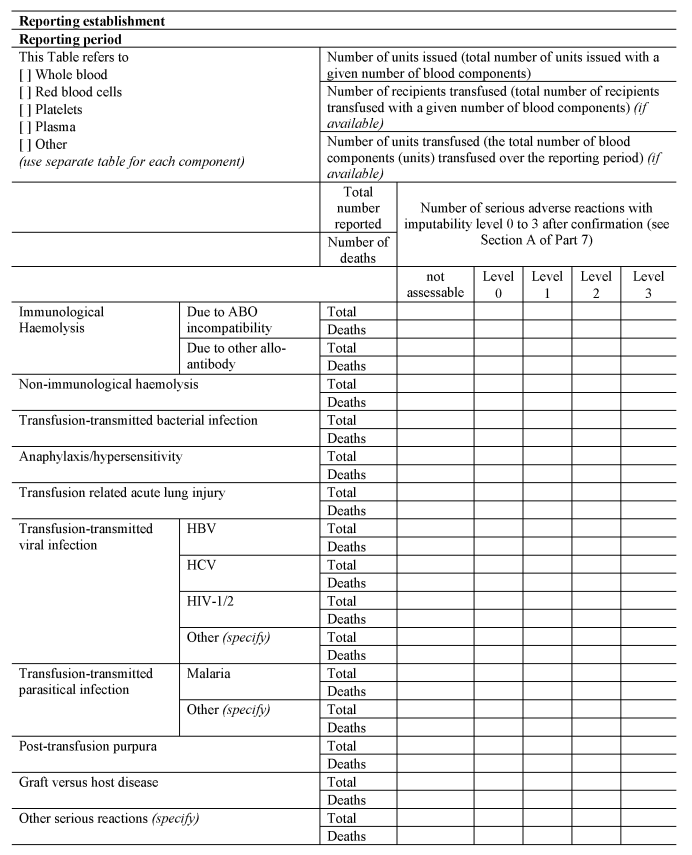
PART 8U.K.NOTIFICATION OF SERIOUS ADVERSE EVENTS
SECTION AU.K.Rapid Notification Format for Serious Adverse Events
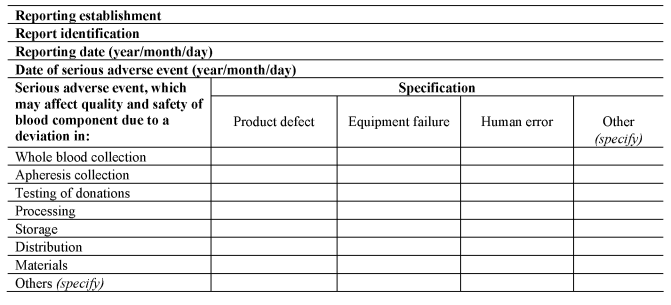
SECTION BU.K.Confirmation Format for Serious Adverse Events

SECTION CU.K.Annual Notification Format for Serious Adverse Events]
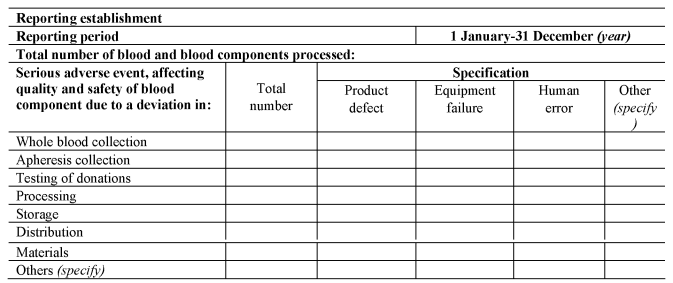
Explanatory Note
(This note is not part of the Regulations)
These Regulations impose safety and quality requirements on human blood collection and storage. The requirements apply to blood transfusion services in England, Scotland, Wales and Northern Ireland. Many of the provisions of the Regulations also apply to hospital blood banks.
The Regulations implement Directive 2002/98/EC of the European Parliament and Council of 27 January 2003 setting out the standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components (“the Directive”) – see OJ L 33, 8.2.2003, p30. They also implement Commission Directive 2004/33/EC – see, OJ L91, 30.3.2004, p25, which contains certain technical requirements relating to blood standards.
Regulation 2 provides that the Secretary of State is to be the competent authority for the purposes of the Directive and outlines the scope of the Regulations.
Regulation 3 prohibits the carrying on of certain activities relating to blood, unless they are a person authorised by the Secretary of State to act as a blood establishment or carried out by hospital blood banks or persons acting on behalf of an authorised blood establishment or a hospital blood bank. Regulation 4 sets out the procedures to be followed in respect of an application for authorisation and regulation 5 sets out the circumstances tin which the Secretary of State may suspend or revoke such authorisation.
Regulations 6 to 8 impose requirements on blood establishments, including requirements relating to “responsible persons” at blood establishments (regulation 6) and the labelling of blood (regulation 8). Regulations 9 and 10 impose requirements on persons responsible for management of hospital blood banks, including requirements to provide information to the Secretary of State (regulation 10). Regulation 11 provides for the service of notices on hospital blood banks by the Secretary of State requiring them to undertake certain actions where they contravene the requirements of these regulations or where there are concerns as to safety.
Regulation 12 makes provision for objections to suspensions and revocations of blood establishment authorisations and to notices served on blood establishments and hospital blood banks by the Secretary of State under regulations 5 and 11.
Regulation 13 prohibits the import of blood or blood components which do not meet the standards of safety and quality equivalent to those specified in Part 5 of the Schedule to the Regulations.
Regulation 14 imposes restrictions on the disclosure of information obtained under the Regulations.
Regulations 15 to 21 provide for enforcement and related matters, including powers of inspection notices to provide information, offences and penalties for breaches of the Regulations.
Regulation 22 provides for fees payable in relation to blood establishment authorisations and inspections of blood establishments and blood banks.
Regulation 23 provides that in the event of a specific epidemiological situation such as a disease outbreak, which necessitates the adoption of deferral criteria additional to those specified in Part 3 of the Schedule, the Secretary of State is to notify the Commission and blood establishments, who are to adopt any additional deferral criteria specified by the Secretary of State.
Regulation 24 makes transitional provision for blood establishments and hospital blood banks so that they may continue to operate under existing provisions until 8th November 2005.
Regulation 25 makes consequential amendments.
A Regulatory Impact Assessment and a Transposition Note have been prepared for these Regulations and a copy of each has been placed in the library of each House of Parliament. Copies of the Regulatory Impact Assessment and the Transposition Note are published on the Department of Health's website (www.dh.gov.uk) and can be obtained from room 631B SKH, Department of Health, Skipton House, 80 London Road, London SE1 6LH.
Options/Help
Print Options
PrintThe Whole Instrument
Legislation is available in different versions:
Latest Available (revised):The latest available updated version of the legislation incorporating changes made by subsequent legislation and applied by our editorial team. Changes we have not yet applied to the text, can be found in the ‘Changes to Legislation’ area.
Original (As Enacted or Made): The original version of the legislation as it stood when it was enacted or made. No changes have been applied to the text.
Point in Time: This becomes available after navigating to view revised legislation as it stood at a certain point in time via Advanced Features > Show Timeline of Changes or via a point in time advanced search.
See additional information alongside the content
Geographical Extent: Indicates the geographical area that this provision applies to. For further information see ‘Frequently Asked Questions’.
Show Timeline of Changes: See how this legislation has or could change over time. Turning this feature on will show extra navigation options to go to these specific points in time. Return to the latest available version by using the controls above in the What Version box.
Opening Options
Different options to open legislation in order to view more content on screen at once
Explanatory Memorandum
Explanatory Memorandum sets out a brief statement of the purpose of a Statutory Instrument and provides information about its policy objective and policy implications. They aim to make the Statutory Instrument accessible to readers who are not legally qualified and accompany any Statutory Instrument or Draft Statutory Instrument laid before Parliament from June 2004 onwards.
More Resources
Access essential accompanying documents and information for this legislation item from this tab. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as enacted version that was used for the print copy
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- correction slips
- links to related legislation and further information resources
Timeline of Changes
This timeline shows the different points in time where a change occurred. The dates will coincide with the earliest date on which the change (e.g an insertion, a repeal or a substitution) that was applied came into force. The first date in the timeline will usually be the earliest date when the provision came into force. In some cases the first date is 01/02/1991 (or for Northern Ireland legislation 01/01/2006). This date is our basedate. No versions before this date are available. For further information see the Editorial Practice Guide and Glossary under Help.
More Resources
Use this menu to access essential accompanying documents and information for this legislation item. Dependent on the legislation item being viewed this may include:
- the original print PDF of the as made version that was used for the print copy
- correction slips
Click 'View More' or select 'More Resources' tab for additional information including:
- lists of changes made by and/or affecting this legislation item
- confers power and blanket amendment details
- all formats of all associated documents
- links to related legislation and further information resources
The data on this page is available in the alternative data formats listed:
 All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.
All content is available under the Open Government Licence v3.0 except where otherwise stated. This site additionally contains content derived from EUR-Lex, reused under the terms of the Commission Decision 2011/833/EU on the reuse of documents from the EU institutions. For more information see the EUR-Lex public statement on re-use.